

Abstract
Alpha synuclein (SYN) is a central player in the pathogenesis of sporadic and familial Parkinson's disease (PD). SYN aggregation and oxidative stress are associated and enhance each other's toxicity. It is unknown whether the redox-sensitive transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) plays a role against the toxicity of SYN. To examine this, mice selectively overexpressing Nrf2 in astrocytes (GFAP-Nrf2) were crossed with mice selectively expressing human mutant SYN (hSYNA53T) in neurons. Increased astrocytic Nrf2 delayed the onset and extended the life span of the hSYNA53T mice. This correlated with increased motor neuron survival, reduced oxidative stress, and attenuated gliosis in the spinal cord, as well as a dramatic decrease in total hSYNA53T and phosphorylated (Ser129) hSYNA53T in Triton-insoluble aggregates. Furthermore, Nrf2 in astrocytes delayed chaperone-mediated autophagy and macroautophagy dysfunction observed in the hSYNA53T mice. Our data suggest that Nrf2 in astrocytes provides neuroprotection against hSYNA53T-mediated toxicity by promoting the degradation of hSYNA53T through the autophagy-lysosome pathway in vivo. Thus, activation of the Nrf2 pathway in astrocytes is a potential target to develop therapeutic strategies for treating pathologic synucleinopathies including PD.
Introduction
Parkinson's Disease (PD) is the second most common neurodegenerative disorder, and is characterized by dopaminergic neuron loss in substantial nigra pars compacta (SNpc). Multiple mechanisms contribute to PD pathogenesis with a clear involvement of oxidative stress and mitochondria dysfunction in disease progression. Recently, accumulating evidence has demonstrated that misfolded proteins and inclusions contribute to the pathology of familial and sporadic PD. Alpha synuclein (SYN) is the main component of these inclusions (Spillantini et al., 1997; Irizarry et al., 1998). Missense mutations (A53T, A30P, E64K) in SYN are associated with autosomal dominant PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Singleton et al., 2003). SYN is natively unfolded but is prone to form fibrils under disease conditions. SYN and phospho-SYN-positive aggregates have been observed in the familial PD patients (Braak et al., 2003; Tamura et al., 2012). Increasing evidence implicates the autophagy-lysosome pathway (ALP) in mediating the clearance of misfolded proteins and impaired organelles in neurodegenerative diseases. A recent report suggests that SYN can be degraded through the ubiquitin-proteasome system (UPS) under normal conditions and ALP is recruited under extra SYN burden (Ebrahimi-Fakhari et al., 2011).
Chaperone-mediated autophagy (CMA) has been shown to clear wild-type SYN. SYN binds with heat shock cognate 70 (Hsc70) that then binds to the lysosomal transmembrane protein Lamp2a facilitating subsequent lysosomal degradation. SYN mutants actually bind to Lamp2a with higher affinity blocking their own degradation as well as other CMA substrates (Cuervo et al., 2004). Macroautophagy results in the formation of autophagic vacuoles with trapped misfolded proteins and damaged organelles. When CMA is impaired, macroautophagy is activated to compensate for reduced protein clearance (Honegger et al., 2006). SYN can also be degraded through macroautophagy in neuronal cells in vitro (Vogiatzi et al., 2008) and overexpression of SYN impairs macroautophagy (Winslow et al., 2010).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is an antioxidant transcription factor; under basal conditions Nrf2 is associated with actin-bound Keap1 in cytoplasm. Keap1 targets Nrf2 to the Cullin3 E3 ligase complex for ubiquitination and subsequent degradation by the UPS. Oxidative stress and/or exposure to electrophilic compounds cause the release and nuclear translocation of Nrf2. In complex with small Maf proteins Nrf2 binds with the antioxidant-responsive elements (ARE) inducing transcriptional activation of downstream genes encoding phase II detoxifying enzymes and antioxidants (Itoh et al., 1997; Motohashi et al., 2002). Previous studies demonstrate that astrocyte-specific overexpression of Nrf2 reduces chemical-mediated neurotoxicity modeling PD and Huntington's disease (Chen et al., 2009; Calkins et al., 2010), as well as genetically induced motor neuron degeneration in models of amyotrophic lateral sclerosis (Vargas et al., 2008).
Reports indicate that oxidative stress can enhance SYN toxicity, induce autophagic cell death, and cause abnormal modification of proteins involved in autophagy (Zheng et al., 2009; Chew et al., 2011). Recent work shows that Nrf2 deficiency can exacerbate SYN aggregation in mice using an adeno-associated viral vector-expressing human SYN (Lastres-Becker et al., 2012). In Drosophila overexpressing SYN, genetically increasing Nrf2 restored locomotor activity (Barone et al., 2011). However, the effect of Nrf2 activation on synucleinopathy and SYN clearance by ALP in rodent models has not been examined. We hypothesized that overexpression of Nrf2 will protect against hSYNA53T toxicity. This was examined by crossing GFAP-Nrf2 mice with Thy1-hSYNA53T mice.
Materials and Methods
Mouse strains.
Thy1-hSYNA53T transgenic mice were purchased from The Jackson Laboratory [Strain Name: B6.Cg-Tg(THY1-SNCA*A53T) M53Sud/J; Stock Number: 008135]. The line was created and characterized by Dr. Thomas C. Südhof's laboratory (Chandra et al., 2005; Gallardo et al., 2008). The ARE-hPAP transgenic reporter mouse line was created using the 51 bp rat NADPH:quinone oxidoreductase (NQO1) promoter containing the core ARE sequence (ARE-hPAP) (Johnson et al., 2002). hSYNA53T mice were bred with ARE-hPAP mice to generate hSYNA53T positive/negative and ARE-hPAP-positive mice [hSYNA53T (+)/hPAP(+), hSYNA53T(−)/hPAP(+)]. Transgenic mice with astrocyte-specific overexpression of Nrf2 were made under the control of the human glial fibrillary acidic protein (GFAP) promoter (GFAP-Nrf2) (Vargas et al., 2008). hSYNA53T mice were bred with GFAP-Nrf2 mice to generate hSYNA53T(+)/GFAP-Nrf2(+) double transgenic mice [AN(+/+)], littermate wild-type hSYNA53T(−)/GFAP-Nrf2(−) mice [AN(−/−)], littermate hSYNA53T(+)/GFAP-Nrf2(−) mice [AN(+/−)], and littermate hSYNA53T(−)/GFAP-Nrf2(+) mice [AN(−/+)]. hSYNA53T heterozygous mice were maintained on C57BL/6J background. GFAP-Nrf2 mice were maintained on B6/SJL mice (The Jackson Laboratory). Thus, these crossbred mice were on a mixed B6/SJL background. Both male and female mice were used. End stage was determined by the inability of the mouse to lift itself within 20 s when placed on its both sides. The disease onset was determined by the peak body weight. Mice were weighed once a week and group housed with food and water ad libitum and with a 12 h light/dark cycle. All animal procedures were approved by the University of Wisconsin-Madison Institutional Animal Care and Use Committee. All experiments were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Western blotting.
The procedures were performed as previously described (Gan et al., 2010). For detergent-soluble and -insoluble fractions: tissues were lysed in lysis buffer (50 mm Tris-HCl, pH7.4, 150 mm NaCl, 2 mm EDTA, 1 mm dithiothreitol, 1 mm Na3VO4, 1 mm NaF, and protease inhibitor mixture tablet from Roche) containing 1% Triton X-100 (Buffer A) as the ratio of 1:10 (1 mg of tissue:10 μl of lysis buffer), and whole lysates were centrifuged at 100,000 g, 30 min at 4°C. The supernatants were taken as Triton-soluble fractions. The pellets were rinsed with Buffer A five times, and then resuspended in lysis buffer containing Buffer A, 1% SDS and 0.5% sodium deoxycholate. After sonication and brief spin down, the lysates were taken as Triton-insoluble (SDS-soluble) fractions. For LC3 Western blot, tissues were directly lysed in lysis buffer containing 1% SDS. The dilutions for primary antibodies: α SYN (4B12) 1:10,000 (Abcam), Ser129-phospho-α SYN 1:2000 (Abcam), CathepsinD (CatD), 1:2000 (Santa Cruz Biotechnology), NQO1 1:1000 (Abcam), β-actin 1:5000 (Abcam), tubulin 1:5000 (hybridoma, University of Iowa), GADPH 1:3000 (Cell Signaling Technology), HSC70 1:3000 (Epitomics), Lamp2a 1:1000 (Abcam), microtuble-associated protein light chain 3 (LC3) 1:2000 (Cell Signaling Technology), ubiquitin 1:1000 (Cell Signaling Technology), sequestosome 1 (SQSTM1, P62) 1:4000 (Abnova), myocyte enhancer factor 2D (MEF2D) 1:2000 (BD Biosciences), glutamate-cysteine ligase catalytic subunit (GCLc) and modifier subunit (GCLm) 1:10000 (provided by Dr. Terry Kavanagh, University of Washington, Seattle, WA).
Immunofluorescence and histochemistry.
Mice were perfused with PBS followed by 4% paraformaldehyde (PFA). Tissues were postfixed for 2 h at 4°C and cryoprotected in 30% sucrose in PBS at 4°C until sinking to the bottom. Coronal sections were prepared using a cryostat (Leica). Sections were permeabilized and blocked with 10% goat serum, 1% bovine serum albumin (BSA), and 0.36% Triton X-100 in PBS for 1 h at room temperature (RT), then incubated with primary antibodies diluted in 10% goat serum, 1% BSA in PBS at 4°C overnight. Appropriate IgG was used for controls. The following day, sections were incubated with secondary antibodies diluted in the same solution for 1 h at RT after intensive wash. For nuclear DNA staining, slides were incubated in 1 μg/ml Hoechst 33258 in PBS for 5 min. Primary antibodies were used at the following dilutions: α SYN (LB509) 1:1000 (Abcam), Ser129-phospho-α SYN 1:500 (Abcam), β-III-tubulin 1:1000 (Abcam), GFAP 1:1000 (Dako), ionized calcium binding adaptor molecular (IBA1) 1:600 (Wako), 4-hydroxynonenal (HNE) 1:500 (Abcam). MEF2D 1:500 (BD Sciences), p62 1:500 (Abnova), and ubiquitin (UV-1) 1:250 (Life Science). Secondary antibodies were using Alex Fluor 488 and 594 (Invitrogen) with dilution of 1:1000. For motor neuron count, cresyl violet acetate staining was performed. Every sixth section (10 μm) was stained and a total of 32–40 sections were counted from each mouse using ImageJ.
Real-time PCR.
mRNA extraction, reverse transcription, real-time PCR performance and data analysis were conducted as previously described (Gan et al., 2010). IBA1 primers: 5′-CGAATGCTGGAGAAACTTGG-3′, 5′-AGAGTAGCTGAACGTCTCCT-3′. The rest of primer sequences were referenced (Johnson et al., 2010).
Glutathione measurement.
To determine the levels of reduced (GSH) and oxidized (GSSG) glutathione, it was measured by normal-phase (ion exchange) HPLC (Fariss and Reed, 1987). Supernatants were alkylated with iodoacetic acid followed by derivatization with 1-flouro-2,4, dinitrobenzene. The S-carboxymethyl-N-dinitrophenyl-derivatized samples were then loaded onto a 3-aminopropyl-Spherisorb column (Waters Corporation) and separated by a sodium acetate/methanol gradient using a Shimadzu Prominence HPLC system. Resolved GSH and GSSG analyte was detected at 365 nm using a Shimadzu Prominence SPD-20A UV/Vis detector. GSH and GSSG concentrations in the samples were determined by reference GSH and GSSG standard curves included in each run and subsequently normalized to respective tissue protein concentrations.
Electron microscopy.
Mice were perfused with PBS followed by 4% PFA, and the spinal cord lumbar area was dissected and postfixed in 2% glutaraldehyde and 2% PFA in PBS for overnight. The primary fixed samples were rinsed five times for 5 min in PBS, and postfixed in 1% osmium tetroxide, 1% potassium ferrocyanide in PBS for 1 h at RT, and then rinsed in PBS as before. After dehydration, samples were infiltrated in the mixtures of PolyBed 812 (Polysciences) and propylene oxide with increasing concentrations. The samples were sectioned on a Leica EM UC6 ultramicrotome at 90 nm, and collected on Cu, pioloform-coated 2 × 1 oval slot grids (EMS) followed with poststaining in uranyl acetate and lead citrate. The sections from different genotypes of mice were viewed at 80 kV on a Philips CM120 transmission electron microscope, equipped with MegaView III camera (Olympus Soft Imaging System).
Statistical analysis.
All data are represented as mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Newman–Keuls multiple paired comparisons (GraphPad prism 4). The Kaplan–Meier survival curves were compared by log rank test. Statistical significance was claimed as if p < 0.05.
Results
Endogenous Nrf2-ARE pathway is activated in hSYNA53T mice
The hSYNA53T mice used in these studies show motor dysfunction, become paralyzed, and die between 6 and 10 months of age (Chandra et al., 2005; Gallardo et al., 2008). To test if the endogenous Nrf2-ARE pathway was activated in hSYNA53T mice and involved in the pathology of this mouse model, mRNA levels of Nrf2 and its regulated genes were measured at 2 and 6 months of age in nonsymptomatic mice. Compared with age-matched hSYNA53T-negative mice, NQO1 increased in spinal cord at 6 months. GCLm increased significantly at 2 months but not at 6 months (Fig. 1A). Correlating with mRNA data, the protein level of NQO1 increased significantly in spinal cord at 6 months, and there were no changes for GCLc and GCLm (Fig. 1B). GSH levels were slightly increased in the spinal cord of 2-month-old mice, but not 6-month-old mice (Fig. 1C). There was no significant change of GSSG in spinal cord and brainstem at both ages (Fig. 1D). hSYNA53T mice were also bred with ARE-hPAP mice and hPAP activity was examined through histochemical staining (Fig. 1E). Increased intensity for hPAP staining was observed in ventral horn of spinal cord of 6-month-old mice (Fig. 1E) but not 2-month-old mice (data not shown). More modest increases in hPAP histochemistry were observed in substantia nigra, amygdala, and brainstem (data not shown).
Figure 1.
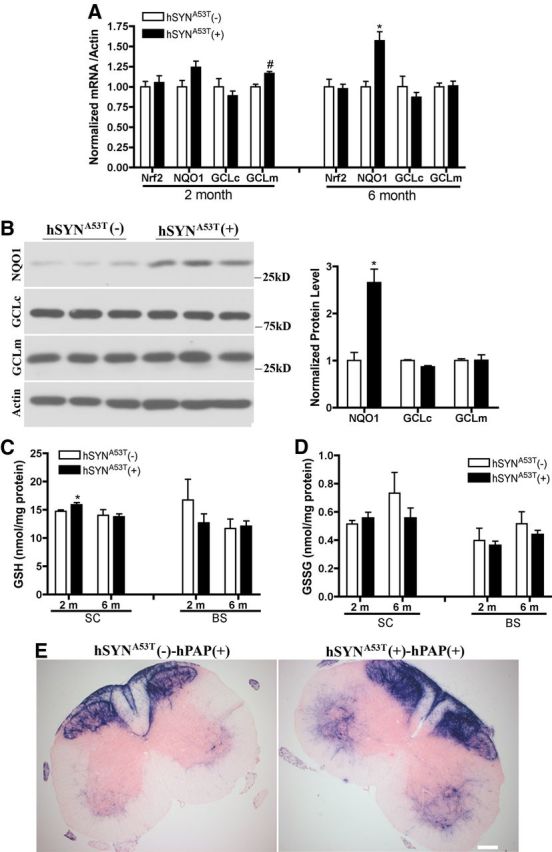
Endogenous Nrf2-ARE pathway is activated in hSYNA53T transgenic mice. A, mRNA levels of Nrf2 and its regulated genes in spinal cord of 2- and 6-month-old nonsymptomatic hSYNA53T mice. n = 4. *p < 0.001, #p < 0.05, hSYNA53T(+) versus hSYNA53T(−). B, Western blots of NQO1, GCLc, and GCLm for 6-month-old hSYNA53T mice. *p < 0.001, hSYNA53T (+) versus hSYNA53T(−). n = 3. C, D, GSH and GSSG levels in spinal cord and brainstem of 2- and 6-month-old nonsymptomatic hSYNA53T mice. n = 3–5. *p < 0.05, hSYNA53T (+) versus hSYNA53T(−). E, hPAP activity represented by histochemical staining in spinal cord of nonsymptomatic 6-month-old hSYNA53T mice; n = 3. Scale bar, 500 μm.
Astrocyte-specific overexpression of Nrf2 extends the life span of hSYNA53T mice and increases motor neuron survival
To enhance this modest endogenous Nrf2 activation, we crossbred hSYNA53T mice with GFAP-Nrf2 transgenic mice to determine whether astrocytic overexpression of Nrf2 could alleviate pathology in the hSYNA53T mice. There was a dramatic and significant extension of life span by 65.5 d in the double transgenic mice (Fig. 2A). The disease onset was also delayed by 63.5 d (Fig. 2B). No significant difference was observed in disease progression between hSYNA53T mice and double transgenic mice (Fig. 2C). Based on the onset data, there appeared to be two different subsets of hSYNA53T [AN(+/−)] mice: early (<200 d) and late (>200 d) onset mice. Thus, we divided these mice into two groups based on onset and re-analyzed the data. For the early onset group, Nrf2 overexpression extended the life span and delayed disease onset and progression significantly by 73 d, 59 d, and 6.2 d, respectively (Fig. 2D–F). For the late onset group the extension in survival (26 d) and delay in onset (13 d) were not statistically significant (Fig. 2G–I). Subsequent biochemical and histochemical analysis was performed on ∼6-month-old mice.
Figure 2.
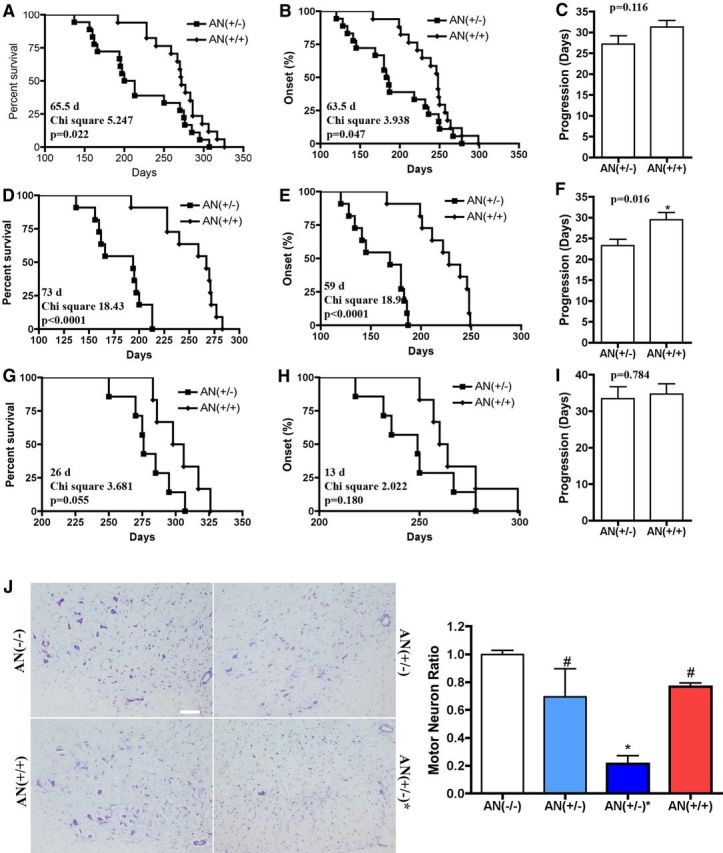
Overexpression of Nrf2 in astrocytes extends the life span of hSYNA53T transgenic mice and increases motor neuron survival in spinal cord. A, Nrf2 overexpression in astrocytes increased the median survival from 206.5 d in hSYNA53Tmice [AN(+/−)] (n = 18) to 272.0 d in hSYNA53T/GFAP-Nrf2 double transgenic mice [AN(+/+)] (n = 17). p = 0.0220, χ2 = 5.247. B, Nrf2 overexpression delayed disease onset from 184.5 d in AN(+/−) mice (n = 18) to 248.0 d in AN(+/+) mice (n = 17). p = 0.047, χ2 = 3.938. C, No difference was observed in disease progression between AN(+/−) mice (27.22 ± 1.93, n = 18) and AN(+/+) mice (31.29 ± 1.60, n = 17). Values are the mean ± SEM. D–F, Early onset (<200 d) cohort. D, The median survival of 194.0 d in AN(+/−) mice (n = 11) was prolonged to 267.0 d in AN(+/+) double transgenic mice (n = 11). p < 0.0001, χ2 = 18.43. E, The disease onset of 169.0 d in AN(+/−) mice (n = 11) was delayed to 228.0 d in AN(+/+) double transgenic mice (n = 11). p < 0.0001, χ2 = 18.92. F, The disease progression of 23.27 ± 1.53 d in AN(+/−) mice (n = 11) was significantly extended to 29.45 ± 1.77 d in AN(+/+) double transgenic mice (n = 11). p = 0.016. Values are the mean ± SEM. G–I, Late onset (>200 d) cohort. G, The median survival of 276.0 d in AN(+/−) mice (n = 7) was prolonged to 302.0 d in AN(+/+) double transgenic mice (n = 6). p = 0.055, χ2 = 3.681. H, The disease onset of 249.0 d in AN(+/−) mice (n = 7) was delayed to 262.0 d in AN(+/+) double transgenic mice (n = 6). p = 0.180, χ2 = 2.022. I, The disease progression in AN(+/−) mice (33.43 ± 3.25 d, n = 7) and AN(+/+) double transgenic mice (34.67 ± 2.86 d, n = 6) was similar; p = 0.784. Values are the mean ± SEM. J, Estimated number of motor neuron in the ventral horn of spinal cord of 6-month-old hSYNA53T transgenic mice without symptoms [AN(+/−)] and with symptoms [AN(+/−)*] along with age-matched littermate wild-type controls [AN(−/−)]. Left, Representative images with Nissl staining. Scale bar, 100 μm. The quantified data are shown on the right. Mean ± SEM; n = 3. *p < 0.01, AN(+/−)* versus AN(−/−); #p < 0.05, AN(+/+), AN(+/−) versus AN(+/−)*.
Another cohort of mice was harvested at 6 months of age and the number of motor neurons in the lumbar spinal cord was quantified blindly (Fig. 2J). Motor neuron numbers in asymptomatic hSYNA53T(+)/GFAP-Nrf2(−) mice [AN(+/−)] were not significantly different from control hSYNA53T(−)/GFAP-Nrf2(−) mice [AN(−/−)] (Fig. 2D). However, the degree of variability within this group does suggest that the range of motor neuron loss between mice is large as would be expected in asymptomatic mice with such a large range in life span. In contrast, symptomatic mice [AN(+/−)*] had a consistent and significant loss of 80% of their motor neurons. This loss was reversed in the double transgenic hSYNA53T(+)/GFAP-Nrf2(+) [AN(+/+)]. Nrf2 overexpression does not affect motor neuron development because there was no difference in motor neuron numbers in GFAP-Nrf2 mice compared with wild-type control mice. (Vargas et al., 2008).
Nrf2 decreases α-synuclein aggregates, gliosis, and oxidative stress in hSYNA53T mice
To ensure that the observed changes in life span and onset were not due to Nrf2-mediated downregulation of the hSYNA53T transgene levels in the mice, a Western blot was performed on 2-month-old spinal cord extracts demonstrating that hSYNA53T levels were not affected by Nrf2 overexpression in astrocytes (Fig. 3A). Compared with 6-month-old mice without symptoms [AN(+/−)], the amount of hSYNA53T in the Triton-soluble fraction from spinal cord decreased 60% in age-matched symptomatic mice [AN(+/−)*] (Fig. 3B,C). This was accompanied by a significant increase in hSYNA53T in the Triton-insoluble/SDS-soluble fraction. This movement of hSYNA53T into Triton-insoluble/SDS-soluble aggregates was completely reversed by overexpression of Nrf2 in astrocytes [AN(+/+)] (Fig. 3B,C). Similar changes were observed for phosphorylated (Ser129) hSYNA53T (p-hSYNA53T) in Triton-soluble and Triton-insoluble/SDS-soluble fractions (Fig. 3B). Fluorescent staining of hSYNA53T on formic acid pretreated sections correlates the results from Western blotting showing a dramatic increase in hSYNA53T aggregates (Fig. 3D). Again, this change was completely reversed by GFAP-Nrf2 [AN(+/+)] (Fig. 3D). In addition, there was a clear colocalization of hSYNA53T and p-hSYNA53T in the aggregates (Fig. 3E). This is consistent with the previous observation that the main form of hSYNA53T in inclusions is the phosphorylated form of hSYNA53T (Fujiwara et al., 2002; Anderson et al., 2006).
Figure 3.
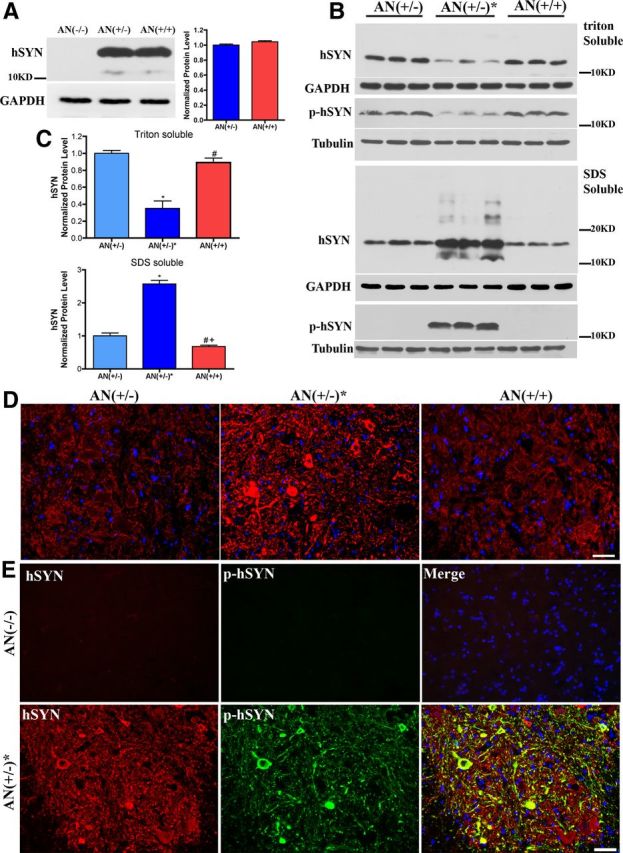
Overexpression of Nrf2 in astrocytes decreases hSYNA53T and p-hSYNA53T aggregates in spinal cord. A, Western blotting of hSYNA53T for Triton-soluble fractions from spinal cords of 2-month-old hSYNA53T mice and age-matched littermates. Quantification is shown in the right, mean ± SEM; n = 4. B, Western blotting of hSYNA53T and p-hSYNA53T in Triton-soluble and -insoluble fractions (SDS-soluble fractions) from the spinal cord of 6-month-old hSYNA53T mice and age-matched littermates. C, Quantificative data for hSYNA53T protein level, mean ± SEM; n = 3. *p < 0.001, AN(+/−)* versus AN(−/−); #p < 0.001, AN(+/+) versus AN(+/−)*; +p < 0.05, AN(+/+) versus AN(+/−). D–E, Fluorescent immunostaining of hSYNA53T and p-hSYNA53T aggregates with 80% formic acid pretreatment in spinal cord of 6-month-old mice. D, Staining for hSYNA53T in asymptomatic, symptomatic, and double transgenic mice. E, Colocalization of hSYNA53T and p-SYNA53T in symptomatic mice; n = 3. Scale bars: 50 μm.
Similar changes for p-hSYNA53T were seen throughout the brain. Fluorescent immunostaining showed increased density of p-hSYNA53T in motor cortex, middle brain, and brainstem in symptomatic hSYNA53T mice and not in double transgenic mice (Fig. 4A,B). There was also a slightly increased density of p-hSYNA53T in striatum and cerebellum of symptomatic hSYNA53T mice (data not shown). There was a striking increase of p-hSYNA53T staining in SN, especially in the pars reticulata (SNpr) (Fig. 4B, middle). There was a modest colocalization of p-hSYNA53T and tyrosine hydrolase (TH) in SNpc (Fig. 4C,D). This is probably due to the low level of SYN expression driven by Thy1 promoter in dopaminergic neurons (van der Putten et al., 2000; Fernagut and Chesselet, 2004). The GFAP-Nrf2 mice completely prevented or significantly reduced the p-hSYNA53T accumulation throughout these different brain regions (Fig. 4A–C).
Figure 4.
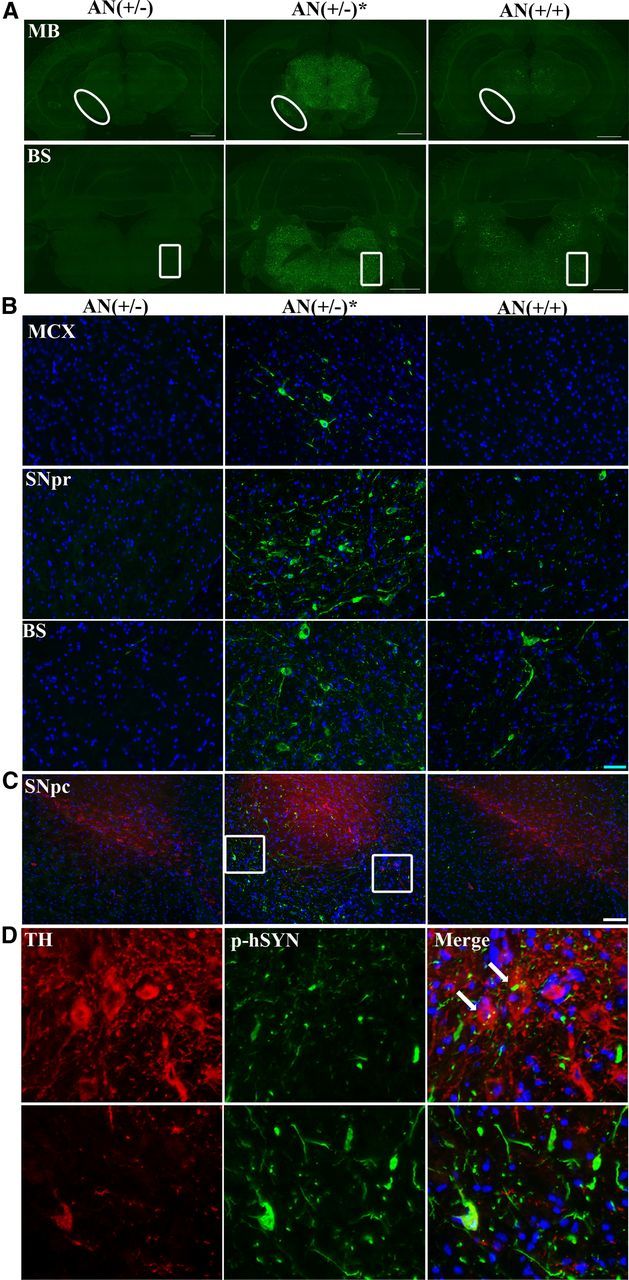
Overexpression of Nrf2 in astrocytes decreases p-hSYNA53T aggregates in the brain. Representative fluorescent immunoimages of p-hSYNA53T (green) with 80% formic acid pretreatment in different brain areas of 6-month-old asymptomatic, symptomatic, and double transgenic mice. A, Middle Brain (MB) and brainstem (BS). Scale bar, 1000 μm. Higher magnification images of the oval and square area are shown in B, middle and bottom, respectively. MCX, motor cortex. Blue: Hoechst; n = 3. Scale bar, 50 μm. C, Double-labeling of p-hSYNA53T (green) and TH (red) in SN. Blue: Hoechst. Scale bar, 100 μm. D, Higher magnification images of the square area. Arrows indicate colocalization of p-hSYNA53T and TH.
Nrf2 overexpression in astrocytes also reduced astrogliosis and microgliosis in symptomatic hSYNA53T mice, as determined by GFAP and IBA1 immunofluorescent staining and mRNA levels (Fig. 5A–C). NADPH oxidase, a potential generator of reactive oxygen species (ROS), increased dramatically in symptomatic hSYNA53T mice as represented by mRNA levels of its subunits gp91-phox, p47-phox, and p67-phox (Fig. 5D). There was also an increased staining density of HNE, a product from lipid peroxidation, especially in neurons in symptomatic hSYNA53T mice (Fig. 5E,F). Again, the GFAP-Nrf2 mice [AN(+/+)] completely prevented or significantly reduced all of these outcome measures (Fig. 5A–F).
Figure 5.
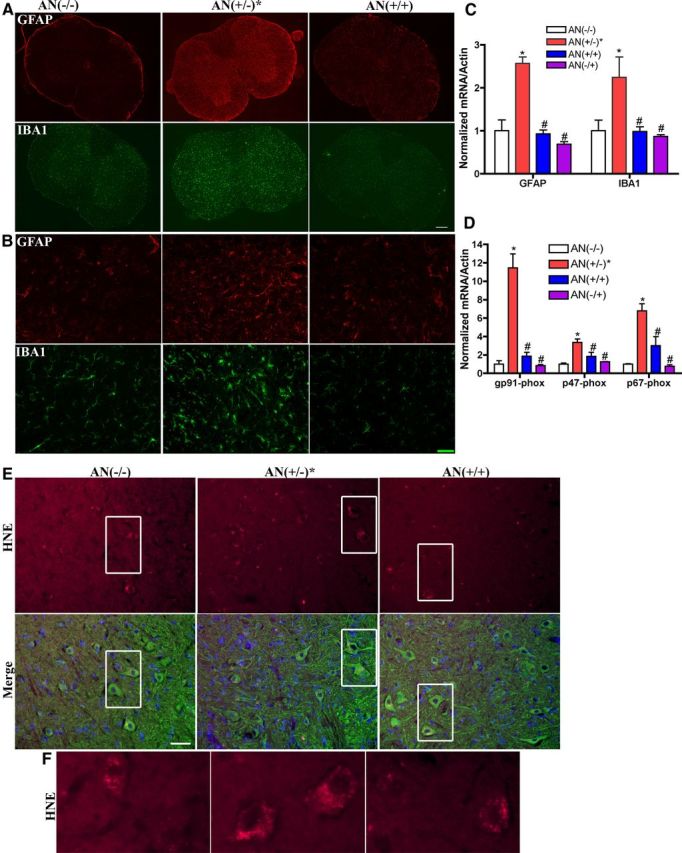
Overexpression of Nrf2 in astrocytes reduces gliosis and oxidative stress in spinal cord. A, Fluorescent staining of GFAP (red) and IBA1 (green); n = 3. Scale bar, 200 μm. B, Representative images of GFAP and IBA1 in ventral horn with higher magnification. Scale bar, 50 μm. C, mRNA levels of GFAP and IBA1 increased in spinal cord of 6-month-old hSYNA53T mice with symptoms [AN(+/−)*] compared with wild-type [AN(−/−)], double transgenic mice [AN(+/+)], and GFAP-Nrf2 littermates [AN(−/+)]. Mean ± SEM; n = 4. *p < 0.05, AN(+/−)* versus AN(−/−); #p < 0.05, AN(+/+), AN(−/+) versus AN(+/−)*. D, mRNA levels of NADPH oxidase subunits gp91-phox, p47-phox, and p67-phox increased in spinal cord of 6-month-old hSYNA53T mice with symptoms compared with wild-type and double transgenic mice. Mean ± SEM; n = 4. *p < 0.001, AN(+/−)* versus AN(−/−); #p < 0.01, AN(+/+), AN(−/+) versus AN(+/−)*. E, Intensity of HNE staining (red) increased in motor neurons in spinal cord of hSYNA53T mice. Green, β-III-tubulin; blue, Hoechst; n = 3. Scale bar, 50 μm. F, Higher magnification images of the square areas.
To determine the effect of hSYNA53T on Nrf2-dependent changes observed in the GFAP-Nrf2 mice, we evaluated mRNA levels of Nrf2 and its regulated genes (Fig. 6A). Compared with AN(−/−) or AN(+/−)* mice, there were dramatic and comparable inductions of Nrf2, NQO1, GCLc, GCLm, and glutathione S-transferase A2 (GSTA2) in the GFAP-Nrf2 [AN(−/+)] and double transgenic [AN (+/+)] mice suggesting that the protection of double transgenic mice was due to Nrf2 overexpression (Fig. 6A). Interestingly, GCLm and GCLc decreased significantly in symptomatic hSYNA53T mice. These data are in contrast to the 2- or 6-month-old nonsymptomatic mouse data (Fig. 1A). In addition, changes in GSH and GSSG were examined in spinal cord (Fig. 6B) and brainstem (Fig. 6C). We have previously published that total GSH (GSH + GSSG) levels are increased by 30–40% in spinal cord and brainstem of GFAP-Nrf2 mice at 1 month of age (Vargas et al., 2008). These data examined a 6-month-old cohort of mice. In both brainstem and spinal cord there was a trend toward decreased GSH and total GSH in the symptomatic hSYNA53T mice [AN(+/−)* vs AN(−/−)]. However, the most striking data are the sustained and significant increases of GSH and total GSH in the double transgenic mice [AN(+/+)] relative to the hSYNA53T mice [AN(+/−)*]. GSSG remained relatively unchanged ranging from 3 to 6% of the total GSH. Thus, the addition of hSYNA53T did not modulate the ability to activate Nrf2 in the astrocyte or sustain Nrf2-dependent changes in the GFAP-Nrf2 mice.
Figure 6.

Overexpression of Nrf2 in astrocytes increases Nrf2-dependent gene expression and GSH levels. A, mRNA levels of Nrf2 and its regulated genes in spinal cord of 6-month-old mice from all four genotypes. Mean ± SEM; n = 4. *p < 0.001, AN(+/+), AN(−/+) versus AN(−/−); #p < 0.001, AN(+/+), AN(−/+) versus AN(+/−)*; +p < 0.05, AN(+/−)* versus AN(−/−). Glutathione levels in spinal cord (B) and brainstem (C) from all four genotypes. Mean ± SEM; n = 3–5. *p < 0.05, AN(+/+) versus AN(−/−); #p < 0.05, AN(+/+) versus AN(+/−)*; +p < 0.05, AN(+/+) versus AN(−/+).
CMA is dysfunctional in hSYNA53T mice, but not in hSYNA53T/GFAP-Nrf2 double transgenic mice
MEF2D, a transcription factor involved in neuronal survival, is a substrate of CMA. Inhibition of CMA lead to increased MEF2D levels and accumulation of inactive MEF2D in cytoplasm (Yang et al., 2009). In addition, an increase in MEF2D has been described in a different line of hSYNA53T mice (Martin et al., 2006). Consistently, we found increased MEF2D (Fig. 7A) and cytoplasmic mislocalization (Fig. 7B) in the hSYNA53T line used in our experiments suggesting dysfunctional CMA. There was no change of protein levels for HSC70 and Lamp2a (Fig. 7C,D), while the protein levels of Lamp1, another lysosomal membrane protein, and both the premature and mature forms of lysosomal aspartyl protease cathepsinD (CatD) increased >4-fold in symptomatic hSYNA53T mice (Fig. 7C,D). The smeared bands for Lamp1 are indicative of increased Lamp1 glycosylation (Lee et al., 2010). All changes were absent in the double transgenic mice [AN(+/+)].
Figure 7.

CMA is impaired in hSYNA53T mice, not in hSYNA53T/GFAP-Nrf2 double transgenic mice. A, Western blotting for MEF2D from total spinal cord lysates in 6-month-old hSYNA53T mice and age-match littermates. Mean ± SEM; n = 3. *p < 0.05, AN(+/−)* versus AN(−/−); #p < 0.01, AN(+/+), AN(−/+) versus AN(+/−)*. B, Fluorescent immunostaining for red, MEF2D; green, β-III tubulin; and blue, Hoechst; n = 3. Scale bar, 20 μm. C, Western blotting for CMA-related chaperone molecular HSC70, lysosomal membrane protein Lamp2a, Lamp1, and lysosomal hydrolase CatD. D, The quantified data for Lamp1, mature CatD (M). Pre, premature CatD. Mean ± SEM; n = 3. *p < 0.001, AN(+/−)* versus AN(−/−); #p < 0.001, AN(+/+), AN(−/+) versus AN(+/−)*.
Macroautophagy is inhibited in hSYNA53T mice, but not in hSYNA53T/GFAP-Nrf2 double transgenic mice
Macroautophagy includes three main steps: initiation (autophagic complex formation), elongation of phagophores, maturation of autophagosomes, and fusion with lysosomes. LC3 is involved in the last two steps. It is synthesized as a precursor form and is cleaved at its COOH terminus to form LC3 I in the cytosol. LC3 II is formed by LC3 I conjugation with phosphatidylethanolamine and recruited to the elongating autophagosome membrane. After fusion with the lysosome, LC3 II on the outer membrane will be delipidated and recycled, while inner membrane LC3 II will be degraded in the autolysosome. LC3 II/LC3 I ratio indicates autophagosome formation. The LC3 II/LC3 I ratio was increased in spinal cord lysates from symptomatic hSYNA53T mice (Fig. 8A). P62 binds with ubiquitinated proteins and protein aggregates, which are trafficked to the autophagosome via p62 association with LC3 II. P62 is also a substrate of macroautophagy. The protein level of p62 increased both in Triton-soluble and -insoluble fractions from symptomatic hSYNA53T mice. The smeared pattern of p62 in the Triton-insoluble fractions indicated conjugation and aggregate formation (Fig. 8B). The increased LC3 II/LC3 I ratio and p62 accumulation in the insoluble fraction demonstrates that there were increased autophagosome vacuoles, but the proteins engulfed inside did not undergo degradation. This speculation was confirmed by colocalization of p62 and p-hSYNA53T in formic acid pretreated sections (Fig. 8D). Ubiquitin was also slightly increased in the Triton-insoluble fraction in symptomatic hSYNA53T mice (Fig. 8C). However, there was no observable histological evidence of ubiquitin accumulation in hSYNA53T mice (Fig. 8E). Ubiquitin staining in hSYNA53T mice was diffuse indicating that the p-hSYNA53T aggregates are p62 positive, but not ubiquitin positive.
Figure 8.

Macroautophagy is dysfunctional in hSYNA53T mice, not in hSYNA53T/GFAP-Nrf2 double transgenic mice. A, Western blots for LC3. Top is overexposed to visualize LC3 II. A shorter exposure to quantify LC3 I is next. The quantification of LC3 II/LC3 I ratio is shown on the left. Mean ± SEM; n = 3. *p < 0.01, AN(+/−)* versus AN(−/−); #p < 0.01, AN(+/+), AN(−/+) versus AN(+/−)*. B, Western blotting of p62. Top, Triton-soluble fractions; bottom, Triton-insoluble fractions; n = 3. C, Western blotting of ubiquitin in Triton-insoluble fractions. Ub, ubiquitin; n = 3. Representative images for p-hSYNA53T and p62 (D) or Ub (E) colocalization; n = 3. Scale bars: 50 μm.
Autophagosome-like vacuoles exist in hSYNA53T mice, but not hSYNA53T/GFAP-Nrf2 double transgenic mice
In symptomatic hSYNA53T mice, neurons with shrunken cell bodies and condensed nuclei were observed compared with neurons with normal morphology in the other three genotypes (Fig. 9A). Lysosomes and lysosome-like structures were rarely observed in the dying neurons of hSYNA53T mice compared with variably shaped lysosomes in the other three genotypes. Autophagosome-like structures with double- and multiple-layered membranes existed in the sick neuronal cell bodies and axons in the spinal cord of hSYNA53T mice with symptoms, but not in hSYNA53T/GFAP-Nrf2 mice. Few to none autolysosome-like vacuoles with single membranes (autolysosome) were observed in hSYNA53T mice (Fig. 9B, top and middle); whereas numerous residual bodies of lysosomes were present in the double transgenic mice (Fig. 9C). Finally, there was an intermediate neuronal phenotype in hSYNA53T mice that contained both vacuolated lysosomes along with normal lysosomes (Fig. 9B, bottom).
Figure 9.

Autophagosome-like vacuoles exist in hSYNA53T mice, not hSYNA53T/GFAP-Nrf2 double transgenic mice. A, Electron microscopic (EM) images of motor neurons in lumbar spinal cord from 8-month-old hSYNA53T mice, hSYNA53T/GFAP-Nrf2 mice, and littermate controls. B, Top and middle, autophagosome-like vacuoles in cell body and axon of neurons from hSYNA53T mouse with symptoms. Scale bars: 5 μm. Higher magnification images of the square areas are shown on the right. Scale bars: 1 μm (top) and 500 nm (middle). Arrows denote the vacuoles. Bottom, A healthier looking neuron from hSYNA53T mouse with vacuolated (arrows) and normal (arrowheads) lysosomes. Scale bars: 5 μm (left) and 1 μm (right). C, EM images for hSYNA53T/GFAP-Nrf2 mouse. Higher magnification images of the square areas are shown on the right. Scale bars: 5 μm (left) and 1 μm (right).
Discussion
SYN is the first gene that was identified in PD patients and is the main component in Lewy bodies. It is believed that SYN is a central player in the pathology of PD and other α-synucleinopathies.
The Nrf2-ARE pathway regulates a cell defense system to fight against oxidative insults. Overexpression of Nrf2 in astrocytes delayed the pathology of hSYNA53T mice and reduced Triton-insoluble aggregates containing hSYNA53T and p-hSYNA53T (Figs. 2–4). The autophagy–lysosome pathway is a highly conserved bulk protein degradation system responsible for the turnover of long-lived proteins, clearance of aggregate-prone proteins, and disposal of damaged organelles. Inactivation of autophagy results in cytoplasmic protein inclusions composed of misfolded proteins and deformed organelles, which could lead to neurodegeneration and other diseases. The level of autophagosome-associated LC3 II or the ratio of LC3 II to LC3 I has been used to evaluate macroautophagy. LC3 II is a substrate of macroautophagy, and the level of LC3 II alone is correlated with the number of phagophores/autophagosomes instead of induction of autophagy (Klionsky et al., 2008). The accumulation of p62, another substrate of macroautophagy, confirmed a dysregulation of macroautophagy. Colocalization of p62 and p-hSYNA53T suggested that p62 and p-hSYNA53T were both localized in aggregates. Increased autophagosome formation could be due to increased autophagic activity or reduced turnover of autophagosomes. This study supports that failed turnover is the cause. Observation of autophagic vacuoles with electronic microscopy is another standard way to monitor autophagy. Autophagosome-like structures not autolysosome-like vacuoles were observed in hSYNA53T mice. All these data confirmed that early autophagy was activated, but the fusion step of autophagy with the lysosome did not proceed.
It is interesting that there was no obvious increase of ubiquitin accumulation, or colocalization of p-hSYNA53T and ubiquitin. There are two possibilities. First, hSYNA53T and p62 could be conjugated together for degradation through ubiquitin-independent macroautophagy. Existing evidence shows that ubiquitinated SYN/p-SYN is not always observed and is not necessary for formation of pathological inclusions or degradation (Liu et al., 2003; Sampathu et al., 2003; Machiya et al., 2010; Ebrahimi-Fakhari et al., 2011). A recent report points out that mono-ubiquitinated SYN is degraded by the UPS and de-ubiquitinated SYN is mainly degraded by ALP (Rott et al., 2011). Additionally, p62 could mediate ALP clearance of nonubiquitinated substrates in an HDAC6-dependent manner (Watanabe and Tanaka, 2011) suggesting that clearance of SYN/p-SYN could occur via a p62-dependent but ubiquitin-independent mechanism. Second, both p-hSYNA53T and p62 could be trapped in vacuoles as separate misfolded proteins. As substrates of macroautophagy, SYN, and p62 may be accumulated together without physical interaction when macroautophagy is dysregulated.
CatD is a soluble lysosomal aspartic endopeptidase. It functions in the lysosome to cleave SYN. The exact outcome of this function is very controversial. Overexpression of CatD prevented SYN aggregation and toxicity in vitro and in a Caenorhabditis elegans model (Qiao et al., 2008). Our data show that there was a dramatic induction of premature and mature CatD that correlates with the formation of aggregates. These observations are consistent with previously published data that phosphorylated SYN induces upregulation of casin kinase 2 and CatD (Takahashi et al., 2007). Inhibition of autophagy could also induce CatD-mediated apoptotic cell death (Carew et al., 2011). H2O2-induced ROS production and endothelial cell apoptosis can be enhanced by CatD overexpression and suppressed by knockdown of CatD (Haendeler et al., 2005). CatD acts as a death executor beyond caspase 3. The CatD death pathway is upstream of or independent of caspase cascades, and autophagosomes appear before DNA fragmentation in vitro (Deiss et al., 1996; Isahara et al., 1999; Kågedal et al., 2001). We observed shrunken cell bodies and condensed nuclear chromatin with electronic microscopy, but could not detect any cleaved caspase 3 by Western blotting (data not shown). These results suggest a CatD-mediated motor neuron loss in hSYNA53T mice.
Together, overexpression of hSYNA53T negatively affected protein clearance pathways including CMA and macroautophagy. This leads us to hypothesize that the increased oxidative stress associated with incomplete degradation of aggregates caused an abnormal modification of lysosome membrane protein Lamp1, and increased permeabilization of the lysosomal membrane (Lee et al., 2010) resulting in lysosomal protease CatD-mediated cell death. The question remains as to how Nrf2 in astrocytes can protect neurons from hSYNA53T-mediated toxicity. The GSH system was negatively affected in symptomatic hSYNA53T mice, whereas increases in GCLc, GCLm, and total GSH were observed in spinal cord of double transgenic mice (Fig. 6). Perhaps overexpression of Nrf2 in astrocytes through secretion of glutathione and other antioxidant proteins could increase the ability of the neurons to combat oxidative stress thereby allowing for maintaining normal function of ALP and hSYNA53T clearance. The data herein and an earlier publication in mouse models of amyotrophic lateral sclerosis (Vargas et al., 2008) provide support for this hypothesis. Alternatively, changes in UPS could also contribute, but we have not seen any change in proteasome activity in the GFAP-Nrf2 mice (data not shown). It is also possible that secreted SYN is taken up and degraded by the Nrf2 astrocytes; however, immunohistochemical double labeling of hSYNA53T and p-hSYNA53T with the astrocyte marker GFAP showed no overlap (data not shown).
A major issue in PD research is the lack of an effective genetic mouse model that develops progressive degeneration of dopaminergic neurons (Hisahara and Shimohama, 2010). This includes the SYN mouse models that primarily develop symptoms of motor neuron diseases (Giasson et al., 2002; Martin et al., 2006). A new SYN line, using the tetracycline inducible system, has been developed and shows progressive dopaminergic neuron degeneration by overexpression of hSYNA53T selectively in dopaminergic neurons (Lin et al., 2012). Determining how Nrf2 modulated dopaminergic pathology in this mouse line may be more relevant to PD. However, this does not dampen the dramatic ability and relevance of Nrf2 to modulate SYN pathology. Indeed, more and more clinical data demonstrate that SYN and p-SYN-positive inclusions exist in the brainstem and spinal cord in asymptomatic (incidental Lewy body disease) and/or sporadic PD patients (Braak et al., 2003; Del Tredici and Braak, 2012; Tamura et al., 2012). Thus, understanding the mechanism as to how Nrf2 overexpressing astrocytes confer protection to neurons overexpressing hSYNA53T is the current focus of our ongoing experiments. Overall, activation of the Nrf2 pathway in astrocytes is a potential target to develop therapeutic strategies for treating pathological synucleinopathies including PD.
Footnotes
This work was supported by the grant from the National Institute of Environmental Health Sciences (NIEHS ES10042). We thank Randall Massey from Medical School Electron Microscope Facility for his technical assistance.
The authors declare no competing financial interests.
References
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Barone MC, Sykiotis GP, Bohmann D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson's disease. Dis Model Mech. 2011;4:701–707. doi: 10.1242/dmm.007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol Sci. 2010;115:557–568. doi: 10.1093/toxsci/kfq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew JS, Espitia CM, Esquivel JA, 2nd, Mahalingam D, Kelly KR, Reddy G, Giles FJ, Nawrocki ST. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J Biol Chem. 2011;286:6602–6613. doi: 10.1074/jbc.M110.151324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci U S A. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew KC, Ang ET, Tai YK, Tsang F, Lo SQ, Ong E, Ong WY, Shen HM, Lim KL, Dawson VL, Dawson TM, Soong TW. Enhanced autophagy from chronic toxicity of iron and mutant A53T alpha-synuclein: implications for neuronal cell death in Parkinson disease. J Biol Chem. 2011;286:33380–33389. doi: 10.1074/jbc.M111.268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Deiss LP, Galinka H, Berissi H, Cohen O, Kimchi A. Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 1996;15:3861–3870. [PMC free article] [PubMed] [Google Scholar]
- Del Tredici K, Braak H. Spinal cord lesions in sporadic Parkinson's disease. Acta Neuropathol. 2012;124:643–664. doi: 10.1007/s00401-012-1028-y. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci. 2011;31:14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariss MW, Reed DJ. High-performance liquid chromatography of thiols and disulfides: dinitrophenol derivatives. Methods Enzymol. 1987;143:101–109. doi: 10.1016/0076-6879(87)43018-8. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Chesselet MF. Alpha-synuclein and transgenic mouse models. Neurobiol Dis. 2004;17:123–130. doi: 10.1016/j.nbd.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–308. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- Gan L, Johnson DA, Johnson JA. Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur J Neurosci. 2010;31:967–977. doi: 10.1111/j.1460-9568.2010.07138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Haendeler J, Popp R, Goy C, Tischler V, Zeiher AM, Dimmeler S. Cathepsin D and H2O2 stimulate degradation of thioredoxin-1: implication for endothelial cell apoptosis. J Biol Chem. 2005;280:42945–42951. doi: 10.1074/jbc.M506985200. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Shimohama S. Toxin-induced and genetic animal models of Parkinson's disease. Parkinsons Dis. 2010;2011:951709. doi: 10.4061/2011/951709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger KJ, Capuano P, Winter C, Bacic D, Stange G, Wagner CA, Biber J, Murer H, Hernando N. Regulation of sodium-proton exchanger isoform 3 (NHE3) by PKA and exchange protein directly activated by cAMP (EPAC) Proc Natl Acad Sci U S A. 2006;103:803–808. doi: 10.1073/pnas.0503562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J Neuropathol Exp Neurol. 1998;57:334–337. doi: 10.1097/00005072-199804000-00005. [DOI] [PubMed] [Google Scholar]
- Isahara K, Ohsawa Y, Kanamori S, Shibata M, Waguri S, Sato N, Gotow T, Watanabe T, Momoi T, Urase K, Kominami E, Uchiyama Y. Regulation of a novel pathway for cell death by lysosomal aspartic and cysteine proteinases. Neuroscience. 1999;91:233–249. doi: 10.1016/s0306-4522(98)00566-1. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kågedal K, Johansson U, Ollinger K. The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J. 2001;15:1592–1594. doi: 10.1096/fj.00-0708fje. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. AlaSOPro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rábano A, Kirik D, Cuadrado A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early stage Parkinson's disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Park MH, Kim HJ, Koh JY. Metallothionein-3 regulates lysosomal function in cultured astrocytes under both normal and oxidative conditions. Glia. 2010;58:1186–1196. doi: 10.1002/glia.20998. [DOI] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H. Conditional expression of parkinson's disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M, Koyama S, Kato T. Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem. 2010;285:40732–40744. doi: 10.1074/jbc.M110.141952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, O'Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Qiao L, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, Engelender S. alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A. 2011;108:18666–18671. doi: 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM. Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol. 2003;163:91–100. doi: 10.1016/s0002-9440(10)63633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ko LW, Kulathingal J, Jiang P, Sevlever D, Yen SH. Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur J Neurosci. 2007;26:863–874. doi: 10.1111/j.1460-9568.2007.05736.x. [DOI] [PubMed] [Google Scholar]
- Tamura T, Yoshida M, Hashizume Y, Sobue G. Lewy body-related alpha-synucleinopathy in the spinal cord of cases with incidental Lewy body disease. Neuropathology. 2012;32:13–22. doi: 10.1111/j.1440-1789.2011.01211.x. [DOI] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20:6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011;124:2692–2701. doi: 10.1242/jcs.081232. [DOI] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009;323:124–127. doi: 10.1126/science.1166088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Kågedal K, Dehvari N, Benedikz E, Cowburn R, Marcusson J, Terman A. Oxidative stress induces macroautophagy of amyloid beta-protein and ensuing apoptosis. Free Radic Biol Med. 2009;46:422–429. doi: 10.1016/j.freeradbiomed.2008.10.043. [DOI] [PubMed] [Google Scholar]