

Abstract
Nicotine is the primary psychoactive substance in tobacco, and it exerts its effects by interaction with various subtypes of nicotinic acetylcholine receptors (nAChRs) in the brain. One of the major subtypes expressed in brain, the α4β2-nAChR, endogenously modulates neuronal excitability and thereby, modifies certain normal as well as nicotine-induced behaviors. Although α4-containing nAChRs are widely expressed across the brain, a major focus has been on their roles within midbrain dopaminergic regions involved in drug addiction, mental illness, and movement control in humans. We developed a unique model system to examine the role of α4-nAChRs within dopaminergic neurons by a targeted genetic deletion of the α4 subunit from dopaminergic neurons in mice. The loss α4 mRNA and α4β2-nAChRs from dopaminergic neurons was confirmed, as well as selective loss of α4β2-nAChR function from dopaminergic but not GABAergic neurons. Two behaviors central to nicotine dependence, reward and anxiety relief, were examined. α4-nAChRs specifically on dopaminergic neurons were demonstrated to be necessary for nicotine reward as measured by nicotine place preference, but not for another drug of addiction, cocaine. α4-nAChRs are necessary for the anxiolytic effects of nicotine in the elevated plus maze, and elimination of α4β2-nAChRs specifically from dopaminergic neurons decreased sensitivity to the anxiolytic effects of nicotine. Deletion of α4-nAChRs specifically from dopaminergic neurons also increased sensitivity to nicotine-induced locomotor depression; however, nicotine-induced hypothermia was unaffected. This is the first work to develop a dopaminergic specific deletion of a nAChR subunit and examine resulting changes in nicotine-related behaviors.
Introduction
Nicotine, the primary psychoactive substance of tobacco, interacts with nicotinic acetylcholine receptors (nAChRs) expressed in many brain pathways. Nicotinic receptors are pentameric ligand-gated ion channels that form several heteromeric receptors. One major class, which has the highest affinity for nicotine, contains α4 and β2 subunits [α4β2*-nAChR (the asterisk indicates the possible presence of additional subunits) (Lukas et al., 1999)]. Widespread expression of α4β2*-nAChRs across the brain suggests involvement in many behavioral responses to nicotine, and studies using mice with global deletions of α4 or β2 subunits have confirmed that α4β2*-nAChRs mediate many of effects observed in response to nicotine (for review, see Picciotto et al., 2001). Null mutant α4 or β2 mice do not exhibit nicotine reward (Picciotto et al., 1998; Pons et al., 2008; Exley et al., 2011). Mice lacking α4*-nAChRs also have elevated levels of basal anxiety, suggesting a role for α4*-nAChR activity in anxiety systems (Ross et al., 2000). Other responses such as locomotor activity show decreased but not complete loss of response to nicotine, indicating that other nAChR subtypes partially mediate these behaviors (Drago et al., 2003).
α4β2*-nAChRs are highly expressed in midbrain dopamine pathways known to be involved in drug reward, mood disorders, stress, movement generation, and learning (Klink et al., 2001; Wise, 2009; Maskos, 2010). Blocking high-affinity α4β2*-nAChRs systemically or within one midbrain dopaminergic area, the ventral tegmental area (VTA), decreases nicotine reward (Corrigall et al., 1994; Watkins et al., 1999), consistent with the loss of nicotine reward observed in α4- or β2-null mutant mice. Reintroduction of the missing subunit into the VTA of β2- or α4-null mutant mice rescued nicotine reward when expressed at high levels, indicating that VTA α4β2*-nAChRs are necessary for nicotine reward (Maskos et al., 2005; Pons et al., 2008). Interestingly, there appears to be a threshold level of β2 necessary, as reintroduction of low levels of β2 within the VTA is not sufficient to rescue nicotine conditioned place preference (CPP) (Mineur et al., 2009). Within the VTA, nAChRs are expressed on diverse populations of neurons and terminal projections, including those that are dopaminergic, GABAergic, glutamatergic, and cholinergic (Kiyatkin and Rebec, 1998; Klink et al., 2001). No previous work has been able to selectively examine α4β2*-nAChRs on distinct neuronal populations in midbrain dopaminergic nuclei (for review, see Maskos, 2010). Although 60–70% of neurons in the VTA are dopaminergic, recent work suggests that nAChRs on GABAergic neurons in the VTA may also play an important role in nicotine dependence (Mansvelder et al., 2002; Nashmi et al., 2007). Nicotinic receptors on GABAergic neurons are also implicated in the anxiolytic effects of nicotine, which are reversed by blocking GABA transmission (O'Neill and Brioni, 1994; Paterson and Nordberg, 2000). Although previous work has suggested that midbrain dopaminergic regions are involved in stress, particularly in relation to prior drug exposure, the role of nAChRs on dopaminergic neurons in the anxiolytic effects of nicotine has yet to be examined (George et al., 2000a,b).
To directly test the hypothesis that α4*-nAChRs specifically on dopaminergic neurons mediate nicotine reward, we generated a mouse lacking α4*-nAChRs in dopaminergic neurons. Data reported here are the first to directly determine that α4*-nAChRs on dopaminergic neurons are necessary for nicotine reward but not for cocaine reward. These data also demonstrate that dopaminergic α4*-nAChRs are involved in the anxiolytic and locomotor-depressant effects of nicotine, but not nicotine-induced hypothermia.
Materials and Methods
Animals
All experiments were conducted in accordance with the guidelines for care and use of animals provided by the National Institutes of Health, and protocols were approved by the Institutional Animal Care and Use Committee at the Salk Institute and/or the Animal Care and Utilization Committee of the University of Colorado, Boulder. Mice were housed two to five animals per cage and kept on a standard 12 h light/dark cycle at 22°C with ad libitum access to food and water. Mice were tested at 75–120 d of age. Animals were transferred to clean cages during weekly cage changing but otherwise were not handled before behavioral testing. Mice for experiments conducted at Institute for Behavioral Genetics, University of Colorado, Boulder, were shipped from the Salk Institute and temporarily housed in Boulder with ad libitum access to food and water and a 12 h light/dark cycle. Mice of either sex were used for all studies, except only males were used for nicotine place preference.
Materials
[3H]Dopamine (7,8-3H at 30–50 Ci/mmol) and [3H]GABA [2,3-3H at 35 Ci/mmol] were obtained from PerkinElmer. 35S-UTP and 125I-epibatidine were obtained from MP Biomedical. HEPES acid and sodium salt were products from BDH Chemicals distributed by VWR. Ultracentrifugation-grade sucrose was obtained from Fisher Chemicals. Optiphase “SuperMix” scintillation fluid was purchased from PerkinElmer Life Sciences. α-Conotoxin MII (α-CtxMII) was obtained from Dr. J. M. McIntosh (University of Utah, Salt Lake City, UT). All other chemicals were reagent grade and, unless specified otherwise, were purchased from Sigma-Aldrich, including ascorbic acid, atropine sulfate, bovine serum albumin (BSA), cocaine hydrochloride, (−)-nicotine tartrate (NIC), nicotine free base, 1-[2-[[(diphenylmethylene)imino]oxy]ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxilic acid (NO-711), nomifensine, and pargyline. For behavioral experiments, nicotine stock (free base; Sigma) was prepared in sterile isotonic saline and neutralized with hydrochloric acid.
Targeted deletion of α4-nAChR subunit
α4 Lox-only mice.
Using established homologous recombination techniques, α4 Lox-only mice were generated with loxP sequences (34 bp) (Sauer and Henderson, 1988; Kilby et al., 1993) inserted into intronic regions on either side of exon V of the α4 gene as seen in Figure 1. Previous work has demonstrated that deletion of exon V disrupts expression of the α4 protein (Marubio et al., 1999; Ross et al., 2000). The neomysin resistance gene was included in the targeting construct to facilitate selection of embryonic stem (ES) cells that had undergone homologous recombination. Diphtheria toxin fragment A gene, inserted beyond the region of homologous DNA as a negative selection marker, was included to cause lethality in cells in which the construct has been aberrantly inserted without undergoing homologous recombination. The targeting construct was transfected into CCE ES cells (derived from 129SvEv mice) by electroporation and grown in the presence of G418 (Roche Diagnostics) to select for neomycin resistance. Correct insertion of the targeted gene was confirmed by PCR and Southern blot analysis. In ES cells in which the targeting construct had been properly inserted, the neomycin resistance gene was removed by Cre-mediated recombination using a Cre-expressing plasmid, pTurbo-Cre (a generous gift from the Embryonic Stem Cell Core, Washington University, St. Louis, MO). As recombination can occur between any two of the three loxP sites, several recombination products were obtained (see Fig. 1). ES cells containing product I with the “floxed” (i.e., flanked by loxP sites) exon V but no neomycin resistance gene were injected into C57BL/6 mouse blastocysts and implanted into pseudopregnant females. Three chimeric mice were obtained that, on the basis of coat color (agouti), contained a substantial amount (70–100%) of tissue derived from the ES cells, indicating a high probability that the mutant DNA from the ES cell was contributing to the germ line. Germ line transmission of the loxP-flanked exon was confirmed by PCR, Southern blot, and sequence analysis of tail DNA amplified by PCR. α4 Lox-only mice are viable with phenotype indistinguishable from wild-type littermates. Mice were backcrossed over 15 generations to C57BL/6 and will be referred to as Lox-only control mice.
Figure 1.

Generation of α4 Lox-only mice. In the targeting construct used to generate α4 Lox-only mice, 34 bp loxP sites (black triangles) were inserted into the intronic regions on either side of exon V of the α4 gene. The neomysin resistance gene (neor) was included in the targeting construct to facilitate selection of ES cells that had undergone homologous recombination. The diphtheria toxin fragment A (DT-A) gene was inserted beyond the region of homologous DNA as a negative selection marker. In ES cells, the neomycin resistance gene was removed by Cre-mediated recombination. As recombination can occur between any two of the three loxP sites, several recombination products were obtained. ES cells containing product I with the floxed exon V but no neomycin resistance gene were injected into C57BL/6 mouse blastocysts and implanted into pseudopregnant females. PGK, Phosphoglucose kinase.
α4-DA mice.
Mice with dopaminergic deletion of α4 (α4-DA) were generated by breeding the α4 Lox-only control mice with knock-in mice expressing Cre recombinase 5′ to the dopamine transporter (DAT) gene (also known as slc6a3-Cre mice) (Zhuang et al., 2005). α4-DA mice were maintained primarily by heterozygous breeding of α4 wt/lox DAT-Cre−/− females with α4 wt/lox DAT-Cre+/− males. This breeding strategy produced the following desired genotypes: α4-DA (α4 lox/lox DAT-Cre+/−), Cre-only control (α4 wt/wt DAT-Cre+/−), Lox-only control (α4 lox/lox DAT-Cre−/−), and wild-type (α4 wt/wt DAT-Cre−/−) littermates. Occasionally, Lox-only control females were mated for a single generation with α4-DA males to increase the number of α4-DA mice.
α4-null mice.
Using Cre-positive females in breeding, we were able to obtain the global deletion of α4, presumably due to expression of Cre in the egg, resulting in recombination before fertilization. α4 heterozygous Cre-positive females (α4 lox/wt DAT-Cre+/−) were used for breeding with wild-type males (α4 wt/wt DAT-Cre−/−). This breeding resulted in the α4 heterozygous Cre-negative mice (α4 wt/− DAT-Cre−/−). Other genotypes produced by this breeding are Cre-only control (α4 wt/wt DAT-Cre+/−), wild-type (α4 wt/wt DAT-Cre−/−), and α4 heterozygous Cre-positive mice (α4wt/− DAT-Cre+/−). The α4 heterozygous Cre-negative mice (α4 wt/− DAT-Cre−/−) were then used in heterozygous mating to produce α4-null mutant mice (α4−/− DAT-Cre−/−) and wild-type controls (α4 wt/wt DAT-Cre−/−).
Genotyping
Genotype was determined by PCR of tail DNA (primers: α4, 5′- CAGTGGGTTTGCTATGGTC-3′, 5′-ATACACCACACCCACGAAC-3′, 5′-GCAGGTGTTAGAATCATAGG-3′; CRE, 5′-ACGCTGTTTTGCACGTTCACC-3′, 5′-GGTTTCCCGCAGAACCTGAA-3′). DNA amplification was achieved through the following protocol: 95°C for 2 min and 29 cycles of 95°C for 25 s, 58°C for 30 s, and 72°C for 45 s, finished with 72°C for 10 min. On a 1.5% agarose gel, the approximate band sizes were 516 bp for wild-type α4, 548 bp for loxP-flanked α4, 478 bp for recombined α4 allele (α4-null), and 210 bp for Cre.
Radioactive in situ hybridization
Coronal sections (15 μm; n = 2–3 mice per genotype for the α4 probe; n = 1–2 mice per genotype for Cre probe) were used for in situ hybridization with a protocol similar to that described previously (Marks et al., 1992). A 600 bp riboprobe complementary to mRNA encoding the intracellular loop of the α4 subunit was synthesized by in vitro transcription using T7 RNA polymerase (Ambion). 35S-UTP was used as the sole source of UTP. The same method was used to produce a riboprobe complementary to the coding sequence of Cre. α4 and Cre probes were hybridized to sequential serial sections.
125I-Epibatidine binding to sections
Coronal sections (15 μm; n = 2–3 mice per genotype) were used for autoradiography using 125I-epibatidine following a protocol nearly identical to that described previously (Whiteaker et al., 2002). Total binding was determined using 10 pm 125I-epibatidine plus 90 pm unlabeled epibatidine (Sigma); nonspecific binding was determine by inclusion of 1 mm unlabeled l-nicotine (Sigma).
Colocalization of α4 with tyrosine hydroxylase and glutamate decarboxylase 67
Mice (n = 2 per genotype) were anesthetized with Nembutal (50 mg/kg, i.p.) and subjected to cardiac perfusion with 20 ml of RNase-free PBS solution (100 mm NaCl, 10 mm sodium phosphate buffer, pH 7.5) followed by 50 ml of 4% paraformaldehyde (PFA). Brains were dissected, cut into 2 mm sections, and incubated in 4% PFA for 2 h at room temperature, and then in 30% sucrose at 4°C for 20 h. Each section was then imbedded in tissue-freezing medium (Triangle Biomedical Sciences) and rapidly frozen using isopentane on dry ice before storage at −80°C. The 2 mm section containing the midbrain was sliced at 15 μm and mounted onto Superfrost Plus slides (VWR). Sections were hybridized with 600 bp digoxigenin-labeled riboprobe complementary to α4 mRNA and a 900 bp fluorescein-labeled riboprobe complementary to the 3′ end of glutamate decarboxylase 67 (GAD67) mRNA at 67°C for 16 h in a buffer containing 50% deionized formamide (Ambion), 0.1 mg/ml tRNA (Ambion), 10% dextran (Millipore Bioscience Research Reagents), and 1× Denhardt's solution (Sigma). The slides underwent 1 h of stringency washes at 63°C in a saline-sodium citrate buffer containing 50% formamide. Slides were then blocked for 1 h in 100 mm maleic acid, 150 mm NaCl, and 0.1% Tween-20 (Roche Diagnostics), pH 7.5 (MABT), containing 2% blocking reagent (Roche Diagnostics) and 10% sheep serum (Jackson ImmunoResearch), and then incubated overnight at 4°C with 1:500 anti-digoxigenin-peroxidase antibody (Roche). Following 50 min of washes in MABT, peroxidase was developed using the Tyramine Signal Amplification (TSA) Cyanine3 kit (PerkinElmer Life Sciences). The reaction was stopped by three 10 min washes in PBS, and then any remaining peroxidase was quenched using 3% H2O2 in PBS for 30 min at room temperature. This protocol was then repeated to stain the fluorescein-labeled probe with overnight antibody incubation using 1:500 anti-fluorescein-peroxidase antibody (Roche), and peroxidase was developed using the TSA Cyanine2 kit. Following development of the GAD67 probe, sections were blocked for 2 h and then incubated overnight with rabbit anti-tyrosine hydroxylase (anti-TH; ab112; AbCam) in a PBS-based blocking solution containing 0.5% donkey serum, 0.25% Triton X-100, and 0.05% cold water fish skin gelatin. The following day, after 1 h of washes in 10% blocking solution, the sections were incubated for 4 h at room temperature with 1:250 Alexa Fluor 488 goat anti-rabbit (Invitrogen). Slides were then rinsed for 20 min with PBS, dehydrated, cleared with xylene, and coverslipped. Slides were examined using a Leica TCS SP2 AOBS laser-scanning confocal microscope with a 63× oil-immersion lens. The Alexa Fluor 488 was excited using the 488 nm line of an argon laser, Cy5 was excited using the 633 nm line of an HeNe laser, and Cy3 was excited by a 561 nm laser. The images presented are projections of 10 sections collected ∼0.5 μm apart using Leica confocal software.
Synaptosome preparation for release experiments
After a mouse was killed by cervical dislocation, the brain was removed and placed immediately on an ice-cold platform, and regions were dissected. Tissues from each mouse (n = 3–6 per genotype) were homogenized in 0.5 ml of ice-cold 0.32 m sucrose buffered with 5 mm HEPES, pH 7.5, and the homogenizer rinsed and combined for final volumes of 2–3 ml. For each experiment, tissues from each mouse were divided as follows: the striatum (ST) into four equal aliquots; the olfactory tubercle (OT) into two aliquots of 25% of suspension and one of 50%; and the cortex (CX) into one aliquot each of 8% and 92% of suspension. A crude synaptosomal pellet was prepared by centrifugation of these aliquots at 12,000 × g for 20 min. One pellet of the ST and the larger pellets of the OT and CX were frozen for future binding experiments. The remaining pellets were used for neurotransmitter release.
Release of [3H]dopamine
All experiments were conducted at room temperature using methods described previously (Grady et al., 1997, 2001), with modifications for collection into 96-well plates. In brief, pellets of 25% of ST or OT synaptosomes were resuspended in uptake buffer [containing the following (in mm): 128 NaCl, 2.4 KCl, 3.2 CaCl2, 1.2 KH2PO4, 1.2 MgSO4 25 HEPES, pH 7.5, 10 glucose, 1 ascorbic acid, and 0.01 pargyline] and incubated at 37°C for 10 min. [3H]dopamine (100 nm; 4 μCi into 0.8 ml of synaptosomes) was then added, and the suspension was incubated for an additional 5 min. Aliquots (80 ml) were pipetted onto filters and perfused at 0.8 ml/min with perfusion buffer (uptake buffer with 0.1% BSA, 1 μm nomifensine, and 1 mm atropine) for 10 min, or buffer for 5 min followed by buffer with 50 nm α-CtxMII for 5 min. Nicotine or high potassium (20 mm) in perfusion buffer for 20 s was used to stimulate the release of [3H]dopamine. Starting from 1 min before stimulation, fractions (∼0.1 ml) were collected every 10 s for 4 min into 96-well plates, using a Gilson FC204 fraction collector. Radioactivity was determined by scintillation counting using a 1450 MicroBeta Trilux scintillation counter (PerkinElmer Life Sciences; Wallac Oy) after addition of 0.15 ml of Optiphase SuperMix scintillation fluid. Instrument efficiency was 40%.
Release of [3H]GABA
Methods described for dopamine release were used with the following modifications: Ascorbate and pargyline were omitted from the uptake buffer, and 1.25 mm aminooxyacetic acid was added. Synaptosomes were incubated at 37°C for 10 min, [3H]GABA (8 mCi into 0.8 ml) was added, and the incubation was continued for another 10 min. Perfusion buffer without nomifensine and with NO-711 (100 nm) was used.
125I-Epibatidine binding to membrane preparations
The methods of Whiteaker et al., (2000a,b) were followed to determine 125I-epibatidine binding to membrane preparations. Briefly, tissue from three to six mice per genotype treated with phenylmethylsulfonyl fluoride (1 mm) were incubated with 125I-epibatidine (200 pm) in binding buffer [30 ml volume including 144 mm NaCl, 1.5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 20 mm HEPES, pH 7.5, with BSA (0.1% w/v), 5 mm EDTA, 5 mm EGTA, and 10 mg/ml each of aprotinin, leupeptin trifluoroacetate, and pepstatin A]. To some samples, 50 nm cytisine was added to selectively block α4β2*-nAChR to allow determination of cytosine-resistant epibatidine binding (the α4β2*-nAChR population) (Grady et al., 2007). To other samples, 100 nm α-CtxMII was added to measure α-CtxMII-sensitive epibatidine binding (the α6β2*-nAChR population) (Grady et al., 2007). Nicotine (1 mm) was used for blank determination. Incubation was for 2 h at room temperature. Using an Inotech Cell Harvester, reactions were terminated by filtration of samples at 4°C onto a two-layer filter consisting of one sheet of GFA/E glass fiber filter (Gelman Sciences) and one sheet of GF/B glass fiber filter (Micro Filtration Systems), both treated with 0.5% polyethylenimine. Samples were subsequently washed six times with ice-cold binding buffer. Radioactivity was measured using a Wallac 1450 Microβ scintillation counter (PerkinElmer Life and Analytical Sciences) after addition of 150 ml of Optiphase SuperMix scintillation fluid (PerkinElmer Life Sciences) to each well of a 96-well counting plate.
Conditioned place preference
A three-chamber conditioning apparatus was used; two conditioning chambers (16.8 × 12.7 cm) with different wall colors and flooring were separated by a smaller neutral chamber (7.2 × 12.7 cm) with retractable doors. An adaptation of previously published protocols was used (Grabus et al., 2006; Brunzell et al., 2009). For the pretraining day, animals received an intraperitoneal saline injection and were placed in the center neutral chamber for a 5 min habituation period. The doors to the conditioning chambers were then opened and the animal was allowed to freely explore all three chambers for 15 min. The animals (n = 8–13 per genotype per dose) had six training sessions in which they were injected with drug (0, 0.04, 0.065, or 0.09 mg/kg nicotine or 0, 5, or 20 mg/kg cocaine; drugs dissolved in sterile isotonic saline; injection volume, ∼0.01 ml/g body weight) or saline and introduced to one of the conditioning chambers. The same animal was later (5 h for nicotine CPP; 24 h for cocaine CPP) injected with the opposite treatment (drug or saline) and introduced to the other chamber. Drug- and saline-paired sides were randomly assigned before testing. Any animal that spent 70% or more of the time in one chamber or 20% or more of the time in either drug-paired chamber on the pretraining day was excluded from the study. The preference score was calculated by the difference in times spent in the drug- and saline-paired chambers on the testing day compared to the pretraining day.
Elevated plus maze
The elevated plus maze consisted of two open arms (30 × 5 cm) and two closed arms (30 × 5 cm) constructed of gray Plexiglas that extended from a center platform. The closed arms had sides of gray Plexiglas (15 cm tall). The entire apparatus was mounted 33 cm above the surface of the floor. Each test began by injecting the animal (n = 7–12 per genotype per dose) with the appropriate dose of nicotine (0, 0.0055, 0.01, 0.05, 0.1, or 0.15 mg/kg/i.p) and placing it in a holding cage for 10 min. After the 10 min holding period, the animal was placed in a cylindrical starting tube (5 cm diameter, 15 cm tall) in the center of the maze for 2 min. The tube was removed, and the subject was observed on the maze for 10 min using the Noldus video tracking system. Subjects were scored for the amount of time spent and the number of entries into the open and closed arms. Total numbers of entries into the closed and open arms were combined as an assessment of activity.
Locomotor activity
A symmetrical Y-maze was constructed of red acrylic plastic and consisted of three enclosed arms that were 26 cm long, 6.1 cm wide, and 10.2 cm high. Crossing and rearing activities were monitored as breaks of infrared beams. The beams for monitoring Y-maze crosses were located in the middle of each arm and at the exit from each arm to the center of the maze. The beams for monitoring rears (when a mouse stands up on its hind legs) passed through the long axis of each arm 6.4 cm above the floor. Animals (n = 4–10 per genotype per dose) were injected with nicotine (0, 0.01, 0.25, 0.5, 1, and 2 mg/kg, i.p) and first placed in a holding cage for 5 min, and then placed in the center of the maze and allowed to explore for 3 min. The total number of crosses between the arms and rears were recorded for the 3 min test.
Body temperature
Initial body temperature was recorded using a rectal thermometer lubricated with mineral oil and inserted 2.5 cm into the mouse's rectum. Following temperature recording, each animal (n = 4–10 per genotype per dose) was injected with the appropriate dose of nicotine (0, 0.01, 0.25, 0.5, 1, or 2 mg/kg i.p) and singly housed in a new cage for 15 min until the final body temperature recording was taken.
Data analysis
All values are presented as the mean ± SEM. Perfusion data were plotted as counts per minute versus fraction number. Fractions collected before and after the peak were used to calculate the baseline as a single exponential decay. The calculated baseline was subtracted from the experimental data. Fractions that exceeded baseline by 10% or more were summed to give the total released counts per minute and then normalized to baseline to give units of release [(counts per minute − baseline)/baseline] (Grady et al., 1997, 2001). One-way ANOVA with Duncan's post hoc analysis was used to determine statistical significance (SPSS software). For behavioral tests, data were analyzed with GraphPad Prism5 software using two-way ANOVA with genotype and drug treatment as variables, followed by Bonferroni's post hoc tests comparing all genotypes.
Results
Deletion of α4 subunit from dopaminergic neurons
To specifically remove α4 from dopaminergic neurons, we first generated a line of mice (Lox-only) in which loxP recognition sequences were inserted on either side of exon V of the α4 gene (which codes for the channel of the receptor). The bacteriophage enzyme Cre recombinase recognizes loxP sites and facilitates recombination between them (Fig. 1). Deletion of exon V has been shown to disrupt transcription of α4 and eliminate expression of the α4 protein (Marubio et al., 1999; Ross et al., 2000). To selectively eliminate α4 expression from dopaminergic neurons, Lox-only control mice (α4 lox/lox DAT-Cre−/−) were bred with knock-in mice expressing Cre under the control of the promoter for the DAT gene (Slc6a3) (α4 wt/wt DAT-Cre+/−) (Zhuang et al., 2005). In addition to the dopaminergic α4 knock-out mice (α4-DA; α4 lox/lox DAT-Cre+/−), the following littermate control mice were tested in the subsequent experiments: Cre-only controls (α4 wt/wt DAT-Cre+/−), Lox-only controls (α4 lox/lox DAT-Cre−/−), and wild types (α4 wt/wt DAT-Cre−/−), as well as mice with global deletion of α4 (α4-null; α4−/− DAT-Cre−/−).
α4-DA mice exhibited a loss of α4 mRNA from dopaminergic midbrain regions [VTA and substantia nigra (SN)], as shown by in situ hybridization, whereas α4 expression was intact in remaining regions (Fig. 2A). Cre expression was restricted to the same dopaminergic midbrain regions (Fig. 2B). A coincident decrease in high-affinity 125I-epibatidine binding in dopaminergic midbrain regions (VTA, SN) and terminal regions [ST (and OT; data not shown)] was observed (Fig. 2C,D), indicating a loss of α4*-nAChRs. α4 mRNA expression was completely eliminated in α4-null mice.
Figure 2.

Deletion of α4 in dopaminergic regions in α4-DA mice. A–C, Expression of α4 mRNA (A), Cre recombinase mRNA (B), and high-affinity 125I-epibatidine binding sites (C) at the level of the dopaminergic cell bodies (VTA/SN) are shown in adjacent brain slices for wild-type, Cre-only control, Lox-only control, α4-DA, and α4-null mice. D, 125I-Epibatidine binding is also shown at the level of the ST that contains the terminal ends of mesolimbic dopaminergic neurons.
To confirm that the regional deletion was specific to dopaminergic neurons, sections were triple labeled for α4 mRNA, the dopaminergic marker TH, and the GABAergic marker GAD67 (Fig. 3). In control animals (only wild-type controls are shown in Fig. 3), α4 mRNA expression colocalized with both TH (arrow) and GAD67 mRNA (underlined arrow). In α4-DA mice, as expected, colocalization of α4 mRNA with TH was not detected (Fig. 3E). Colocalization of α4 mRNA with GAD67 mRNA remained intact, demonstrating that α4 expression was selectively eliminated in dopaminergic (arrow) but not GABAergic neurons (underlined arrow). Of the 150 TH-positive neurons examined from α4-DA mutants, only three cells expressed α4 mRNA, indicating that the α4 gene was mutated with high efficiency by Cre recombinase (data not shown).
Figure 3.

Selective deletion of α4 in dopaminergic neurons. A–H, Brain slices from wild-type controls (A–D) and α4-DA (E–H) mice were labeled for α4 mRNA (red, B, F), a marker for dopaminergic neurons (TH; green, C, G), and a marker of GABAergic neurons (GAD67; blue, D, H). Dopaminergic neurons are indicated by arrows, and GABAergic neurons by underlined arrows. Scale bars: 15 μm.
Functional deletion of α4*-nAChRs from dopaminergic neurons and preservation of α6(non-α4)-nAChRs
To verify the functional deletion of α4*-nAChRs specifically from dopaminergic neurons, we examined nicotine-stimulated dopamine and GABA release. Nicotine-stimulated dopamine release is mediated by several subtypes of nAChRs in two brain regions (ST and OT) that receive dopaminergic projections from the SN and VTA. These nAChRs fall into two main categories, an α-CtxMII-resistant component (α4β2- and α4α5β2-nAChRs) and an α-CtxMII-sensitive component (α4α6β2β3-, α6β2β3-, and α6β2-nAChRs) (Salminen et al., 2007). Consistent with the presence of the α4 subunit in all receptors mediating α-CtxMII-resistant dopamine release (Fig. 4A), this release was abolished in the α4-null and α4-DA mice at both low (0.1 μm) and moderate (1.0 μm) concentrations of nicotine (0.01 μm, ST, F(4,17) = 10.18, p < 0.001; OT, F(4,16) = 9.001, p = 0.001; 1.0 μm, ST, F(4,17) = 32.39, p < 0.001; OT, F(4,16) = 29.874, p < 0.0010). The α-CtxMII-sensitive dopamine release (Fig. 4B) was reduced at 0.1 μm nicotine (ST, F(4,17) = 2.963, p = 0.05; OT, F(4,16) = 3.262, p = 0.039) but remained at control levels at 1.0 μm nicotine for both α4-null and α4-DA mice (ST, F(4,17) = 0.193, p = 0.938; OT, F(4,15)=.439, p = 0.779). This rightward shift may be explained by the loss of the α4α6β2β3*-nAChR (the receptor with the lowest EC50 value) leaving the α6β2- and α6β2β3-nAChRs intact (Salminen et al., 2007). These findings are consistent with the loss of α4*-nAChRs in dopaminergic pathways that originate in midbrain dopaminergic regions. Nicotine-stimulated GABA release (Fig. 4C) from the ST and CX (a control region not receiving dopaminergic input from the VTA and SN) was eliminated in the α4-null mice, as expected (1 μm, ST, F(4,14) = 14.721, p < 0.001; CX, F(4,14) = 6.948, p = 0.003; 10 μm, ST, F(4,14) = 21.687, p = 0.001; CX, F(4,14) = 8.994, p = 0.001). The α4-DA mice retain the control level of nicotine-stimulated GABA release activity in the CX, showing no loss of the α4 subunit in GABA neurons. Although the α4-DA mice showed significantly lowered nicotine-stimulated GABA release activity in ST, the level was comparable to the LoxP-only control, indicating a possible effect of the lox sites on expression in this region. This decrease in Lox-only control mice was not detected by ligand binding (Fig. 4D–F), indicating an alteration in expression below the level of detection using ligand binding. These biochemical studies confirm that deletion of the α4 subunit in the α4-DA mice is restricted to dopaminergic neurons.
Figure 4.
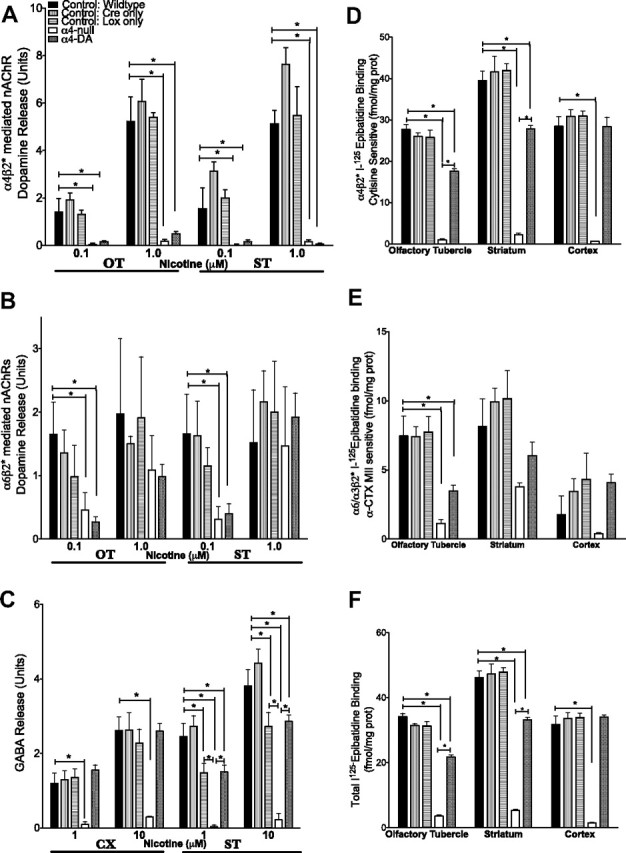
Functional confirmation of α4 deletion from dopaminergic neurons. A, B, α4β2*-nAChR-mediated (α-CtxMII-resistant; A) and α6β2*-nAChR-mediated (α-CtxMII-sensitive; B) dopamine release from two dopaminergic projection regions, OT and ST. C, α4β2*-nAChR-mediated GABA release from ST and CX. For both dopamine and GABA release, full dose–response curves were measured; representative concentrations are reported here (units normalized to baseline). K+-stimulated neurotransmitter release did not differ across genotypes (data not shown). D–F, 125I-Epibatidine binding (femtomoles per milligram of protein) in OT, ST, and CX. D, Cytistine-sensitive component (representing α4β2*-nAChRs). E, α-CtxMII-sensitive component (α6β2*-nAChRs). F, Total (representing all high-affinity heteromeric nAChRs). *p < 0.05.
Quantification of α4β2*-nAChRs and α6β2*-nAChRs
The functional changes were supported by membrane binding experiments conducted with 125I-epibatidine in samples of the same tissue used for the neurotransmitter release assays. Consistent with autoradiography results presented in Figure 2, α4-null mice showed a decrease in total 125I-epibatidine binding in dopaminergic regions (OT, F(4,19) = 183.095, p < 0.001; ST, F(4,18) = 96.559, p < 0.001) (Fig. 4F). This decrease results from an almost complete loss of the cytisine-sensitive component (analogous to the α-CtxMII-resistant component of dopamine release, i.e., α4β2*-nAChR subtypes; OT, F(4,19) = 112.128, p < 0.001; ST, F(4,18) = 65.489, p < 0.001) (Fig. 4D) and a partial loss of the α-CtxMII-sensitive component (i.e., a loss of the α4α6β3β2-nAChR subtype; OT, F(4,19) = 9.593, p < 0.001; ST, F(4,18) = 2.488, p = 0.08) (Fig. 4E). In CX, all 125I-epibatidine binding was absent in the α4-null mice (F(4,19) = 59.475; p < 0.001); however, in the α4-DA mutants, 125I-epibatidine binding in CX remained at control levels. In ST and OT from α4-DA mice, 125I-epibatidine binding (cytisine sensitive, α-CtxMII sensitive, and total) was reduced but not abolished, consistent with deletion of α4 in dopaminergic neurons with continued expression in GABAergic neurons (Fig. 4D–F). The decrease in nicotine-stimulated GABA release seen in the Lox-only control was not detectable in these binding experiments.
α4*-nAChRs on dopaminergic neurons mediate nicotine reward
To examine the role of α4 in nicotine reward, we used the CPP paradigm. Nicotine reward was measured as an increase in time spent in the nicotine-paired chamber over the saline-paired chamber (positive preference score). This method produced an inverted-U-shaped dose–response curve (Fig. 5A) in all control lines (wild type, Cre-only control, and Lox-only control) with a significant increase in preference for the nicotine-paired chamber at 0.065 mg/kg (i.p.) nicotine (F(3,169) = 5.04; p = 0.0023). This change in preference score was not seen in either α4-DA or α4-null mice at any dose tested, indicating that they did not develop preference (F(4,169) = 2.55; p = 0.0410).
Figure 5.
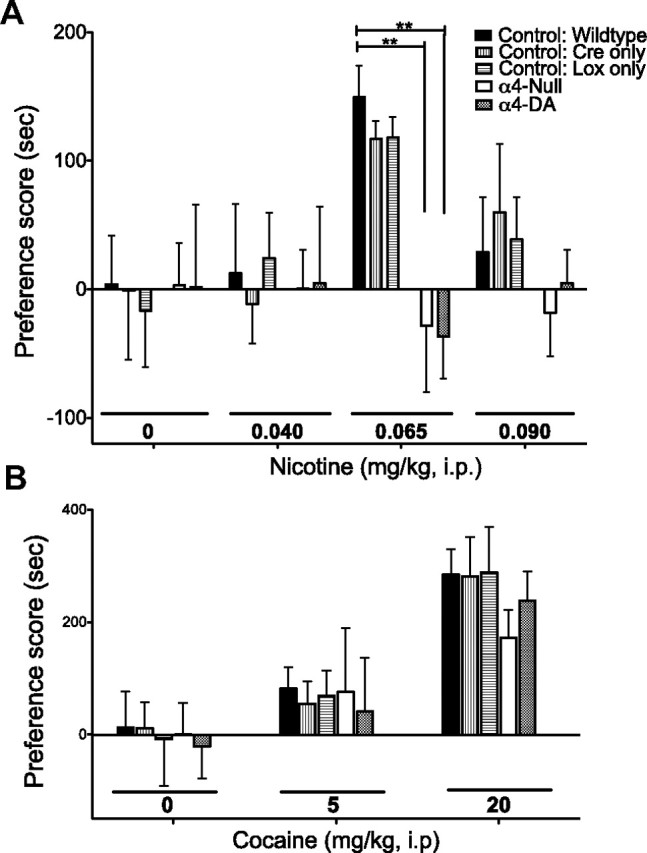
α4-containing nAChRs on dopaminergic neurons mediate nicotine reward but not cocaine reward. A, Nicotine elicited significant place preference at 0.065 mg/kg (i.p.) in all three control groups (wild type, Cre-only control, and Lox-only control). Neither the α4-null mice nor the α4-DA mice developed a preference for the nicotine-paired side over a wide range of doses (0.04, 0.065, and 0.09 mg/kg). **p = 0.01. B, Cocaine conditioned place preference. There was no difference between mice at any dose studied.
α4*-nAChRs on dopaminergic neurons do not mediate cocaine reward
The same method of CPP was used to investigate whether α4*-nAChRs modulate reward for another common drug of abuse, cocaine. Again, mice did not develop a consistent preference for either side when saline was administered; however, all three control lines as well as α4-DA and α4-null mice developed cocaine place preference with 20 mg of cocaine (Fig. 5B) [indicated by a treatment effect (F(2,116) = 22.01; p < 0.0001) but lack of a genotype effect (F(4,116) = 0.9043; p = 0.9043)]. Similar trends were observed for all genotypes with 5 mg of cocaine, although there was no significant difference from saline treatment. These data indicate that loss of the α4 subunit globally or selectivity in dopaminergic neurons does not prevent acquisition of cocaine preference.
α4*-nAChRs on dopaminergic neurons are involved in anxiolytic effects of nicotine
The elevated plus maze was used to examine anxiety changes in response to acute injections of nicotine. In this paradigm, there are four equally sized arms; two are gray, enclosed arms, and are therefore less anxiogenic than the two open arms, which are brighter and have no encasement. The number of times the mouse is willing to enter the more anxiogenic open arms and the amount of time spent in the open arms are interpreted as decreased anxiety. Using these measures of anxiety, we observed an inverted-U-shaped dose–response curve in control animals (wild type, Cre-only control, and Lox-only control), where low doses of nicotine elicited an anxiolytic response demonstrated by increases in the time spent in (Fig. 6A) and the number of entries into the open arm (Fig. 6B) of the maze (time, F(5,208) = 5.75, p < 0.0001; entries, F(5,208) = 4.35, p = 0.0009). The α4-null mice, unlike control mice, were insensitive to the anxiolytic effects of nicotine over these doses and did not increase the amount of time in or entries into the open arm (time, F(4,208) = 3.45, p = 0.0093; entries, F(4,208) = 3.56, p = 0.0078). The α4-DA mice were also insensitive to nicotine at the 0.01 mg/kg dose (Bonferroni's p < 0.05). Though α4-DA mice tended to increase the amount of time spent on the open arm at the 0.05 mg/kg nicotine dose, this point was not statistically significantly different from α4-null mice or controls. As a measure of general activity, the total number of entries into closed and open arms was also evaluated. A genotype difference in activity was detected, with Cre-only control mice showing increased activity as measured by more total entries than other groups of mice (F(4,208) = 7.89; p < 0.0001); however, this was not an effect of nicotine (F(5,208) = 1.28; p = 0.2753).
Figure 6.
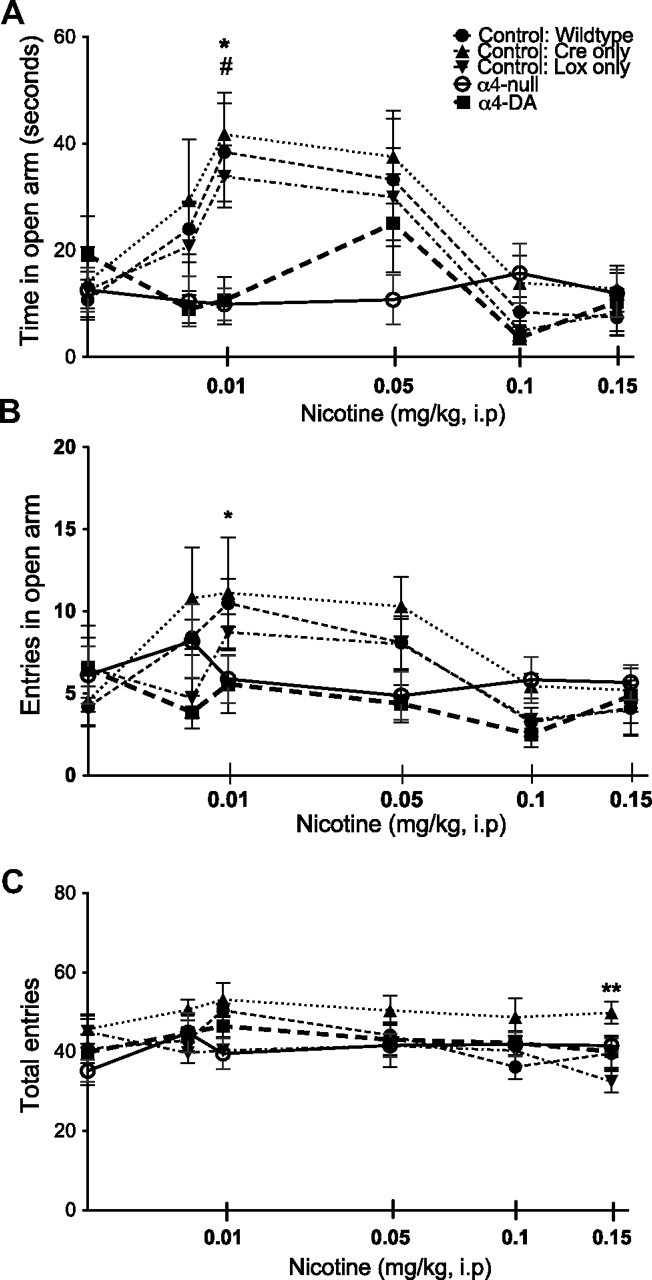
α4-containing nAChRs on dopaminergic neurons partially mediate the anxiolytic effects of nicotine. A, B, Nicotine at 0.01 mg/kg significantly decreased anxiety in all three control groups (wild type, Cre-only control, and Lox-only control) as measured by increased time (A) and entries (B) into the open arm. α4-null mice did not show a change in anxiety as measured by time and entries into the open arm (*p < 0.05). α4-DA mice showed a decreased sensitivity to nicotine, with significantly less time spent in the open arm (#p < 0.05) than control mice. C, There were no differences between wild-type control, Lox-only control, α4-null, and α4-DA mice in activity (total entries into open and closed arms); however, Cre-only control mice showed increased activity (**p < 0.01). Data are scaled as the square root of dose.
Deletion of α4*-nAChRs from dopaminergic neurons increases sensitivity to locomotor-depressing effects of nicotine
Previous work has shown that α4-null mice have decreased sensitivity to the locomotor-depressing effects of nicotine (Ross et al., 2000). We replicated these findings using a different paradigm, a Y-maze, that minimizes anxiogenic stimuli. For the Y-maze test, all animals were injected intraperitoneally with the selected nicotine dose, and the number of crosses and rears were recorded (Figure 7A,B). We found that nicotine produced a significant dose-dependent decrease in the number of crosses and rears for all genotypes tested. A significant dose-dependent depression of locomotor activity occurred at 1 mg/kg nicotine in all three control lines (wild type, Cre-only control, and Lox-only control) for both measures of locomotor activity (crosses, F(5,168) = 129.85, p < 0.0001) and exploratory activity (rears, F(5,168) = 62.19, p < 0.0001). Similar to previously published work (Ross et al., 2000), α4-null mice showed decreased sensitivity to nicotine, with minimal depression of locomotor activity at 1 mg/kg nicotine (crosses, F(4,168) = 6.61, p = 0.0001, Bonferroni's p < 0.001; rears, F(4,168) = 3.38, p = 0.0109) but full suppression at 2 mg/kg nicotine. α4-DA mice, however, showed increased sensitivity to nicotine-induced locomotor depression, with a significant decrease in locomotor activity at 0.5 mg/kg nicotine (crosses, Bonferroni's p < 0.001; rears, Bonferroni's p < 0.05). There was a significant interaction between genotype and nicotine in crosses (F(20,168) = 3.04; p < 0.0001).
Figure 7.
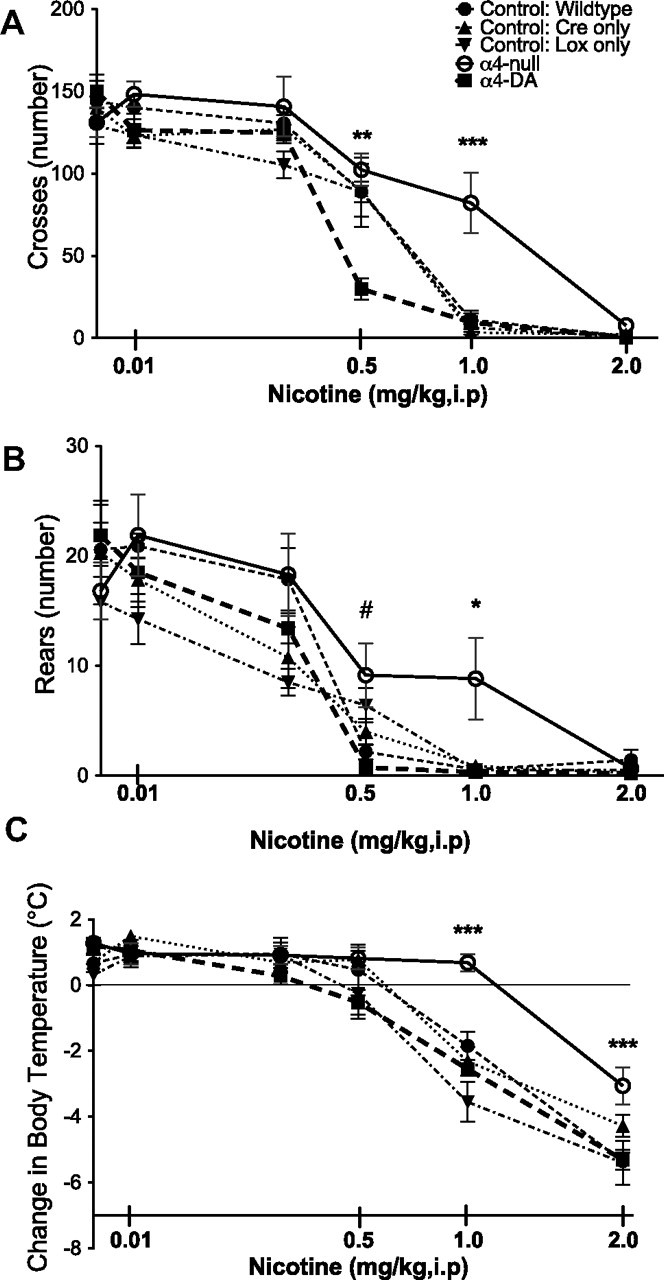
α4-containing nAChRs on dopaminergic neurons are involved in locomotor suppression but not thermoregulatory effects of nicotine. A, B, Activity [crosses (A) and rears (B)] were recorded over 3 min in the Y-maze. All groups of mice showed locomotor suppression; however, α4-null mice showed decreased sensitivity (***p < 0.001, wild type by α4-null), whereas α4-DA mice showed increased sensitivity as measured by crosses (**p < 0.05, wild type by α4-DA). α4-null mice also showed decrease sensitivity to nicotine as measure by rears (*p < 0.05, wild type by α4-null). Although α4-DA mice did not show increased sensitivity relative to control mice in rears, they did show a significant difference from α4-null mice (#p < 0.05, α4-DA by α4-null). C, Similarly, all mice showed decrease in body temperature in response to increasing doses of nicotine. α4-null mice showed decreased sensitivity (***p < 0.0001, wild type by α4-null), whereas α4-DA mice did not differ from control groups. Data are scaled as the square root of dose.
Of note, the range of nicotine doses that depress locomotor activity is considerably higher than the range of those that elicit changes in CPP or anxiety, and no effect on locomotion was detected in the range used for CPP or anxiety studies. Additionally, with this minimal anxiogenic method of locomotor testing, we did not detect the increased locomotor activity in the Cre-only control line that was seen in the elevated plus maze experiments.
α4*-nAChRs on dopaminergic neurons are not involved in nicotine-induced hypothermia
Given that previous work has suggested that α4β2*-nAChRs mediate nicotine-induced hypothermia (Tapper et al., 2004; Tritto et al., 2004), we examined whether mice lacking the α4 subunit globally or only within dopaminergic neurons have altered responses to nicotine in regard to change in body temperature. In contrast to control mice, α4-null mice did not show decreases in body temperature at 1 mg/kg nicotine (genotype effect, F(4,158) = 12.37, p < 0.0001; dose effect, F(5,158) = 145.54, p < 0.0001), consistent with reports for β2-null mice (Tritto et al., 2004). α4-DA mice responded just as control mice, indicating that dopaminergic α4*-nAChRs are not involved in nicotine-induced hypothermia. A significant interaction of dose and genotype was seen (F(20,158) = 3.15; p < 0.0001).
Discussion
Nicotine-related diseases remain at epidemic proportions around the world and are responsible for nearly six million deaths per year (Mathers and Loncar, 2006; Samet and Wipfli, 2010). Understanding the biochemistry behind nicotine-related behaviors may provide insight into novel therapies and molecular targets for treating nicotine dependence. α4β2*-nAChRs were identified as a major target of nicotine binding in the brain, and previous work has implicated these nAChRs in mediating multiple behavioral responses to nicotine.
Midbrain dopaminergic neurons are generally divided into two circuits, the mesolimbic dopaminergic pathway implicated in drug reward and stress responses (Pierce and Kumaresan, 2006; Krishnan et al., 2007), and nigrostriatal dopaminergic pathways necessary for the initiation of movement (Lima et al., 2009). α4β2*-nAChRs are highly expressed in these brain regions, which are predominantly dopaminergic but that also contain GABAergic and other cell types. α4β2*-nAChRs have been implicated in behaviors mediated by midbrain nuclei; however, it has not previously been possible to elucidate whether the α4β2*-nAChRs involved in those behaviors are on dopaminergic or other types of neurons. This work demonstrated the role of α4*-nAChRs present on dopaminergic neurons in various nicotine-related behaviors including drug reward, anxiety responses, and locomotor activity.
Selective deletion of dopaminergic α4*-nAChRs
Mice engineered with a deletion of the α4*-nAChR subunit only in dopaminergic neurons (α4-DA) were characterized extensively. In the α4-DA mice, expression of α4 mRNA and protein were eliminated selectively in dopaminergic neurons, as measured by in situ hybridization, high-affinity 125I-epibatidine binding, and colocalization of mRNA with TH and GAD67. A specific loss of all function of α4*-nAChRs at dopaminergic terminals, but not GABAergic terminals, confirmed the localized deletion.
Interestingly, both the Lox-only control and the α4-DA mice had a decrease in nicotine-stimulated GABA release in striatum but not in cortex. While insertion of loxP sites around exon V had no other detectable phenotypic or expression changes, this decrease may indicate that interruption of an intron by insertion of a loxP site may alter gene expression in specific neuronal populations. This illustrates the necessity of examining mice of all possible genotypes (wild type, Cre-only control, Lox-only control) in addition to the desired mutation (Schmidt-Supprian and Rajewsky, 2007), here the α4-DA mutant. In this case, the partial decrease in nicotine-stimulated GABA release did not alter any behavioral measurements or ligand binding, as the Lox-only control behavioral responses and ligand binding were comparable to those of wild-type and Cre-only control mice.
Nicotine CPP is mediated by dopaminergic α4*-nAChRs
Previous studies examining nicotine reward in mice with global deletions of individual nAChR subunits and restricted reintroduction of the missing subunit into the VTA have implicated α4β2*-nAChRs in dopaminergic regions in nicotine reward (Pons et al., 2008). In the work presented here, selective deletion of α4 subunits from dopaminergic neurons abolished nicotine CPP, providing direct evidence that dopaminergic α4*-nAChRs are necessary for nicotine reward. Mineur et al. (2009) found that low levels of expression of β2*nAChRs within the VTA (in dopamine and GABA neurons) is not sufficient to rescue nicotine reward, suggesting a threshold level of expression is required for nicotine reward. Our work suggests that normal expression of high-affinity nAChRs in other neuronal populations is not sufficient to maintain nicotine reward. Adequate dopaminergic expression of α4*-nAChRs is required. Interestingly, the remaining low level of activity by the α6β2β3 nAChR in dopaminergic neurons is apparently insufficient to maintain reward in the absence of α4*-nAChRs.
While previous work has suggested that α4*-nAChR-induced changes in GABAergic tone by desensitization were essential for nicotine reward (Wooltorton et al., 2003; Mansvelder et al., 2009), this current work clearly demonstrates the requirement of nAChRs on dopaminergic neurons, as α4 subunit expression in GABAergic neurons remained intact. This does not, however, eliminate the possibility that α4*-nAChRs on other neuronal populations, or other receptors on dopaminergic neurons, may play a role in nicotine reward. Nicotine-stimulated glutamate-driven burst firing of dopamine neurons is an important action of nicotine in the midbrain (Mansvelder et al., 2002, 2009) that requires α4β2*nAChRs (Exley et al., 2011). The necessity of glutamatergic (NMDA-dependent) synaptic plasticity in nicotine reward was confirmed when mice with a selective deletion of the NMDA receptor subunit NR1 in dopaminergic neurons did not develop nicotine CPP (Wang et al., 2010). Further work is required to identify whether α4-nAChRs on dopaminergic neurons are involved in this plasticity or are essential for another mechanism involved in nicotine reward.
This synaptic plasticity and other commonalties exist between nicotine and another common drug of abuse, cocaine (Zweifel et al., 2008). Previous work has suggested an endogenous role for nAChRs in cocaine reward (Bechara and van der Kooy, 1989; Zachariou et al., 2001). The common dopaminergic circuitry mediating reward for all drugs of abuse, including cocaine and nicotine (Di Chiara and Imperato, 1988; Dani and Heinemann, 1996; Nestler, 2005), and our finding that α4*-nAChRs are necessary for nicotine CPP led us to examine the role of α4*-nAChRs in cocaine reward. Our data demonstrate that while a4*-nAChRs on dopaminergic neurons are necessary for nicotine reward, they are not required for cocaine CPP. This suggests that while dopaminergic neurons are considered a final common pathway for all drugs of abuse, α4*-nAChRs on dopaminergic neurons are uniquely important for nicotine reward, rather than for the reward system in general. A previous study reported reduced cocaine CPP at a low dose (5 mg/kg) in β2-null mutant mice, whereas they displayed normal cocaine CPP at higher doses (Zachariou et al., 2001). Although a slightly different CPP protocol was used in the study reported here, α4-DA mice showed no difference in preference for cocaine at 5 or 20 mg/kg compared to control littermates. The lack of effect of α4 deletion on cocaine CPP at the low (5 mg/kg) dose may suggest that non-α4*-nAChRs that contain the β2 subunit are necessary for cocaine reward at low doses.
Anxiolytic effects of nicotine are partially mediated by dopaminergic α4*-nAChRs
We also examined whether α4*-nAChRs are involved in another key feature of nicotine dependence, anxiety changes. Smokers report anxiety relief as a key reason for continued smoking and relapse from abstinence (Pomerleau, 1986; Gilbert et al., 1989; McKee et al., 2011). Additionally, people with panic disorders have higher rates of smoking than the general population (Ziedonis et al., 2008). Previous pharmacological studies have implicated nAChRs in mediating the anxiolytic effects of nicotine (Brioni et al., 1993), and mutations in α4*-nAChRs result in alterations of basal anxiety in animals (Ross et al., 2000; Labarca et al., 2001). Additionally, previous work has implicated dopaminergic neurons in stress behaviors (George et al., 2000a,b). In this study, we identified a range of nicotine doses that decrease anxiety on the elevated plus maze in control mice. After developing this method, we were able to demonstrate that α4*-nAChRs are necessary for this response, as mice lacking α4 (α4-null) did not increase the time spent on the open (stressful) arm of the maze when given nicotine. In addition, mice specifically lacking α4*-nAChRs in dopaminergic neurons had decreased sensitivity to nicotine, suggesting that α4*-nAChRs are necessary for the anxiolytic effects of nicotine and that dopaminergic neurons are a component of the circuitry needed for this response. The lack of difference in total entries confirmed that the decreased sensitivity to the anxiolytic effects of nicotine in the elevated plus maze could not be attributed to changes in locomotor activity. Previous work has implicated many brain regions in the anxiolytic effects of nicotine, including the dorsal hippocampus and the septum (Cheeta et al., 2000); however, this is the first work to demonstrate that dopaminergic α4*-nAChRs are involved in the anxiolytic effects of nicotine.
The role of α4*-nAChRs on dopaminergic neurons in other nicotine-mediated behaviors
Previous work reported that α4-null mice have decreased sensitivity to the locomotor-depressing effects of nicotine (Ross et al., 2000), which we confirmed with our α4-null mice. Interestingly, elimination of α4*-nAChRs only on dopaminergic neurons increased the sensitivity to nicotine for the locomotor-depressing effects of nicotine. One explanation may be a change in the balance between nicotine stimulation of α4*-nAChRs on dopaminergic neurons versus GABAergic neurons, leading to an increase in inhibitory tone and thus an increased sensitivity to the locomotor-depressing effects of nicotine. The doses at which this altered sensitivity to nicotine is detected are an order of magnitude larger than those that elicit CPP or changes in anxiety. Previous groups have also examined nicotine-induced locomotor activation, which requires chronic nicotine administration, and found that while mice lacking β2*nAChRs did not have nicotine-induced locomotor activation, reexpression of low levels of β2*nAChRs were sufficient to rescue this behavior (Mineur et al., 2009). Taken with data presented here, this suggests that dopaminergic regions may control both the inhibitory and stimulatory effects of nicotine on locomotion, although further work is required to identify if α4*-nAChRs on dopaminergic neurons are necessary.
We also examined a behavior that we did not expect to be altered by dopaminergic deletion of α4*-nAChRs, nicotine-induced hypothermia. Null mutant mice lacking β2*-nAChRs were reported previously to have decreased sensitivity to nicotine-induced hypothermia, suggesting that activation of α4β2*-nAChRs are central to this behavior (Tritto et al., 2004). In the study reported here, α4-null mice showed decreased sensitivity to nicotine-induced hypothermia, whereas deletion of dopaminergic α4*-nAChRs do not influence this behavior.
Conclusions
Here we report that dopaminergic α4*-nAChRs are involved in nicotine CPP, anxiety reduction, and locomotor suppression; however, dopaminergic α4*-nAChRs are not involved in cocaine CPP or nicotine-induced hypothermia. To our knowledge, this is the first mouse with a deletion of an nAChR subunit within a single neuronal population that identifies the role of a specific nAChR subtype in nicotine-related behaviors.
Identification of the subunit composition of the nAChRs in the neuronal pathway uniquely necessary for two key features of nicotine dependence, reward and anxiety, may provide an extraordinary opportunity to understand the circuitry and biochemistry that lead to nicotine dependence. While further studies are needed to examine the role of α4*-nAChRs in other nicotine-dependence-related behaviors such as tolerance to and withdrawal from nicotine, this work implicates dopaminergic α4*-nAChRs in major acute nicotine behaviors. This work also begins to distinguish specific neuronal populations involved in α4*-nAChR-mediated nicotine-related behaviors.
Footnotes
This work was supported by NIH Grants NS10830, AA13018, DA018247, DA003194, and DA012242. We thank Dr. Michael McIntosh (University of Utah, Salt Lake City, UT) for the generous donation of α-conotoxin MII, Dr. Henry Lester (California Institute of Technology, Pasadena, CA) for the use of the CPP apparatus and targeting construct backbone, Dr. Jim Boulter (University of California, Los Angeles) for mouse a4 clones, and the Embryonic Stem Cell Core at Washington University (St. Louis, MO) for pTurbo-Cre. We are also grateful to Dr. Allan C. Collins (University of Colorado, Boulder, CO) for his helpful discussions and revisions of this manuscript.
The authors declare no competing financial interests.
References
- Bechara A, van der Kooy D. The tegmental pedunculopontine nucleus: a brain-stem output of the limbic system critical for the conditioned place preferences produced by morphine and amphetamine. J Neurosci. 1989;9:3400–3409. doi: 10.1523/JNEUROSCI.09-10-03400.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brioni JD, O'Neill AB, Kim DJ, Decker MW. Nicotinic receptor agonists exhibit anxiolytic-like effects on the elevated plus-maze test. Eur J Pharmacol. 1993;238:1–8. doi: 10.1016/0014-2999(93)90498-7. [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Mineur YS, Neve RL, Picciotto MR. Nucleus accumbens CREB activity is necessary for nicotine conditioned place preference. Neuropsychopharmacology. 2009;11:1993–2001. doi: 10.1038/npp.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeta S, Kenny PJ, File SE. Hippocampal and septal injections of nicotine and 8-OH-DPAT distinguish among different animal tests of anxiety. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24:1053–1067. doi: 10.1016/s0278-5846(00)00129-9. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago J, McColl CD, Horne MK, Finkelstein DI, Ross SA. Neuronal nicotinic receptors: insights gained from gene knockout and knockin mutant mice. Cell Mol Life Sci. 2003;60:1267–1280. doi: 10.1007/s00018-003-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Maubourguet N, David V, Eddine R, Evrard A, Pons S, Marti F, Threlfell S, Cazala P, McIntosh JM, Changeux JP, Maskos U, Cragg SJ, Faure P. Distinct contributions of nicotinic acetylcholine receptor subunit α4 and subunit α6 to the reinforcing effects of nicotine. Proc Natl Acad Sci U S A. 2011;108:7577–7582. doi: 10.1073/pnas.1103000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George TP, Verrico CD, Xu L, Roth RH. Effects of repeated nicotine administration and footshock stress on rat mesoprefrontal dopamine systems: evidence for opioid mechanisms. Neuropsychopharmacology. 2000a;23:79–88. doi: 10.1016/S0893-133X(99)00165-7. [DOI] [PubMed] [Google Scholar]
- George TP, Verrico CD, Picciotto MR, Roth RH. Nicotinic modulation of mesoprefrontal dopamine neurons: pharmacologic and neuroanatomic characterization. J Pharmacol Exp Ther. 2000b;295:58–66. [PubMed] [Google Scholar]
- Gilbert DG, Robinson JH, Chamberlin CL, Spielberger CD. Effects of smoking/nicotine on anxiety, heart rate, and lateralization of EEG during a stressful movie. Psychophysiology. 1989;26:311–320. doi: 10.1111/j.1469-8986.1989.tb01924.x. [DOI] [PubMed] [Google Scholar]
- Grabus SD, Martin BR, Brown SE, Damaj MI. Nicotine place preference in the mouse: influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology (Berl) 2006;184:456–463. doi: 10.1007/s00213-006-0305-7. [DOI] [PubMed] [Google Scholar]
- Grady SR, Grun EU, Marks MJ, Collins AC. Pharmacological comparison of transient and persistent [3H]dopamine release from mouse striatal synaptosomes and response to chronic L-nicotine treatment. J Pharmacol Exp Ther. 1997;282:32–43. [PubMed] [Google Scholar]
- Grady SR, Meinerz NM, Cao J, Reynolds AM, Picciotto MR, Changeux JP, McIntosh JM, Marks MJ, Collins AC. Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: a function mediated by a different nAChR than dopamine release from striatum. J Neurochem. 2001;76:258–268. doi: 10.1046/j.1471-4159.2001.00019.x. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74:1235–1246. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilby NJ, Snaith MR, Murray JA. Site-specific recombinases: tools for genome engineering. Trends Genet. 1993;9:413–421. doi: 10.1016/0168-9525(93)90104-p. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Heterogeneity of ventral tegmental area neurons: single-unit recording and iontophoresis in awake, unrestrained rats. Neuroscience. 1998;85:1285–1309. doi: 10.1016/s0306-4522(98)00054-2. [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–1463. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, Nashmi R, Kofuji P, Dang H, Shi W, Fidan M, Khakh BS, Chen Z, Bowers BJ, Boulter J, Wehner JM, Lester HA. Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci U S A. 2001;98:2786–2791. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima MM, Reksidler AB, Vital MA. The neurobiology of the substantia nigra pars compacta: from motor to sleep regulation. J Neural Transm Suppl. 2009;73:135–145. doi: 10.1007/978-3-211-92660-4_11. [DOI] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Mertz M, Role LW. Nicotinic modulation of synaptic transmission and plasticity in cortico-limbic circuits. Semin Cell Dev Biol. 2009;20:432–440. doi: 10.1016/j.semcdb.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- Maskos U. Role of endogenous acetylcholine in the control of the dopaminergic system via nicotinic receptors. J Neurochem. 2010;114:641–646. doi: 10.1111/j.1471-4159.2010.06798.x. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee SA, Sinha R, Weinberger AH, Sofuoglu M, Harrison EL, Lavery M, Wanzer J. Stress decreases the ability to resist smoking and potentiates smoking intensity and reward. J Psychopharmacol. 2011;25:490–502. doi: 10.1177/0269881110376694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Brunzell DH, Grady SR, Lindstrom JM, McIntosh JM, Marks MJ, King SL, Picciotto MR. Localized low-level re-expression of high-affinity mesolimbic nicotinic acetylcholine receptors restores nicotine-induced locomotion but not place conditioning. Genes Brain Behav. 2009;8:257–266. doi: 10.1111/j.1601-183X.2008.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- O'Neill AB, Brioni JD. Benzodiazepine receptor mediation of the anxiolytic-like effect of (−)-nicotine in mice. Pharmacol Biochem Behav. 1994;49:755–757. doi: 10.1016/0091-3057(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol. 2000;61:75–111. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Caldarone BJ, Brunzell DH, Zachariou V, Stevens TR, King SL. Neuronal nicotinic acetylcholine receptor subunit knockout mice: physiological and behavioral phenotypes and possible clinical implications. Pharmacol Ther. 2001;92:89–108. doi: 10.1016/s0163-7258(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev. 2006;30:215–238. doi: 10.1016/j.neubiorev.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Pomerleau OF. Nicotine as a psychoactive drug: anxiety and pain reduction. Psychopharmacol Bull. 1986;22:865–869. [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J. Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, Grady SR. Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol. 2007;71:1563–1571. doi: 10.1124/mol.106.031492. [DOI] [PubMed] [Google Scholar]
- Samet JM, Wipfli HL. Globe still in grip of addiction. Nature. 2010;463:1020–1021. doi: 10.1038/4631020a. [DOI] [PubMed] [Google Scholar]
- Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8:665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Tritto T, McCallum SE, Waddle SA, Hutton SR, Paylor R, Collins AC, Marks MJ. Null mutant analysis of responses to nicotine: deletion of beta2 nicotinic acetylcholine receptor subunit but not alpha7 subunit reduces sensitivity to nicotine-induced locomotor depression and hypothermia. Nicotine Tob Res. 2004;6:145–158. doi: 10.1080/14622200310001656966. [DOI] [PubMed] [Google Scholar]
- Wang LP, Li F, Shen X, Tsien JZ. Conditional knockout of NMDA receptors in dopamine neurons prevents nicotine-conditioned place preference. PLoS One. 2010;5:e8616. doi: 10.1371/journal.pone.0008616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SS, Epping-Jordan MP, Koob GF, Markou A. Blockade of nicotine self-administration with nicotinic antagonists in rats. Pharmacol Biochem Behav. 1999;62:743–751. doi: 10.1016/s0091-3057(98)00226-3. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol. 2000a;131:729–739. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ. 125I-alpha-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol. 2000b;57:913–925. [PubMed] [Google Scholar]
- Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ. Involvement of the alpha3 subunit in central nicotinic binding populations. J Neurosci. 2002;22:2522–2529. doi: 10.1523/JNEUROSCI.22-07-02522.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Roles for nigrostriatal—not just mesocorticolimbic—dopamine in reward and addiction. Trends Neurosci. 2009;32:517–524. doi: 10.1016/j.tins.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Caldarone BJ, Weathers-Lowin A, George TP, Elsworth JD, Roth RH, Changeux JP, Picciotto MR. Nicotine receptor inactivation decreases sensitivity to cocaine. Neuropsychopharmacology. 2001;24:576–589. doi: 10.1016/S0893-133X(00)00224-4. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Ziedonis D, Hitsman B, Beckham JC, Zvolensky M, Adler LE, Audrain-McGovern J, Breslau N, Brown RA, George TP, Williams J, Calhoun PS, Riley WT. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10:1691–1715. doi: 10.1080/14622200802443569. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–496. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]