

Abstract
Live attenuated vaccines for prevention of congenital cytomegalovirus infections encode numerous immune evasion genes. Their removal could potentially improve vaccine safety and efficacy. To test this hypothesis, three genes encoding MHC class I homologs (presumed NK evasins) were deleted from the guinea pig cytomegalovirus genome and the resulting virus, 3DX, was evaluated as a live attenuated vaccine in the guinea pig congenital infection model. 3DX was attenuated in vivo but not in vitro. Vaccination with 3DX produced elevated cytokine levels and higher antibody titers than wild type virus (WT) while avidity and neutralizing titers were similar. Protection, assessed by maternal viral loads and pup mortality following pathogenic viral challenge during pregnancy, was comparable between 3DX and WT and significant compared to naïve animals. These results suggest that the safety and perhaps efficacy of live attenuated human cytomegalovirus vaccines could be enhanced by deletion of viral immunomodulatory genes.
Keywords: Guinea pig cytomegalovirus, cytomegalovirus vaccines, congenital cytomegalovirus infection, animal models, viral immune evasion
1. Introduction
Human cytomegalovirus (HCMV) is ubiquitous in the human population and is estimated to infect ~60% of women of childbearing age in the developed world. Primary infection during pregnancy results in infection of approximately 30–40% of fetuses [1]. Consequently, in the U.S. approximately 40,000 babies are born infected with HCMV each year and of these 5–18% (up to 18,000/year) display sequelae of infection, such as hearing loss or mental retardation [2–5]. Prior immunity to HCMV provides some protection against congenital infection, reducing the incidence of infection to less than 2% [6,7], and in those infants that are infected, prior immunity reduces disease severity [8].
At present there is no licensed vaccine for prevention of HCMV infections. Protective immunity may require both B and T cell-mediated responses [9–12], which makes live-attenuated vaccine strategies attractive insofar as they express antigens that stimulate both the humoral and cellular arms of host immunity. A live vaccine based on the attenuated Towne strain has been extensively tested in humans and has demonstrated efficacy in preventing HCMV disease post-renal transplantation [13–17]. However, the Towne vaccine did not protect mothers from acquiring primary infection from their children who were actively shedding virus [18]. Hence, in this setting Towne appears deficient at eliciting immune responses sufficient to confer protection. Moreover, there remains a theoretical concern that a live attenuated vaccine administered inadvertently to a pregnant woman might itself lead to congenital HCMV infection.
Current live virus vaccine candidates such as Towne encode numerous immune evasion genes that may enhance pathogenesis or impair the vaccine's ability to induce robust host immune responses [19]. That removal of immune evasion genes can promote safety without impairing protection was recently shown by a vaccine/challenge study in which murine cytomegalovirus (MCMV) was rendered avirulent by deletion of 32 immune evasion genes yet induced levels of immunity and protection similar to the parental wild type virus [20].
The ability to evade or impair natural killer (NK) functions may be particularly relevant to congenital infection, as evasion of NK control may be critical for viral dissemination to or transmission across the placenta [21]. Cytomegaloviruses aggressively down-regulate host MHC I from the infected-cell surface [22], and although this would normally result in potent activation of NK cells to secrete IFN-γ and to kill the MHC Ilow infected cells, viral factors termed “NK evasins” attenuate this activation and allow infected cells to escape NK-mediated killing. Moreover, NK evasins presumably diminish the NK-mediated IFN-γ response that occurs at the earliest times of viral infection [23, 24]. Hence, removal of NK evasin genes from a live attenuated vaccine might improve safety by restoring effective NK restriction of viral dissemination. At the same time, unopposed activation of NK cells by MHC Ilow virus-infected cells would increase early IFN-γ levels, which, by enhancing downstream adaptive responses, might ultimately improve vaccine efficacy.
Testing the hypothesis that removal of NK evasin genes will enhance both the efficacy and safety of live attenuated vaccines will initially require in vivo studies using animal models. Since HCMV does not replicate in species other than humans, related cytomegaloviruses such as MCMV, rat cytomegalovirus (RCMV), and guinea pig cytomegalovirus (GPCMV) have been used as in vivo models. HCMV and MCMV encode proteins that have been shown to impair NK activation in vitro [25–30] and/or to play important roles in viral pathogenesis in vivo [31, 32]. Among these are viral-encoded MHC I homologs that are postulated to block NK activation by providing inhibitory signals to NK cells in a manner similar to that of host MHC I. Unfortunately, MCMV and RCMV cannot be used to evaluate vaccines intended to prevent fetal infection or disease as they do not cross the placenta [33–35]. In this respect, GPCMV, which can cross the placenta [36–38], provides an important model system for testing vaccines or other intervention strategies aimed at preventing congenital cytomegalovirus infection or for elucidating the roles of specific viral factors in congenital transmission and pathogenesis [39–45].
In order to identify potential NK evasins encoded by GPCMV, the sequence of the GPCMV genome [46] was examined for open reading frames (ORFs) that could encode proteins having either sequence or predicted structural homologies to MHC I. Three ORFs designated gp147, gp148, and gp149 were identified as encoding potential MHC I homologs, designated gp147, gp148, and gp149, respectively [46]. These ORFs lie adjacent to one another near the right terminus of the GPCMV genome (Fig. 1a).
Fig. 1.

(a) Hind III map of the GPCMV genome (top) with the Hind III L - M region enlarged below to show the locations of ORFs encoding MHC I homologs gp147, gp148 and gp149 in the WT genome and their replacement with kanr/lacZ in virus 3DX. Thick bars indicate sequences used as hybridization probes. (b) Southern hybridizations of Hind III- and EcoR I-restricted BAC DNAs hybridized with kanr/lacZ or gp148 probes. Positions of DNA size markers are shown on the left. (c) Hind III- and Nhe I-restricted WT and 3DX virion DNAs separated on 0.6% agarose and visualized with ethidium bromide and UV light. Arrows indicate diagnostic fragments; sizes of DNA markers are shown on the right.
In this study the importance of gp147, gp148, and gp149 was examined in vivo. A recombinant virus, 3DX, deleted of gp147–149, was found to replicate with wild-type kinetics in vitro but was essentially avirulent in vivo. Even so, immunization with the 3DX vaccine induced potent humoral immunity that protected pups from congenital infection, with efficacy similar to that observed following immunization with parental wild-type (WT) virus. These results demonstrate that preconception immunization with a live, highly attenuated CMV vaccine, engineered to delete putative immune modulation genes, is capable of conferring protection against congenital infection and disease in the guinea pig model.
2. Methods
2.1. Animal studies
Hartley guinea pigs were purchased from Harlan Laboratories (Indianapolis, IN) or Elm Hill Laboratories (Chelmsford, MA). All animals were determined to be GPCMV seronegative prior to vaccination by ELISA screening [42]. Animals were housed under conditions approved by the American Association of Accreditation of Laboratory Animal Care, in accordance with institutional animal use committee approved protocols at the University of Minnesota, Minneapolis.
2.2. Virus and cells
GPCMV strain 22122 (ATCC VR682) was propagated in guinea pig lung fibroblast (GPL) cells (ATCC CCL 158) and maintained in F-12 media supplemented with 10% fetal calf serum (FCS; HyClone Laboratories), 10,000 IU/L penicillin, 10 mg/L streptomycin and 7.5% NaHCO3. BAC-derived viruses were also propagated in GPL cells, maintained in minimum essential media (Gibco-BRL) supplemented as described above.
2.3. Construction of mutant virus 3DX lacking the gp148 – gp149 region
Linear recombination was used to delete the gp148 – gp149 region from BAC pN13R10, which contains the GPCMV strain 22122 genome [47]. Two regions of the GPCMV genome, approximately 500 bp upstream and downstream of the gp147–gp149 gene cluster, were PCR-amplified using primers 3D1F (5'GTCGGGCGATAACATGTAAGG) (forward, left side) and 3D2R (5'AGATGCAGTACTGCGGCCGCAACGACAGAGACTATGAGGGA) (reverse, left side) or 3D3F (5'CGCCGGCGAGTACTGCATCTCATCGAGGACAACTTTTGGGT) (forward, right side) and CIM-R (5'GCTAGCAAGAATCCTTGAAGAAGAAT) (reverse, right side). The two products were then annealed through homologous non-viral sequences that were added to create a Not I restriction site (underlined in 3D2R and 3D3F) and PCR-amplified using primers 3D1F and CIM-R to generate a 1-kb product comprised of a Not I restriction site flanked by the two regions of viral sequence homology. This product was T/A cloned into pCR®8/GW/TOPO® (Invitrogen) to produce plasmid pGP238. A marker gene cassette encoding kanamycin-resistance and lacZα(kanr/lacZ) was excised from pYD-Tn1721 [48] (kindly provided by Dong Yu) by Not I digestion and ligated into Not I-digested pGP238 to create pGP239. SW102 E. coli cells (the kind gift of Søren Warming) [49] containing pN13R10 were induced to express phage λ recombinases by growth at 42 °C for 5 min, then transformed with the PCR product generated by amplification of pGP239 using primers 3D1F and CIM-R. Candidate clones were selected as blue kanamycin-resistant colonies and screened for the predicted Hind III restriction pattern (data not shown). The loss of the gp148 – gp149 sequences (nucleotides 223464 – 230728 as numbered in: Accession number FJ355434) [46] was confirmed by Southern blot hybridization (Fig. 1b) and PCR (data not shown). One BAC clone was selected and designated pN13R10-3DX.
2.4. Viral reconstitution and growth curve analysis
Infectious viruses were reconstituted by transfection of GPL cells with BAC DNAs as described previously [47]. In both BACs the BAC origin of replication can be excised by co-transfection with plasmid pCre (constructed by Wolfram Brune and kindly provided by Gabriele Hahn). As a green fluorescent protein (GFP) marker gene lies within the excised sequences, properly excised viruses can be isolated by limiting-dilution and screening for lack of GFP expression [47]. Virus 3DX was obtained by co-transfection of pN13R10-3DX with pCre and isolation of a GFP-negative virus, while a GFP-negative wild type virus (WT) was similarly derived from the parental BAC pN13R10. For neutralizing studies a GFP-positive 3DX virus (3DX-GFP) was derived by transfection of pN13R10-3DX without pCre. Virion DNAs were prepared as previously described [47] and digested with Hind III and Nhe I to confirm the expected restriction patterns for WT and 3DX (Fig. 1c) and for 3DX-GFP (not shown). Multi-step growth curves were conducted using an MOI of 0.01 as described previously [47].
2.5. Southern Hybridization
Hind III- or EcoR I-digested pN13R10 and pN13R10-3DX BAC DNAs were subjected to electrophoretic separation on a 0.6% agarose gel, stained with ethidium bromide, and the fragments visualized by UV light. DNA fragments were then transferred to a nitrocellulose membrane (Schleicher and Schuell), UV cross-linked (0.12 J/cm2), and hybridized as described previously [50]. Probes kanr/lacZ and gp148 consisted of gel purified EcoR I fragments that were derived from plasmids pGP238 and pGP258 (respectively) and labeled with [32P]-dCTP by random hexamer priming (Boehringer Mannheim) [50]. Plasmid pGP258 was constructed by PCR-amplification of a 500-bp region internal to ORF gp148 from pN13R10 BAC DNA using primers L1-F (5' TCTGAACATCGCGACGATC) and L1-R (5' TCAGATGGTTCG CGATGAC) and TOPO-cloning the product into vector pCR-TOPO-XL (Invitrogen). Approximately 106 CPM of each probe were used for hybridization.
2.6 FACSCAN analysis of MHC class I surface expression
GPL cells were infected with either WT-GFP or 3DX-GFP (MOI=0.1) viruses and were analyzed for class I surface expression at 72 h post infection as described previously [51] except that the murine monoclonal antibody B640, a gift from Hubert Schafer, was used to detect guinea pig MHC I (1/100 dilution). For MHC I down regulation analysis a GFP-positive wild-type GPCMV was used as a control [51]. Mouse IgG isotype control (Biolegend) was used as a control for B640 MHC I antibody.
2.7. Vaccine/challenge study outline
Vaccinations were administered both intraperitoneally (1 ml) and subcutaneously (neck, 1 ml) with 5×107 plaque forming units (pfu) at each site. Following immunization, animals were bled by toenail clip on days 5, 8, 13, 18, 25, 33 and 64 post immunization. Female guinea pigs were mated 64 days post immunization with seronegative proven breeder males and were subsequently monitored for pregnancy by palpation [52]. Pregnant guinea pigs were challenged in the third trimester with subcutaneous administration of 1×105 pfu of a pathogenic salivary gland passaged stock of GPCMV shown in prior studies to result in a 50–70% pup mortality rate when administered to GPCMV- naïve pregnant dams [53]. Females were bled on day 7 post-challenge for DNAemia analysis. Pups were sacrificed within three days of delivery and blood was collected by cardiac puncture for DNAemia analysis.
2.8. Quantitative Real-Time PCR analysis
DNA was extracted from 200 μL citrated whole blood using the MagNA Pure LC System (Roche) on days 5, 8, 13, 18, 25 and 33 following vaccination. The GPCMV gB specific primer pair LCF1 (5'CTTCGTGGTTGAACGGG) and LCR1 (5'GTAGTCGAAAGGACGTTGC) was utilized for the real-time PCR assay. The PCR reaction was performed in a 20 μl volume reaction as specified by the LightCycler® FastStart DNA Master HybProbe reaction mix (Roche Diagnostics). PCR was performed using the LightCycler 480 Real-Time PCR System (Roche) under the following conditions: initial denaturation at 95 °C for 10 min, followed by 95 °C for 10 s, 54 °C for 15 s, 72 °C for 15 s for a total of 50 cycles, followed by melting curve analysis at 95 °C for 1 min and 45 °C for 1 min and ending at 85 °C, then a final hold step at 40 °C. Data were analyzed with the LightCycler Data Analysis Software (version 1.0; Roche) using standard curves generated using serial dilutions of plasmid and viral DNA at known concentrations. The magnitude of DNAemia was expressed as the total number of genome copies per ml of blood, with results representing the average of 2 independently performed assays.
2.9. Serologic assays
Serum was analyzed by ELISA for the presence of anti-GPCMV IgG antibodies as described previously [42] using sonicated, clarified GPCMV-infected GPL cell lysates as the coating antigen. ELISA titers were calculated as the reciprocal of the highest dilution that produced an absorbance of at least 0.1 and twice the absorbance produced by negative control antigen prepared from uninfected GPL cells.
IgG avidity indices were determined by measuring ELISA OD450 values in the presence or absence of 4 mM urea, then calculating the OD450 (with urea)/OD450 (without urea) ratio and multiplying by 100.
Neutralizing titers were determined by preparing triplicate 2-fold dilutions (1:16 to 1:32,786) of each serum in 96-well plates, adding 100 μL culture medium containing 2×103 pfu 3DX-GFP to each well, incubating at 37 °C for 1 h, then transferring 100 μL of each virus/serum mixture to confluent GLF cells in white-walled flat-bottomed 96-well plates (Costar). GFP fluorescence was measured at 7 days post-infection using a PerkinElmer Victor2 1420 Multilable Counter. Fifty percent inhibitory concentration (IC50) values were calculated from GFP values as described previously [54].
2.10. Reverse transcriptase PCR
Reverse transcriptase-PCR was performed as described previously [55]. RNA was extracted from 200 μL citrated blood using the MagNA Pure LC System. Primer pairs gpIFN-γ1 (5' GACCTGAGCAAGACCCTGAG) and gpIFN-γ2 (5' GCCATTTCGCCTGACATATT) specific for guinea pig IFN-γ, gpIL-12 (5' TCTGAGCCGGTCACAACTGC) and gpIL-12-2 (5'AGGCGCTGTCCTCCTGACAC) specific for the guinea pig IL-12 p40 subunit, or gpGAPDH (5' GGGCAAGGTCATCCCAGAG) and gpGAPDH2 (5' TGGAAGAATGGCTGTCACTGTT) specific for guinea pig GAPDH, were used in a 20 μL One Step RT-PCR reaction (Qiagen). Two μL (10 ng) of RNA was used in each reaction. Each PCR reaction was initiated with a first step at 50 °C for 35 min followed by 15 min at 95 °C followed by 37 cycles of 94 °C for 20 s, 56 °C for 20 s and 72 °C for 40 s. For the final step, the reaction mixture was heated at 72 °C for 10 min. Amplified DNA was separated on a 2% agarose gel containing ethidium bromide. Relative units for IFN-γ or IL-12 mRNA expression were determined by measuring signal sum intensities using KODAK Molecular Imaging software and dividing the IFN-γ or IL-12 sum intensities by the GAPDH sum intensities.
2.11. Statistical analyses
Proportions of live-born pups and GPCMV PCR+ dams were compared using Fisher's exact test. Nonparametric variables were compared by Mann-Whitney U test. Statistics were analyzed by the InStat program (version 3.0; GraphPad software).
3. Results
3.1. Construction and in vitro characterization of virus 3DX
Analyses conducted in silico using the predicted primary amino acid sequences of gp147, gp148, and gp149 determined that each has sequence as well as potential structural homology to MHC I [46; Table 1]. The MHC class I heavy (α) chain is made up of α1, α2 and α3 domains, where α1 and α2 form the extracellular peptide binding cleft and α3 associates with β2-microglubin, which makes up membrane proximal portion of MHC class I. Sequence alignment and secondary structure prediction software (Jpred) was used to compare Cavia porcellus MHC class (gp MHC I) gp147 and gp149 amino acid sequences (Fig. 2). The gp148 was not included because it is not predicted to have string structural similarity based on 3D-PSSM (Table 1). Alignment of gp147 amino acid sequence with gp MHC I demonstrates that gp147 appears to encode for α1, α2, and α3 extracellular domains similar to MHC class I (Fig 2a). Alignment of gp149 amino acid sequence with gp MHC I demonstrates that gp149 appears to encode for α1, α2, and has the potential to encode for the α3 domain, which contains a string of β-sheets (Fig 2b). These alignments suggest that gp147 and gp149 have the potential to be structurally similar to MHC class I based on analysis of primary amino acid sequence. Importantly, there are several conserved cysteine residues as well as several conserved tyrosines suggesting similarities in tertiary structure. Additionally, NetNGlyc analysis predicts 10 potential N-linked glycosylation sites in the extracellular domain in both gp147 and gp149 suggesting that these proteins have potential to be heavily glycosylated.
Table 1.
E-values* for viral MHC-I homologs
| MHC-I homolog | size (aa) | BLASTP† (sequence homology) | 3D-PSSM‡ (structural homology) | ||
|---|---|---|---|---|---|
|
| |||||
| E-value | MHC-I (sequence) | E-value | MHC-I (structure) | ||
| UL18§ | 368 | 4e−18 | human (CAL85437.2) | 6e−6 | human (ABA41587) |
| m144§ | 383 | 6e−9 | mouse (AAA39553) | 1e−4 | mouse (AAR11190 |
| UL142∥ | 306 | n.s. | not applicable | 0.15 | human (ABV71649) |
| m157∥ | 331 | n.s. | not applicable | 0.089 | mouse (AAR11192) |
| gp147 | 520 | 0.4 | tuatara (ABA42599) | 0.83 | human (AAA98727.1) |
| gp148 | 483 | 8e−8 | rhesus (BAG69572) | 2.3 | mouse (CAA26586.1) |
| gp149 | 627 | n.s. | not applicable | 0.49 | mouse (AAA53200.1) |
| cI-M (aa 133–339) | 207 | 0.049 | platypus (XP001511789) | 0.04 | human (ABY26532.1) |
E-values reflect the probability that sequence or structural similarity between two proteins is random. E-values < 1 indicate a significant probability that a similarity is non-random and this probability increases with decreasing E-value; n.s., no significant similarity
known MHC-I sequence homologs
known MHC-I structural homologs
Fig. 2.

Structure-based alignment of gp147 and gp149 with Cavia porcellus MHC I predicting α1, α2, and α3 domains in the GPCMV MHC I homologs. ClustalW alignments of gp147 (ACJ35890) and gp149 (ACJ35892) with Cavia porcellus MHC I (gp MHC I, Ensemble protein # ENSCPOP00000002441). Secondary structure of (a) gp147 and (b) gp149 was predicted using the Jpred online server. Predicted β-strands are depicted by arrows and α-helices are depicted by solid line. The conserved cysteines are bold and underlined, conserved tyrosines are marked with an asterisk, and potential N-linked glycosylation sites are underlined. The single N-linked glycosylation site in gp MHC I is underlined.
As MHC I homologs encoded by HCMV and MCMV have been shown to impair NK activation in vitro [25–30], we hypothesized that gp147, gp148, and gp149 represent GPCMV NK evasins. To determine if deleting these genes would alter pathogenesis of the virus in vivo or its immunogenicity or efficacy when used as a live attenuated vaccine, we constructed a mutant GPCMV lacking all three MHC I homologs.
BAC mutagenesis in E. coli was used to delete the gp148 – gp149 region from the pN13R10 BAC clone of the GPCMV genome [47] and replace it with a kanr-lacZ cassette to produce BAC pN13R10-3DX (Fig. 1a). The correct structure of pN13R10-3DX was verified by Hind III digestion and PCR (data not shown) and by Southern hybridization (Fig. 1b). A kanr/lacZ probe specific to the inserted sequences hybridized to 5.5- and 1.3-kb Hind III or 6.0- and 30.7-kb EcoR I fragments from pN13R10-3DX but did not hybridize to fragments from pN13R10. These results are consistent with the predicted structure of pN13R10-3DX, as in the circular BAC the 5.5-kb Hind III fragment represents a fusion of the left (2.6-kb) and right (2.9-kb) terminal Hind III fragments expected for linear viral DNA, while the 6.0-kb EcoR I fragment represents a fusion of the left (2.6-kb) and right (3.4-kb) terminal EcoR I fragments. Rehybridization of the same membrane with the gp148 probe detected the internal 6.1-kb Hind III L fragment and a 42.2-kb fusion of the left (2.6-kb) and right (39.6-kb) EcoR I terminal fragments from pN13R10 but failed to hybridize to any sequences in pN13R10-3DX. These results indicate that pN13R10-3DX has the predicted structure and that coding sequences for gp147, gp148, and gp149 have been deleted.
Transfection of pN13R10-3DX DNA into GPL cells reconstituted a virus that appeared to replicate efficiently. Thus, gp147, gp148, and gp149 are not essential for in vitro replication. Stocks of WT (derived from pN13R10) and 3DX (derived from pN13R10-3DX), in which the BAC origin was excised, were prepared as described in materials and methods. The Hind III and Nhe I restriction patterns of WT and 3DX virion DNAs are shown in Fig. 1c. As predicted, Hind III L (6.1 kb) and M (4.9 kb) fragments containing the three homologs in WT were replaced by 1.3-kb and 2.9-kb Hind III fragments in 3DX, and a 10.5-kb Nhe I fragment in WT was replaced by 2.9- and 2.1-kb fragments in 3DX (Fig. 1c). These data confirmed that the predicted modifications occurred and that gp147, gp148, and gp149 were deleted from the 3DX genome.
It was of interest to examine the impact of the gp147–149 deletion on the ability of GPCMV to down-regulate class I expression. To verify that 3DX retained the ability to downregulate MHC I from the surface of infected cells, GPL cells were infected with either WT-GFP (Fig 3a) or 3DX-GFP (Fig 3b) at a multiplicity of infection of 0.1 and harvested 72 hours post infection. Cells were analyzed by FACSCAN for surface expression of MHC-I (PE) and GFP, which indicates infected cells. These results demonstrate that gp147, gp148 and gp149 are not involved in MHC I downregulation.
Fig. 3.
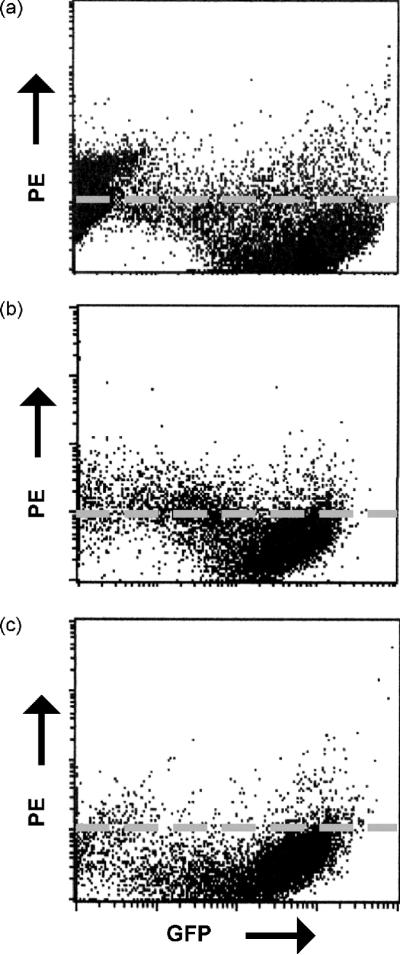
ORFS encoding gp147, gp148 and gp149 do not appear to be involved in MHC-I down-regulation. GPL cells were infected with WT-BAC (a) or 3DX (b) at an MOI of 0.1 for 72 h. Control in panel (c), mouse IgG isotype control for B640 antibody. Cells were analyzed by FACSCAN for surface expression of MHC-I (PE) and GFP. No impact of deletion of the gp147-149 region is noted on MHC-1 down-regulation.
To determine if gp147, gp148, and gp149 are important viral replication in vitro, replicate cell cultures were infected at a multiplicity of infection of 0.01 using carefully titer-matched inocula of WT and 3DX and culture supernatants were titered up to 18 days post infection. The resulting growth curves for WT and 3DX were essentially superimposable, demonstrating that in cell culture, deletion of gp147, gp148, and gp149 has no detectable impact on viral growth efficiency or kinetics (Fig. 4). These results are compatible with a putative role for gp147, gp148, and gp149 in immune evasion insofar as immune evasion genes in other cytomegaloviruses are often dispensable for replication in cultured cells.
Fig. 4.
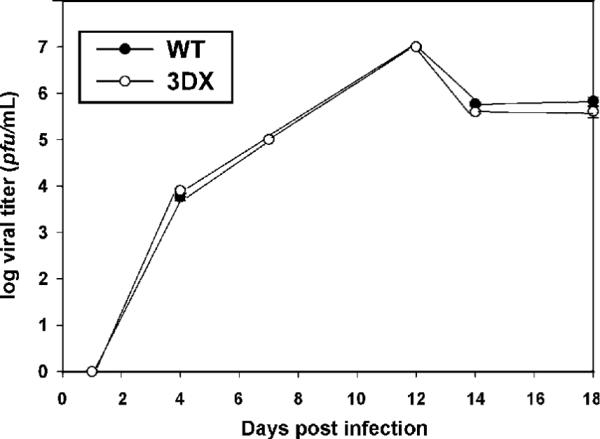
Sequences encoding gp148, gp147 and gp149 are not required for efficient viral growth in vitro. Cells were infected with WT (25CF) or 3DX (25EF) at an MOI of 0.01. Viral titers in the culture supernatants were determined on the days indicated post infection. Results represent the means of triplicate titers and error bars indicate ± one standard deviation.
3.2. 3DX is attenuated in vivo
To assess the effects that removal of the three MHC I homologs has on viral pathogenesis, seronegative female outbred Hartley guinea pigs were inoculated with 5×107 pfu subcutaneously and 5×107 pfu intraperitoneally (WT, n=7; 3DX, n=8). Guinea pigs were bled on days 5, 8, 13, 18 and 25 post-inoculation. DNAemia was quantified by qPCR with primers specific to the GPCMV gB gene. The magnitude of DNAemia following inoculation with 3DX was below the 102 genome copies per ml of blood limit of detection at all times post inoculation, whereas up to 1×105 genome copies/ml were detected in all WT-inoculated animals (Fig. 5). These results suggest that the inability to express the three MHC I homologs severely attenuates the ability of 3DX to disseminate in the blood. We have also determined that following infection with either WT or 3DX, both were detected at similar levels in the liver and spleen of infected animals (data not shown).
Fig. 5.
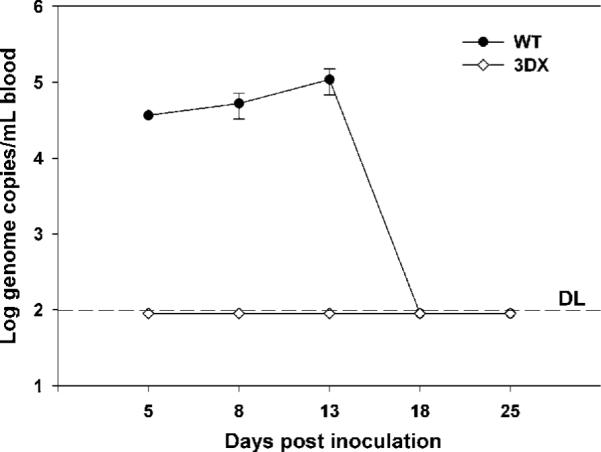
Genes encoding gp147, gp148 and gp149 are important for DNAemia. Seronegative outbred Hartley strain female guinea pigs were inoculated IP and SC with 1×108 total pfus of WT or 3DX. Blood viral loads were measured by real-time PCR and DNAemia expressed as mean genome copies per ml. Data points are means of data from duplicate blood samples drawn from 7 WT-infected animals or 8 3DX-infected animals. Error bars indicate ± one standard deviation. DL, detection limit.
Weight loss or gain was used as an indirect measure of pathogenesis. Five days post-inoculation WT-inoculated guinea pigs had lost an average of 6.14 ± 3.6 g whereas 3DX-inoculated animals had gained an average of 9.6 ± 3.4 g (p<0.02). On subsequent days WT-inoculated animals recovered and began gaining weight but remained below those of the 3DX inoculated group. The latter differences, however, were not statistically significant.
Taken together these results suggest that removal of MHC class I homologs renders GPCMV less pathogenic in vivo.
3.3 Inoculation with 3DX results in altered expression kinetics of type 1 cytokines
The early stages of viral infection are marked by activation of NK cells and dendritic cells to produce IFN-γ and IL-12, respectively. The degree of this response has been shown to impact down-stream events, including a second phase of IFN-γ release by T cells and NK cells and ultimately the development of protective adaptive immunity [56]. Therefore, if viral MHC I homologs function like host MHC I to send inhibitory signals that counteract the activating signals that are received by NK cells upon encountering infected MHC Ilow cells, a virus that retains the ability to down-regulate host MHC I but is deleted of its MHC I homologs might induce robust NK activation. We have determined that 3DX retains the ability to down regulate MHC I in vitro (Fig. 3). The resulting elevated IFN-γ and/or IL-12 responses at early times might, in the long-term, promote more robust adaptive responses. To test this hypothesis, RT-PCR was used to measure IFN-γ and IL-12 mRNA levels in the blood of animals inoculated with WT or 3DX (Fig 6). Blood mRNA levels for IFN-γ were elevated in 3DX- vs. WT-infected animals on days 5 (0.95 vs 0.86; p<0.05) and 8 (0.71 vs 0.61, p<0.05) post inoculation (Fig. 6a). Although peak IL-12 levels were similar for the two groups and both returned to baseline by day 33 post inoculation, peak IL-12 mRNA levels occurred much earlier in 3DX-infected animals (day 8 for 3DX vs. day 18 for WT) (Fig. 6b). These results are consistent with the above hypothesis in that removal of the three MHC I homologs increased IFN-γ levels and accelerated the kinetics of IL-12 expression during acute infection.
Fig. 6.

Deletion of gp147, gp148 and gp149 alters IFN-γ and IL-12 responses. Levels of IFN-γ, IL-12, and GAPDH mRNA in whole blood from 3DX- and WT- inoculated animals were measured by RT-PCR. Relative units were calculated as ratios of IFN-γ (a) or IL-12 (b) sum intensities to GAPDH sum intensities. Data shown represent means ± SEM (standard error of the mean) (uninfected, n=5; WT, n = 7; 3DX, n=8). *p < 0.05 or **p < 0.005, Mann-Whitney U test.
3.4. 3DX induces robust antibody responses
The above hypothesis further predicts that deletion of MHC I homologs may enhance the development of adaptive responses. Hence humoral responses induced by 3DX and WT were evaluated. All animals in both groups seroconverted following viral inoculation. On day 64 post-inoculation the mean antibody ELISA titers were 3.5-fold higher for the 3DX group (1:5,495 for 3DX vs. 1:1,548 for WT) and this difference was statistically significant (p< 0.02, Mann-Whitney U test) (Fig. 7a). Mean avidity indices for sera from 3DX-inoculated animals matured from 43.2% on day 25 to 54.5% on day 64, while the avidity of sera from WT-inoculated animals matured with similar avidities from 42.8% on day 25 to 48.7% on day 64 (Fig. 7b). Finally, both 3DX and WT elicited high neutralizing antibody titers that were indistinguishable (1:576 for 3DX vs. 1:592 for WT) (Fig. 7c). Together these results indicate that although 3DX is attenuated for pathogenesis and dissemination in vivo, it retains the ability to induce antibody responses that are comparable, even superior, to those induced by WT virus.
Fig. 7.

Anti-GPCMV IgG ELISA titers (a), avidity (b), or neutralizing titers (c) were measured in sera obtained from 3DX- and WT-inoculated animals on the indicated days post inoculation. **p<0.02, Mann-Whitney U test.
3.5. Pregnancy outcomes in viral-challenge study
A challenge experiment was performed to assess the efficacy of 3DX as a vaccine to protect against adverse pregnancy outcomes associated with GPCMV infection. Two months after the animals described above were immunized by inoculation with WT or 3DX the dams were mated and challenged in mid-third trimester of pregnancy with 1×105 pfu of virulent salivary gland-passaged GPCMV. Dams that were seronegative at the time of challenge had a pup mortality of 44%, compared to 29% for dams that were naturally seropositive. The protection provided by prior immunization with either WT or 3DX was on par with that provided by natural infection, with resulting pup mortalities of 22% (3DX) and 23% (WT). Although the size for this study was not sufficient to achieve statistical significance for either vaccine alone, when data from WT and 3DX immunization groups were combined the mean pup mortality of 22.6% was highly significant compared to the seronegative controls (Fisher's exact test; p < 0.02; see Table 2).
Table 2.
Pup mortality in vaccine study
| Vaccine group | Live | Dead | Mortality (%) |
|---|---|---|---|
| Control (n = 12 dams) | 26 | 20 | 43 |
| WT (n = 7 dams) | 20 | 6 | 23 |
| 3DX (n = 7 dams) | 21 | 6 | 22 |
| WT + 3DX (n = 14 dams) | 41 | 12 | 22.6* |
| Preconception infection (GPCMV seropositive; n = 5 dams) | 10 | 4 | 29 |
p < 0.02 vs. control Fisher's exact test
3.6. Impact of 3DX vaccination on maternal and pup viral load
Previous studies in the guinea pig model have demonstrated that the magnitude of maternal DNAemia is one of the best predictors of pup outcome following maternal GPCMV challenge [52, 57]. To assess the impact of prior immunization with 3DX or WT on post-challenge DNAemia, qPCR was performed on maternal blood 7 days after challenge. Immunization with WT virus significantly reduced the incidence of DNAemia seven days post challenge (Table 3). While 83% of dams in the seronegative control group were positive for viral DNA in the blood, 14% in the WT- and 57% in 3DX group were positive. While the effect on prevalence of DNAemia was statistically significant only for the WT group (p < 0.005; Fisher's exact test), immunization with either virus dramatically reduced the magnitude of viral load. For both WT and 3DX the effect was highly significant (Table 3). Thus both immunization strategies are effective in reducing viral load in pregnant GPCMV-challenged dams. The incidence of DNAemia in live born pups was also similar for the two immunization groups (Table 3).
Table 3.
DNAemia in dams and pups in vaccine study
| Dams | Live born pups | ||||
|---|---|---|---|---|---|
| Group | +/total (%) | DNAemiaa | litters | Total | + (%) |
|
|
|
||||
| Control | 10/12 (83) | 1.95 × 105 | 10 | 26 | not tested |
| WT | 1/7 (14)** | 4.12 × 102* | 7 | 20 | 0 (0) |
| 3DX | 4/7 (57) | 9.12 × 102** | 7 | 21 | 2 (9) |
p < 0.02 vs. control; Fisher's exact test
p < 0.005 vs. control; Fisher's exact test
logarithmic means of genome copies/ml blood
4. Discussion
Cytomegaloviruses commit a considerable portion of their genetic capacity to expression of proteins involved in immune evasion. These functions may be highly relevant to live attenuated CMV vaccines as they have the potential to influence both vaccine safety and efficacy. In particular, viral dissemination to the placenta may depend on the ability of the virus to escape NK control by expressing NK evasins, and attenuation of NK activation may lessen subsequent development of adaptive immunity [58]. Although NK evasins encoded by HCMV and MCMV have been characterized [25–30, 32], the species specificity of HCMV and the lack of congenital transmission in the mouse and rat prevents investigation of the roles that such NK evasins may play in congenital infection or the induction of protective immunity by live vaccines.
Although GPCMV is congenitally transmitted, the molecular biology of GPCMV and the immunology of guinea pigs are poorly developed. At the present time we lack adequate tools to determine if certain GPCMV genes function as NK evasins. However, given that other viral MHC I homologs function as NK evasins, it is a reasonable (though unproven) hypothesis that one or more of the GPCMV MHC I homologs serves a similar function. Both sequence and predicted structural comparisons of the GPCMV gp147–149 ORFs suggested that these represent bona fide MHC I homologs (Table 1, Fig. 2). Thus, the guinea pig model provided an opportunity to determine if MHC I homologs (as putative NK evasins) are important for vaccine efficacy or viral pathogenesis.
Many viral immune evasion genes are non-essential for viral growth in vitro; it was therefore not surprising that in vitro the replication of 3DX is indistinguishable from that of WT. In vivo, however, one or more of the MHC I homologs is clearly important for pathogenesis. 3DX infection failed to induce the transient weight loss that was evident in WT-infected animals, and, in contrast to WT, 3DX DNA was never detected in the blood. This latter result is potentially important for a live vaccine as it implies that removal of NK evasins could render a live vaccine less likely to disseminate to the placenta and subsequently to infect the fetus. We cannot at present confirm that improved NK control is responsible for the reduced virulence of 3DX; however, our results are consistent with the hypothesis that enhanced NK activation by 3DX-infected cells at the initial site of infection limits the subsequent spread of virus to the blood.
A second area of considerable interest for live vaccines is the role that NK evasins may have in attenuating host immune responses by reducing IFN-γ levels produced at early times of infection. Recent studies have demonstrated that early activation of NK cells to release IFN-γ plays an important role in the long-term development of protective adaptive immunity [59, 60]. Hence, removal of viral genes that impair NK activation could enhance adaptive immunity induced by live vaccines. While we cannot as yet determine if, as MHC I homologs, gp147, gp148, and gp149 function as NK evasins, we did observe elevated IFN-γ expression at early times following 3DX infection. Moreover, that this elevation was evident by day 5 post-infection, prior to the typical time required for T cell activation, suggests that the elevated IFN-γ was NK-derived [61]. This is consistent with the hypothesis that NK activation (and consequently IFN-γ expression) will be elevated after 3DX infection because 3DX can down-regulate host MHC I but cannot counteract the resulting NK activation signals by expressing viral MHC I homologs. Alternatively, the IL-12 response also occurred earlier in 3DX- vs. WT-infected animals. It is possible that impaired dissemination increased the local replication of 3DX, which then stimulated dendritic cells to produce an earlier IL-12 response. This may then have activated NK cells, and perhaps at later times T cells, to produce IFN-γ [62]. Additional studies will be needed to dissect the mechanism of altered cytokine responses to 3DX.
Assessing the downstream impact of removing MHC I homologs on adaptive immunity focused on humoral immunity as assays to measure virus-specific T cell responses in the guinea pig are still under development. Given that 3DX appears to be highly attenuated in vivo, it was perhaps surprising that 3DX infection elicited virus-specific antibody titers that were comparable and at a late time 3.5-fold higher than those induced by WT virus. While virus/host interactions in vivo are complex, it is reasonable to speculate that elevated IFN-γ levels shortly after infection had a long-term impact on B cell development that resulted in elevated antibody titers two months later. The quality of the antibody response was also unaffected by the attenuation of 3DX in vivo. Neutralizing titers induced by 3DX were equal to those induced by WT virus, while the avidity of antibodies induced by both viruses matured with similar kinetics. In this respect 3DX resembles the Towne vaccine, as both induce antibody responses similar to wild type infection in the absence of apparent systemic infection [18]. With respect to vaccine efficacy, a robust antibody response is clearly important, as prior studies have shown that passive administration of GPCMV-specific antibodies with high neutralizing activity can prevent pup infection and reduce pup mortality [39], while in humans efficacy of CMV-hyperimmune globulin has been reported for both prevention and treatment of congenital disease [63].
As a vaccine, immunization with the attenuated 3DX was comparable to immunization with WT virus. Vaccination with either 3DX or WT had a similar and significant effect of reducing maternal DNAemia following challenge. Both vaccines had a similar effect on reducing pup mortality after challenge. Although the study size was too small to show statistical significance for either vaccine alone, the efficacy of vaccination (irrespective of virus strain) was highly significant in reducing pup mortality.
That gp147, gp148, or gp149 function as NK evasins remains to be determined. Such studies await development of assays to measure guinea pig NK cell responses to suitable target cells that can either be infected with wild type or mutant viruses or engineered to express the viral proteins in question. Methods to detect, isolate, and propagate guinea pig NK cells are also currently inadequate. Further development of guinea pig NK biology would greatly facilitate studies to determine the impact of GPCMV MHC I homologs on NK functions both in vitro and in vivo. Although studies in non-pregnant guinea pigs demonstrate a high degree of attenuation of the 3DX virus, further studies are also needed to determine if 3DX is defective in placental infection or congenital transmission.
These results are the first to examine the impact of removing immune evasion genes on the efficacy of vaccines to protect against congenital CMV disease. They imply that live attenuated vaccines that express NK evasins may, by attenuating the initial NK response, give rise to suboptimal adaptive responses, and conversely, that removal of NK evasin genes can potentiate NK activation and IFN-γ release, giving rise to more robust adaptive immune responses and perhaps increasing vaccine efficacy. The results support further studies of this novel approach to live vaccine development and suggest that live HCMV vaccines altered to remove NK evasin genes should be considered as potential candidates for clinical evaluation.
Acknowledgments
The authors are grateful to Dong Yu for providing plasmid pYD-Tn1721, to Søren Warming for providing SW102 E. coli cells, and to Jodi Anderson and Michael Leviton for their technical assistance. We also thank Julie McVoy for providing the Excel program used for curve fitting and IC50 determinations. This work was supported by grant R01HD044864 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Stagno S, Pass RF, Cloud G, Britt WJ, Henderson RE, Walton PD, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. Jama. 1986;256(14):1904–1908. [PubMed] [Google Scholar]
- [2].Bradford RD, Cloud G, Lakeman AD, Boppana S, Kimberlin DW, Jacobs R, et al. Detection of cytomegalovirus (CMV) DNA by polymerase chain reaction is associated with hearing loss in newborns with symptomatic congenital CMV infection involving the central nervous system. J Infect Dis. 2005;191(2):227–233. doi: 10.1086/426456. [DOI] [PubMed] [Google Scholar]
- [3].Damato EG, Winnen CW. Cytomegalovirus infection: perinatal implications. J Obstet Gynecol Neonatal Nurs. 2002;31(1):86–92. doi: 10.1111/j.1552-6909.2002.tb00026.x. [DOI] [PubMed] [Google Scholar]
- [4].Nelson CT, Demmler GJ. Cytomegalovirus infection in the pregnant mother, fetus, and newborn infant. Clin Perinatol. 1997;24(1):151–160. [PubMed] [Google Scholar]
- [5].Wong A, Tan KH, Tee CS, Yeo GS. Seroprevalence of cytomegalovirus, toxoplasma and parvovirus in pregnancy. Singapore Med J. 2000;41(4):151–155. [PubMed] [Google Scholar]
- [6].Stagno S, Reynolds DW, Huang ES, Thames SD, Smith RJ, Alford CA. Congenital cytomegalovirus infection. N Engl J Med. 1977;296(22):1254–1258. doi: 10.1056/NEJM197706022962203. [DOI] [PubMed] [Google Scholar]
- [7].Schopfer K, Lauber E, Krech U. Congenital cytomegalovirus infection in newborn infants of mothers infected before pregnancy. Arch Dis Child. 1978;53(7):536–539. doi: 10.1136/adc.53.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326(10):663–667. doi: 10.1056/NEJM199203053261003. [DOI] [PubMed] [Google Scholar]
- [9].Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202(5):673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Elkington R, Walker S, Crough T, Tammik C, Frascaroli G, Landini MP, et al. Ex vivo profiling of CD8+-T-cell responses to human cytomegalovirus reveals broad and multispecific reactivities in healthy virus carriers. J Virol. 2003;77(9):5226–5240. doi: 10.1128/JVI.77.9.5226-5240.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Varani S, Cederarv M, Feld S, Tammik C, Frascaroli G, Landini MP, et al. Human cytomegalovirus differentially controls B cell and T cell responses through effects on plasmacytoid dendritic cells. J Immunol. 2007;179(11):7767–7776. doi: 10.4049/jimmunol.179.11.7767. [DOI] [PubMed] [Google Scholar]
- [12].Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29(4):650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Plotkin SA, Smiley ML, Friedman HM, Starr SE, Fleisher GR, Wlodaver C, et al. Towne-vaccine-induced prevention of cytomegalovirus disease after renal transplants. Lancet. 1984;1(8376):528–530. doi: 10.1016/s0140-6736(84)90930-9. [DOI] [PubMed] [Google Scholar]
- [14].Balfour HH, Jr, Sachs GW, Welo P, Gehrz RC, Simmons RL, Najarian JS. Cytomegalovirus vaccine in renal transplant candidates: progress report of a randomized, placebo-controlled, double-blind trial. Birth Defects Orig Artic Ser. 1984;20(1):289–304. [PubMed] [Google Scholar]
- [15].Brayman KL, Dafoe DC, Smythe WR, Barker CF, Perloff LJ, Naji A, et al. Prophylaxis of serious cytomegalovirus infection in renal transplant candidates using live human cytomegalovirus vaccine. Interim results of a randomized controlled trial. Arch Surg. 1988;123(12):1502–1508. doi: 10.1001/archsurg.1988.01400360072012. [DOI] [PubMed] [Google Scholar]
- [16].Plotkin SA, Starr SE, Friedman HM, Brayman K, Harris S, Jackson S, et al. Effect of Towne live virus vaccine on cytomegalovirus disease after renal transplant. A controlled trial [see comments] Ann Intern Med. 1991;114(7):525–531. doi: 10.7326/0003-4819-114-7-525. [DOI] [PubMed] [Google Scholar]
- [17].Plotkin SA, Higgins R, Kurtz JB, Morris PJ, Campbell DA, Jr, Shope TC, et al. Multicenter trial of Towne strain attenuated virus vaccine in seronegative renal transplant recipients. Transplantation. 1994;58(11):1176–1178. [PubMed] [Google Scholar]
- [18].Adler SP, Starr SE, Plotkin SA, Hempfling SH, Buis J, Manning ML, et al. Immunity induced by primary human cytomegalovirus infection protects against secondary infection among women of childbearing age. J Infect Dis. 1995;171(1):26–32. doi: 10.1093/infdis/171.1.26. [DOI] [PubMed] [Google Scholar]
- [19].Powers C, DeFilippis V, Malouli D, Fruh K. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol. 2008;325:333–359. doi: 10.1007/978-3-540-77349-8_19. [DOI] [PubMed] [Google Scholar]
- [20].Cicin-Sain L, Bubic I, Schnee M, Ruzsics Z, Mohr C, Jonjić S, et al. Targeted deletion of regions rich in immune-evasive genes from the cytomegalovirus genome as a novel vaccine strategy. J Virol. 2007;81(24):13825–13834. doi: 10.1128/JVI.01911-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cauda R, Prasthofer EF, Grossi CE, Whitley RJ, Pass RF. Congenital cytomegalovirus: immunologic alterations. J Med Virol. 1987;23:41–9. doi: 10.1002/jmv.1890230106. [DOI] [PubMed] [Google Scholar]
- [22].Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3(11):1019–1025. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- [23].Braud VM, Tomasec P, Wilkinson GW. Viral evasion of natural killer cells during human cytomegalovirus infection. Curr Top Microbiol Immunol. 2002;269:117–129. doi: 10.1007/978-3-642-59421-2_8. [DOI] [PubMed] [Google Scholar]
- [24].Lopez-Botet M, Angulo A, Guma M. Natural killer cell receptors for major histocompatibility complex class I and related molecules in cytomegalovirus infection. Tissue Antigens. 2004;63(3):195–203. doi: 10.1111/j.1399-0039.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- [25].Cretney E, Degli-Esposti MA, Densley EH, Farrell HE, Davis-Poynter NJ, Smyth MJ. m144, a murine cytomegalovirus (MCMV)-encoded major histocompatibility complex class I homologue, confers tumor resistance to natural killer cell-mediated rejection. J Exp Med. 1999;190(3):435–444. doi: 10.1084/jem.190.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kubota A, Kubota S, Farrell HE, Davis-Poynter N, Takei F. Inhibition of NK cells by murine CMV-encoded class I MHC homologue m144. Cell Immunol. 1999;191(2):145–151. doi: 10.1006/cimm.1998.1424. [DOI] [PubMed] [Google Scholar]
- [27].Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, et al. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med. 1998;187(10):1681–1687. doi: 10.1084/jem.187.10.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Reyburn HT, Mandelboim O, Vales-Gomez M, Davis DM, Pazmany L, Strominger JL. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature. 1997;386(6624):514–517. doi: 10.1038/386514a0. [DOI] [PubMed] [Google Scholar]
- [29].Vales-Gomez M, Browne H, Reyburn HT. Expression of the UL16 glycoprotein of Human Cytomegalovirus protects the virus-infected cell from attack by natural killer cells. BMC Immunol. 2003;4:4. doi: 10.1186/1471-2172-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, et al. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J Immunol. 2005;175(11):7457–7465. doi: 10.4049/jimmunol.175.11.7457. [DOI] [PubMed] [Google Scholar]
- [31].Bubic I, Wagner M, Krmpotic A, Saulig T, Kim S, Yokoyama WM, et al. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78(14):7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, et al. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature. 1997;386(6624):510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- [33].Schleiss MR. Animal models of congenital cytomegalovirus infection: an overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV) J Clin Virol. 2002;25(Suppl 2):S37–49. doi: 10.1016/s1386-6532(02)00100-2. [DOI] [PubMed] [Google Scholar]
- [34].Loh HS, Mohd-Lila MA, Abdul-Rahman SO, Kiew LJ. Pathogenesis and vertical transmission of a transplacental rat cytomegalovirus. Virol J. 2006;3:42. doi: 10.1186/1743-422X-3-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Woolf NK, Jaquish DV, Koehrn FJ. Transplacental murine cytomegalovirus infection in the brain of SCID mice. Virol J. 2007;4:26. doi: 10.1186/1743-422X-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kumar ML, Nankervis GA. Experimental congenital infection with cytomegalovirus: a guinea pig model. J Infect Dis. 1978;138(5):650–654. doi: 10.1093/infdis/138.5.650. [DOI] [PubMed] [Google Scholar]
- [37].Griffith BP, McCormick SR, Fong CK, Lavallee JT, Lucia HL, Goff E. The placenta as a site of cytomegalovirus infection in guinea pigs. J Virol. 1985;55(2):402–409. doi: 10.1128/jvi.55.2.402-409.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Johnson KP. Mouse cytomegalovirus: placental infection. J Infect Dis. 1969;120(4):445–450. doi: 10.1093/infdis/120.4.445. [DOI] [PubMed] [Google Scholar]
- [39].Chatterjee A, Harrison CJ, Britt WJ, Bewtra C. Modification of maternal and congenital cytomegalovirus infection by anti-glycoprotein b antibody transfer in guinea pigs. J Infect Dis. 2001;183(11):1547–1553. doi: 10.1086/320714. [DOI] [PubMed] [Google Scholar]
- [40].Bravo FJ, Cardin RD, Bernstein DI. Effect of maternal treatment with cyclic HPMPC in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis. 2006;193(4):591–597. doi: 10.1086/499603. [DOI] [PubMed] [Google Scholar]
- [41].Schleiss MR, Anderson JL, McGregor A. Cyclic cidofovir (cHPMPC) prevents congenital cytomegalovirus infection in a guinea pig model. Virol J. 2006;3:9. doi: 10.1186/1743-422X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schleiss MR, Stroup G, Pogorzelski K, McGregor A. Protection against congenital cytomegalovirus (CMV) disease, conferred by a replication-disabled, bacterial artificial chromosome (BAC)-based DNA vaccine. Vaccine. 2006;24(37–39):6175–6186. doi: 10.1016/j.vaccine.2006.06.077. [DOI] [PubMed] [Google Scholar]
- [43].Schleiss MR. Nonprimate models of congenital cytomegalovirus (CMV) infection: gaining insight into pathogenesis and prevention of disease in newborns. Ilar J. 2006;47(1):65–72. doi: 10.1093/ilar.47.1.65. [DOI] [PubMed] [Google Scholar]
- [44].Schleiss MR, Lacayo JC, Belkaid Y, McGregor A, Stroup G, Rayner J, et al. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis. 2007;195(6):789–798. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- [45].Schleiss MR. Comparison of vaccine strategies against congenital CMV infection in the guinea pig model. J Clin Virol. 2008;41(3):224–230. doi: 10.1016/j.jcv.2007.10.008. [DOI] [PubMed] [Google Scholar]
- [46].Schleiss MR, McGregor A, Choi KY, Date SV, Cui X, McVoy MA. Analysis of the Nucleotide Sequence of a BAC-Derived Clone of the Guinea Pig Cytomegalovirus (GPCMV) Genome. Virology Journal. 2008;5:139. doi: 10.1186/1743-422X-5-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cui X, McGregor A, Schleiss MR, McVoy MA. Cloning the complete guinea pig cytomegalovirus genome as an infectious bacterial artificial chromosome with excisable origin of replication. J Virol Methods. 2008;149(2):231–239. doi: 10.1016/j.jviromet.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu D, Smith GA, Enquist LW, Shenk T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol. 2002;76(5):2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33(4):e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].McVoy MA, Adler SP. Human cytomegalovirus DNA replicates after early circularization by concatemer formation, and inversion occurs within the concatemer. J Virol. 1994;68(2):1040–51. doi: 10.1128/jvi.68.2.1040-1051.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lacayo J, Sato H, Kamiya H, McVoy MA. Down-regulation of surface major histocompatibility complex class I by guinea pig cytomegalovirus. J Gen Virol. 2003;84:75–81. doi: 10.1099/vir.0.18675-0. [DOI] [PubMed] [Google Scholar]
- [52].Schleiss MR, Lacayo J. The Guinea-Pig Model of Congenital CMV Infection. In: Reddehase MJ, Lemmermann N, editors. Cytomegaloviruses: Molecular Biology and Immunology. Horizon Scientific Press; 2006. pp. 525–50. [Google Scholar]
- [53].Schleiss MR, Bourne N, Stroup G, Bravo FJ, Jensen NJ, Bernstein DI. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein B vaccine. J Infect Dis. 2004;189(8):1374–1381. doi: 10.1086/382751. [DOI] [PubMed] [Google Scholar]
- [54].Cui X, Meza BP, Adler SP, McVoy MA. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine. 2008;26:5760–5766. doi: 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Allen SS, McMurray DN. Coordinate cytokine gene expression in vivo following induction of tuberculous pleurisy in guinea pigs. Infect Immun. 2003;71:4271–4277. doi: 10.1128/IAI.71.8.4271-4277.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mailliard RB, Son YI, Redlinger R, Coates PT, Giermasz A, Morel PA, et al. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol. 2003;171(5):2366–2373. doi: 10.4049/jimmunol.171.5.2366. [DOI] [PubMed] [Google Scholar]
- [57].Schleiss MR, Bourne N, Bernstein DI. Preconception vaccination with a glycoprotein B (gB) DNA vaccine protects against cytomegalovirus (CMV) transmission in the guinea pig model of congenital CMV infection. J Infect Dis. 2003;188(12):1868–1874. doi: 10.1086/379839. [DOI] [PubMed] [Google Scholar]
- [58].Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–32. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- [59].Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124(4):849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- [60].Zingoni A, Sornasse T, Cocks BG, Tanaka Y, Santoni A, Lanier LL. NK cell regulation of T cell-mediated responses. Mol Immunol. 2005;42(4):451–454. doi: 10.1016/j.molimm.2004.07.025. [DOI] [PubMed] [Google Scholar]
- [61].Bukowski JF, Woda BA, Welsh RM. Pathogenesis of murine cytomegalovirus infection in natural killer cell-depleted mice. J Virol. 1984;52:119–28. doi: 10.1128/jvi.52.1.119-128.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Biron CA, Brossay L. NK cells and NKT cells in innate defense against viral infections. Curr Opin Immunol. 2001;13(4):458–464. doi: 10.1016/s0952-7915(00)00241-7. [DOI] [PubMed] [Google Scholar]
- [63].Adler SP, Nigro G, Pereira L. Recent advances in the prevention and treatment of congenital cytomegalovirus infections. Semin Perinatol. 2007;31(1):10–18. doi: 10.1053/j.semperi.2007.01.002. [DOI] [PubMed] [Google Scholar]