

Abstract
The immaturity of the CNS at birth greatly affects injury after stroke but the contribution of the blood–brain barrier (BBB) to the differential response to stroke in adults and neonates is poorly understood. We asked whether the structure and function of the BBB is disrupted differently in neonatal and adult rats by transient middle cerebral artery occlusion. In adult rats, albumin leakage into injured regions was markedly increased during 2–24 h reperfusion but leakage remained low in the neonates. Functional assays employing intravascular tracers in the neonates showed that BBB permeability to both large (70 kDa dextran) and small (3 kDa dextran), gadolinium (III)-diethyltriaminepentaacetic acid tracers remained largely undisturbed 24 h after reperfusion. The profoundly different functional integrity of the BBB was associated with the largely nonoverlapping patterns of regulated genes in endothelial cells purified from injured and uninjured adult and neonatal brain at 24 h (endothelial transcriptome, 31,042 total probe sets). Within significantly regulated 1266 probe sets in injured adults and 361 probe sets in neonates, changes in the gene expression of the basal lamina components, adhesion molecules, the tight junction protein occludin, and matrix metalloproteinase-9 were among the key differences. The protein expression of collagen-IV, laminin, claudin-5, occludin, and zonula occludens protein 1 was also better preserved in neonatal rats. Neutrophil infiltration remained low in acutely injured neonates but neutralization of cytokine-induced neutrophil chemoattractant-1 in the systemic circulation enhanced neutrophil infiltration, BBB permeability, and injury. The markedly more integrant BBB in neonatal brain than in adult brain after acute stroke may have major implications for the treatment of neonatal stroke.
Introduction
Breakdown of the blood–brain barrier (BBB) is an important contributing factor to injury in many brain diseases, including stroke. A number of different, partially independent components, including the extracellular matrix, tight junctions (TJs), pericytes, and astrocyte endfeet, together with adherens junctions, form junctional complexes and play a central role in the control of BBB permeability and maintenance of cell polarity (Bazzoni et al., 2000; Zlokovic, 2008). After stroke in the adult, BBB disruption can either occur transiently, in two distinct phases (Belayev et al., 1996; Rosenberg et al., 1998), or be continuous (McColl et al., 2008) during the acute injury phase, with the extent and timing dependent on age, genetic background, and gender. Stroke severity is also exacerbated by predisposing factors, such as infection or systemic inflammation, that affect various components of the BBB (Denes et al., 2010).
Strikingly, the incidence of arterial stroke in term human babies is similar to that in the elderly (Lynch, 2009). Although basic mechanisms of neurodegeneration after stroke are shared across age groups, immaturity critically affects brain susceptibility and response to ischemia-related insults, including modes of neuronal cell death, inflammation, leukocyte-mediated injury, and enhanced susceptibility to reactive oxygen species (ROS) (Vexler and Yenari, 2009). However, the role of the immature BBB in the differential response to ischemic-reperfusion injury in mature and neonatal brain remains largely unknown.
Emerging evidence suggests that the early postnatal BBB is not as permeable as once thought. Although mechanisms of the BBB's function in the fetus are different from those in the adult (Saunders et al., 1999), TJs are present early in embryonic development (Kniesel et al., 1996), restricting entrance of proteins into the brain in a controlled fashion. Furthermore, expression of the influx and efflux transporters during midgestation is even higher than in the adult (Ek et al., 2010). At birth, the BBB is functional with no fenestrations (Engelhardt, 2003) but gene expression of BBB endothelial proteins undergoes changes in normal brain from postnatal period to adulthood (Daneman et al., 2010b). However, BBB permeability does not decrease linearly with age under injurious conditions as was evident from the profoundly higher BBB permeability after intrastriatal injections of inflammatory cytokines in postnatal day 21 (P21) rats than in 2 h-old rat pups (Anthony et al., 1997, 1998).
We asked whether arterial stroke affects BBB permeability in neonatal and young adult brain differently. Using an age-appropriate model of neonatal focal stroke that we developed and characterized (Derugin et al., 1998, 2000; Manabat et al., 2003)—a transient middle cerebral artery occlusion (MCAO) in P7 rats—and a similar model in the adult, we examined both the structural and functional aspects of the BBB after acute stroke. We discovered that BBB was markedly more intact in neonatal rats than in adult rats after acute MCAO, in part because of the differential expression of the basal lamina and TJ proteins and neutrophil behavior.
Materials and Methods
Animal preparation.
All experimental procedures were performed, with prior approval from the Committee of Animal Research at the University of California San Francisco, in accordance with NIH guidelines for humane handling of animals. Female Sprague Dawley rats with a 5–6 d-old litter (10 pups per litter) and adult male Sprague Dawley rats 250–280 g were obtained from Simonsen Laboratories. Rats were given food and water ad libitum and housed in a temperature/light-controlled animal care facility.
MCAO.
P7 rats underwent a transient 3 h suture MCAO as we initially described (Derugin et al., 1998), with modifications (Derugin et al., 2005), using a coated monofilament Dermalon suture (6-0). Adult male rats were subjected to a similar procedure (Longa et al., 1989). Briefly, the right common carotid artery was identified, and the external carotid artery (ECA) coagulated and cut. A 4-0 Dermalon suture with the tip rounded by heat was inserted through the ECA, and advanced through the internal carotid artery (ICA) 17 mm past the ICA–ECA bifurcation. Reperfusion was achieved by retracting the occluding filament. The proximal end of the ECA was then tied off, and the filament suture completely removed.
MRI.
MRI was performed using a 2 tesla magnet with a Bruker Omega system equipped with actively shielded gradients that provide ±200 mTm−1 gradient amplitude. The instrument settings for T2-weighted, diffusion-weighted imaging (DWI) and perfusion-sensitive sequences were as previously described (Derugin et al., 2005). Spin echo (SE) DWI was performed in all animals ∼2 h after MCAO to confirm the desired injury pattern, as previously described (Derugin et al., 2000; Manabat et al., 2003). Integrity of the BBB was evaluated using T1-weighted MRI in conjunction with gadolinium (III)-diethyltriaminepentaacetic acid (Gd-DTPA, 0.3mmol/kg, Magnevist, i.v.), as described previously (Dzietko et al., 2011).
Perfusion-sensitive MRI.
Perfusion-sensitive MRI was performed in subgroups of rats of each age. SE-EPI, a sequence sensitive to perfusion, was used to measure the MRI signal changes during the first pass of a magnetic susceptibility MRI contrast agent, dysoprosium DTPA-bis-(methylamide) (DyDTPA-BMA, 0.25mmol/kg, Nicomed-Salutar, i.v.), as first described for adults (Kucharczyk et al., 1993) and then for neonates (Derugin et al., 2000). Contrast agent was administered via jugular vein of P7 rats or tail vein of adult rats (n = 3–6 per group) 2.5 h after MCAO, 30 min and 24 h postreperfusion. Since the contrast agent effect, characterized by ΔR2*, has been shown to be approximately proportional to its concentration (Moseley et al., 1991), the ΔR2* was used to estimate relative cerebral blood volume (rCBV).
MRI image analysis.
MRI image analysis was performed using commercially available software (MRVision). All images were subjected to a combination of threshold and segmentation operations to eliminate all but brain pixels. The size of hyperintense region on the DWI during occlusion (region of slow water diffusion) was quantified by selecting pixels with signal greater than mean ± 1.5 SD of that in adjacent tissue, as previously described (Manabat et al., 2003). On SE-EPI, boundaries were drawn based on information from DWI.
Evans blue injection and visualization.
At 2 h after reperfusion, 2% Evans blue (EB) solution in 0.1 m PBS was slowly injected into the jugular vein of P7 rats (0.04 ml/15 g of rat) or tail vein of adult rats (0.8 ml/200 g of rat) and let circulate for 20–22 h. Trunk blood and brain tissue from injured and matching contralateral regions were collected and flash-frozen. EB was extracted by incubating plasma or tissue samples with 50% TCA solution (1:3), spinning at 10,000× and incubating in ethanol (1:3). Fluorescence (590/645) was measured using a multiplate reader (Synergy, BioTek) equipped with a K4 Software and calibration curve. The fluorescence intensities were normalized to wet tissue weight.
Intravascular injection and detection of fluorescence tracers.
Alexa-555 conjugate bovine serum albumin (1.85 mg/20 g rat, Invitrogen), tetramethylrhodamine conjugated 70 kDa dextran (2 mg/20 g rat, Invitrogen), or fluorescein conjugated 3 kDa dextran (2 mg/20 g of rat, Invitrogen) were injected into the jugular vein of neonatal rats 24 h after reperfusion. One hour later, animals were perfused transcardiacally with HBSS followed by ice-cold 4% PFA in 0.1 m PBS. Postfixed (4% PFA for 16–18 h) and flash-frozen brains were cut on cryostat (thickness, usually 12 μm, in some cases 50 μm). Vessels were labeled with mouse anti-rat endothelial cell antigen 1 (anti-RECA-1, 1:200, AbD Serotec) and isolectin (IB4; 1:100, Invitrogen), and nuclei were counterstained with DAPI. Stained sections were visualized and digitalized using a Zeiss AxioImager Z-2 microscope coupled to a digital camera (Hammamatsu) and equipped with Volocity Software and a confocal grid (Optigrid, Improvision).
Endothelial transcriptome.
Endothelial cells were purified from ischemic regions based on DWI images and from the contralateral hemisphere of neonatal and adult rats 24 h post-MCAO (n = 4 per group), as previously published (Daneman et al., 2009, 2010b). Briefly, rats were euthanized by decapitation with a guillotine, and cerebral cortex was dissected. Brain tissue was then dissociated enzymatically with papain followed by mechanical trituration, and endothelial cells were purified from the cell suspension by sequential negative [mouse anti-rat CD45, Serotec; goat anti-PDGF receptor β (anti-PDGFRβ), R&D Systems; and secondary antibody only] and positive (mouse anti-rat CD31, Fitzgerald) immunopanning steps. RNA purification and amplification was performed as previously described (Daneman et al., 2010a,b) and hybridized to Affymetrix Rat 230 2.0 Array chip. The data was normalized using the robust multiarray average algorithm and statistical analysis was performed using the ArrayStar program from DNASTAR with a Student's t test and Benjamini–Hochberg false discovery rate correction. Genes were filtered with differential expression of two with 95% confidence.
Western blot analysis.
Protein expression was determined in lysates from injured and contralateral tissue using rabbit anti-collagen IV (Col-IV; 1:1000, Abcam), rabbit anti-laminin (1:1000, Novus Biologicals), mouse anti-occludin (1:500, Invitrogen), mouse anti-claudin-5 (1:500, Invitrogen), goat anti-PDGFRβ (1:500, R&D Systems), and rabbit anti-zonula occudens protein 1 (anti-ZO1, 1:200, Invitrogen) diluted in blocking buffer (5% milk in 0.2% Tween 20/TBS, 4°C, overnight). The results were normalized to actin expression (mouse anti-β-actin, 1:5000, Sigma-Aldrich).
Immunofluorescence and Image analysis.
Rats were anesthetized, perfused transcardiacally with 4% PFA in 0.1 m PBS, the brains were removed, postfixed (4% PFA, 4°C, 24 h), and cryoprotected. Slides containing 12 μm serial sections (500 μm apart) were either stained with Nissl or incubated with blocking solution (10% normal goat serum, 0.1% TX-100 in 0.1 m PBS) for 1 h at room temperature and with mouse anti-RECA-1 (1:200, AbD Serotec), rabbit anti-Col-IV (1:1000, Abcam), rabbit anti-laminin (1:1000, overnight at 4°C, Novus Biologicals). Sections were then incubated with secondary goat anti-mouse Alexa 488, goat anti-rabbit Alexa 568, and IB4 Alexa 657 (1:100, Invitrogen) for 1 h at room temperature. Nuclei were counterstained with DAPI. For antigen retrieval of Col-IV and laminin, slides were incubated with Digest All 3 pepsin solution (Invitrogen) in 0.2N HCl (1:4 for adult brain sections, 1:20 for neonate brain sections, 10 min, 37°C before the incubation with blocking solution).
For quantitative analysis of Col-IV, laminin, and RECA-1-positive brain vessels, three 10 μm-thick confocal-like Z-stacks (1 μm z-step, 25× objective) per injured and contralateral caudate were acquired in three consecutive serial sections (using Optigrid). Images were thresholded based on the signal intensity 2 SDs over the background. The sum volume (mm3) of selected objects was determined in three 3D-voxel fields-of-view per region, and volume density was normalized to the total sampled volume.
ZO1 immunoflurescence was determined in perfusion-fixed paraffin-embedded sections. Paraffin-embedded sections were deparaffinized in 100% Citrusolve for 1 h and placed through a series of alcohols with decreasing concentration. Antigen retrieval was then performed by incubating slides in 1 mg/ml Protease Streptomyces Griseus (Sigma-Aldrich) in 0.1 m PBS for 10 min at 37°C. Slides were blocked for 1 h at room temperature in 10% donkey serum/0.2% TX-100/0.1 m PBS, incubated in the rabbit anti-rat ZO1 antibody (Zymed, 1:60, overnight, 4°C) and then incubated with FITC-conjugated IB4 (1:100, Invitrogen) and goat anti-rabbit Cy-3 secondary antibody (1:100, Jackson) for 1 h at room temperature. Slides then were incubated in bis-benzimide (1×, Sigma-Aldrich) for 5 min. Confocal microscopy was used to delineate the 3D distribution of ZO-1 in injured and matching contralateral tissue at 24 h postreperfusion (n = 4 animals per group; Zeiss Confocal Microscope and LSM 510 software). Planes for image acquisition were chosen based on the appearance of bis-benzimide-stained nuclei. Multiple fields-of-view were captured within the ischemic core and normal tissue, using the 20× and 63× objectives. Z-stacks were obtained using a 0.5 μm step at a 63× magnification (15–20 planes per slice).
Administering cotton rat cytokine-induced neutrophil chemoattractant-1 (crCINC-1), neutralizing anti-cytokine-induced neutrophil chemoattractant antibody (anti-CINC-1), and identifying neutrophils.
The presence of neutrophils was determined on H&E-stained brain sections at 1, 4, 8, 24, and 72 h and at 7 d after MCAO or at 4, 18–22 h after stereotactic injection of recombinant cotton rat cytokine-induced neutrophil chemoattractant-1 (CINC-1)/growth-regulated oncogene α (GROα) [rrCINC-1; 1 μg/2 μl or 2 μg/2 μl, dissolved in 0.4% bovine serum albumin (BSA) in 0.1 m PBS, R&D Systems] or vehicle (0.4% BSA in 0.1 m PBS) slowly injected into the striatum (2.5 mm anterior and 2.5 mm lateral from bregma, 3 mm deep). A neutralizing anti-CINC-1 antibody (2.5 μg/15 g of pup in PBS, R&D Systems, intrajugular), or equivalent volumes of IgG or PBS were administered 15 min before MCAO. Pups were sacrificed either by perfusion-fixation through the heart for identification of neutrophils by an anti-polymorphonuclear (anti-PMN) serum (1:100, Cedarlane) or on H&E or by flash-freezing the brain, with or without saline perfusion, for immunofluorescence using an anti-granulocyte antibody (His48 antibody). For immunostaining, flash-frozen brain sections (12 μm) were fixed in acetone at −20°C, blocked (1 h, room temperature, 10% goat serum) and the slides were incubated in mouse anti-rat His48 antibody (1:100, 2% goat serum, overnight, 4°C, BD Biosciences) and then goat anti-mouse IgM antibody (1:100, 1 h, room temperature, 2% goat serum, Jackson ImmunoResearch), and incubated in bis-benzimide (1×, Sigma-Aldrich).
Statistical analysis.
Data were analyzed by ANOVA with post hoc testing (Bonferroni) for multiple groups or paired t test when changes in injured and uninjured tissue were compared between two groups. Differences were considered significant at p < 0.05. Results are shown as mean ± SD.
Results
Extravasation of EB dye is markedly increased in the adult brain but not in the neonatal brain within 24 h after reperfusion following MCAO
We first asked if BBB permeability to high molecular weight proteins is increased after acute focal stroke in adult and neonatal rats. To determine albumin extravasation in the tissue, we used EB, a 961Da dye that becomes strongly bound to the albumin fraction of proteins and makes a high molecular weight complex (68.5 kDa). EB does not permeate the intact BBB but easily permeates the compromised BBB after brain injury, such as stroke in adult rats (Belayev et al., 1996).
Injured P7 and adult rats were identified on DWI during MCAO (Fig. 1C) (Derugin et al., 2000; Manabat et al., 2003) and leakage of EB was determined in rats of both ages following its circulation between 2 and 24 h after reperfusion. As expected, EB leakage was minimal in the contralateral hemisphere in both age groups. Leakage was markedly increased in the injured cortex (4.7-fold) and the caudate (14.4-fold) of adult rats, whereas only a 2.1-fold increase in leakage was observed in the injured neonatal brain (Fig. 1A,B). The concentration of circulating EB was similar in both age groups—∼10-fold higher than that in the contralateral hemisphere of the same animals—indicating that the observed difference in its brain accumulation between adult and neonatal rats was not due to insufficient availability of the dye.
Figure 1.
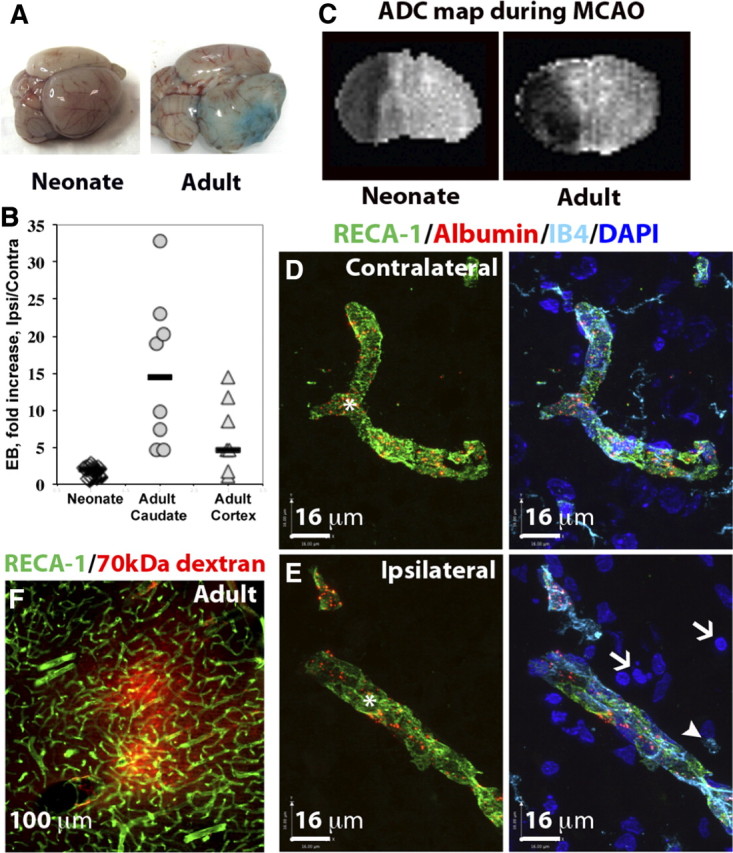
Blood–brain barrier permeability is markedly increased in injured brain regions in the adult but is largely preserved in the neonate after acute MCAO. A, Representative whole brains showing Evans blue extravasation and accumulation in neonatal and adult brain 24 h after reperfusion. B, Quantification of intravenously administered Evans blue in the brain between 2 and 24 h after reperfusion. Evans blue accumulation is profoundly increased in the adult but not in the neonatal rat. Numbers show how many times greater the accumulation of Evans blue occurred in the injured region as compared to the contralateral region in the same rat. Shown are data for individual rats; horizontal bars indicate medians. C, Apparent diffusion coefficient (ADC) maps showing a similar extension of brain edema during MCAO in neonate and adult brains. D, E, The spatial distribution of intravenously administered Alexa-555-conjugated albumin in contralateral (D) and injured (E) cortical regions in neonates 24 h after reperfusion. Injured areas in the ipsilateral hemisphere (E) were identified by the presence of pyknotic nuclei (DAPI, white arrows) and round, ameboid-like IB4+/RECA-1− microglia/macrophages (white arrowheads). In the injured areas, intravenous tracers colocalized with brain vessels (RECA1+/IB4+, asterisk) and were not observed in the extravascular spaces or in phagocytic microglia/macrophages (white arrowheads). Green, RECA-1; turquoise, IB4; blue, DAPI. Sections are 12 μm thick. F, Extravasation of Alexa-555-conjugated albumin into the injured cortex of adult rats 24 h after reperfusion (section thickness, 50 μm).
Distribution of intravenously administered albumin, 70 kDa dextran, and 3 kDa dextran is restricted to the brain vasculature in injured neonatal brain 24 h after reperfusion
To further characterize the BBB's functional integrity after acute neonatal stroke, we intravenously administered fluorescently labeled albumin (65 kDa) and tracers of various sizes, including 70 kDa and 3 kDa dextran, and determined the spatial tracer distribution within contralateral and injured caudate and cortex.
Albumin distribution was limited to the vessels in contralateral tissue (Fig. 1D, red, asterisk). A similar distribution pattern was observed in injured tissue at 24 h: limited to the vessels and not found in injured parenchyma (Fig. 1E, red, asterisk), which was identified by the presence of condensed and irregular nuclei appearance (DAPI, white arrows) and round (ameboid) IB4+/RECA-1− microglia/macrophages (Fig. 1E, arrowhead). A similar intravascular distribution pattern was observed in both injured and matching contralateral tissue after injection of 70 kDa dextran at 4 h (data not shown) and 24 h (Fig. 2A,B, red). In all cases, the intensity of the extravascular fluorescence in injured regions was similar to that in the corresponding contralateral regions and in negative controls (data not shown), indicating that permeability of the neonatal BBB to large tracers (∼70 kDa) of differing chemical structure is minimal in injured regions. Moreover, both tracers showed a characteristic vesicular pattern, with vesicles colocalized with the endothelial cell marker RECA-1 (Fig. 2B). Vesicles were absent in IB4+/RECA-1− cells, indicating that tracers are not internalized by activated microglia/macrophages (white arrows). In contrast, in adult rats, extravasation of 70 kDa dextran was apparent in injured regions (Fig. 1F).
Figure 2.
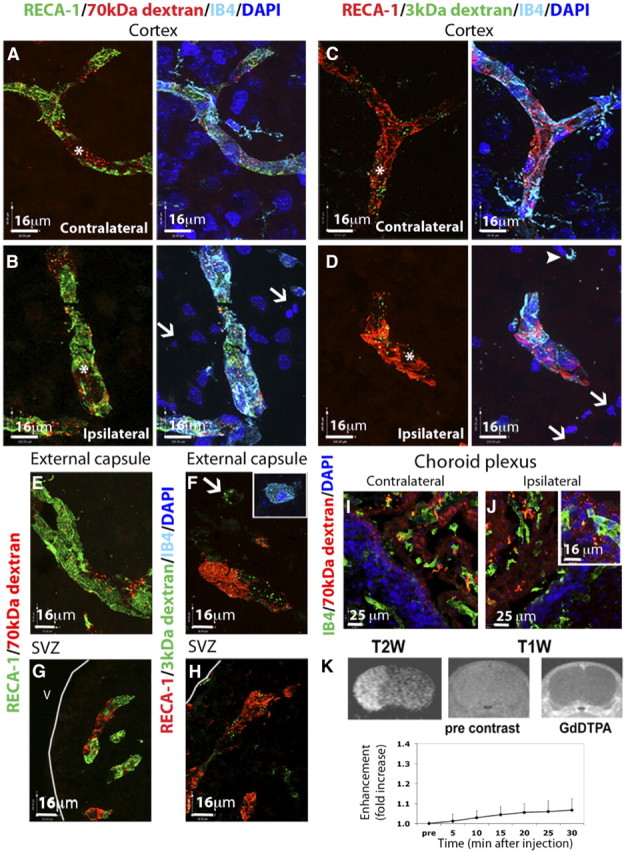
Extravasation of intravascular tracers of different size is low in the injured regions of the neonate after acute MCAO. A–D, The distribution of TRITC-conjugated 70 kDa dextran (A, B) and fluorescein-conjugated 3 kDa dextran (C, D) was restricted to the vasculature (RECA-1+/IB4+ vessels, asterisks) in the contralateral (A, C) brain hemisphere. In the injured areas (B, D), intravenous tracers colocalized with brain vessels (RECA1+/IB4+, asterisks) and were not observed in the extravascular spaces. E, F, The distribution of 3 and 70 kDa dextran was restricted to the brain vasculature in the ipsilateral external capsule 24 h after reperfusion (E, F), although occasional 3 kDa signal was detected in microglia/macrophages located in this brain region (F, arrow and inset). G, H, No leakage of 70 kDa dextran (G) or 3 kDa dextran (H) was observed in the ipsilateral SVZ 24 h after reperfusion. I, J, Leakage of 70 kDa dextran was detected in the choroid plexus in both contralateral (I) and ipsilateral (J) ventricles. K, Coronal T1-weighted images showing contrast imaging of Gd-DTPA 24 h after neonatal MCAO. Contrast was increased in areas outside the brain (right) but was minimal in the brain up to 30 min after injection of Gd-DTPA (right vs middle, bottom graph). Injured areas were identified in the same animals by T2-weighted imaging (left). V, ventricle.
Injection of a much smaller tracer, a 3 kDa dextran, also showed the intravascular pattern of tracer distribution (Fig. 2C,D, green), with essentially unchanged signal intensity outside the vessels in injured regions, thus demonstrating the largely preserved BBB impermeability to a smaller tracer in acutely injured regions in neonates.
The BBB remained largely impermeable to 70 kDa dextran (Fig. 2E, green) in the white matter, a region with occasional activated microglia/macrophages in naive neonates. Meanwhile, 3 kDa dextran also remained intravascular and was only occasionally detected in IB4+ macrophages in this region (Fig. 2F, arrow and insert). No tracer extravasation was seen in the subventricular zone (Fig. 2G,H). However, extravasation of 70 kDa dextran was apparent bilaterally in the choroid plexus, where capillaries are fenestrated (Fig. 2I,J).
Gd-DTPA enhancement is minimal 24 h after reperfusion in neonatal rats
To independently determine the extent of the BBB disruption in living neonatal rats, T1-weighted MRI enhanced with Gd-DTPA was used. While the presence of injury was evident on T2-weighted MRI 24 h after MCAO (Fig. 2K, left), and Gd-DTPA enhancement was clearly seen in the tissue surrounding the brain (Fig. 2K, right), no enhancement was observed in brain tissue contralateral to MCAO, and only minimal Gd-DTPA enhancement (<10%) was seen in the injured tissue up to 30 min after Gd-DTPA injection (Fig. 2K, bottom). Taken together, these results demonstrate a limited permeability of the BBB after acute cerebral ischemia in the neonate to tracers of various size and chemical structure.
The patterns of the disrupted cerebral perfusion in ischemic-reperfused tissue are similar in both adult and neonatal rats
Considering that insufficient cerebral microcirculation may adversely affect BBB integrity and tracer distribution, we then used perfusion-sensitive MRI to noninvasively determine the severity of perfusion deficits during MCAO and the extent of restored perfusion 30 min or 24 h after suture retraction. Using SE-EPI and a bolus of DyDTPA-BMA in subgroups of adult and neonates, we determined the rCBV, relative cerebral blood flow (rCBF), and delay in contrast transit in a region with abnormal DWI compared to that in the matching region within contralateral hemisphere in the same animal.
A relative change in the ΔR2* during the first pass of the DyDTPA-BMA passage showed, as expected (Derugin et al., 2000), a transient signal intensity change through the normal but not the ischemic brain regions in the neonatal and adult brain. Quantification of the ratio of the peak ΔR2* of the ipsilateral versus the contralateral cortex showed residual perfusion of 16 ± 5% in P7 rats (n = 4) and 15 ± 7% in adult rats (n = 3). Therefore, the severity of perfusion deficits was similar during MCAO in both age groups.
Retraction of the suture filament was associated with a partial restoration of perfusion in previously occluded cortical regions in neonatal and adult rats (data not shown). At 24 h after reperfusion, a time point when intravascular tracers were injected (Figs. 1, 2), T2-weighted hyperintensity was observed in both age groups (Fig. 2K), and T2-weighted-enhanced cortical regions remained well perfused: the relative peak ΔR2*, which is proportional to rCBF, was 55 ± 6% in injured tissue compared to matching contralateral regions of neonates (n = 4) and was 83 ± 13% in injured tissue of adults compared to matching contralateral regions (n = 3). There was no significant delay in contrast arrival to injured regions compared to contralateral regions (1.08 ± 0.06 in neonates vs 1.00 ± 0.01 in adults), and the relative transit time of the first pass of the DyDTPA-BMA was 1.28 ± 0.63 in the neonates and 0.95 ± 0.12 in adults. Therefore, the leakage of EB in the adult but a lack of leakage in neonatal rats cannot be explained by the age-associated deficiency of cerebral perfusion during reperfusion.
Stroke differentially affects endothelial gene expression in adults and neonates
Given that many of the properties of the BBB are manifested in the endothelial cells (Daneman et al., 2010b), we compared the transcriptional profiles in endothelial cells isolated by immunopanning through negative and positive selection from adult and neonatal injured brain regions, along with cells isolated from matching contralateral brain tissue, 24 h after MCAO.
With a total of 31,042 probe sets used to determine endothelial gene expression and the chosen significance threshold of >2-fold change, the endothelial transcriptome data sets revealed significant upregulation of 877 probes and downregulation of 389 probe sets in injured regions in adult (Fig. 3, green areas), whereas only an upregulation of 219 probe sets and downregulation of 142 probe sets in injured regions in neonates (Fig. 3, red areas). The patterns of both upregulated and downregulated genes were largely nonoverlapping between the two ages, with only 70 upregulated probe sets overlapping and 20 downregulated genes overlapping (Fig. 3, yellow areas).
Figure 3.

Differential effect of acute focal stroke on endothelial gene expression in adult and neonatal rats. The endothelial transcriptome data sets (31,042 probe sets) were obtained in endothelial cells purified by sequential negative and positive selection from injured and contralateral tissue of adults and neonates 24 h after reperfusion. Shown are genes with >2-fold change in expression compared to that in contralateral hemisphere.
Table 1 shows expression of several groups of genes directly related to the BBB function, including TJ components, adhesion molecules, extracellular matrix components, angiogenesis regulators, molecular transporters, and mediators of Wnt (wingless-type mouse mammary tumor virus integration site family) signaling required for BBB development. Expression of many genes in uninjured tissue was different in immature and adult brain, and the response of many genes to ischemia reperfusion differed in the neonate and adult. The response of the extracellular matrix and basal lamina proteins was rather complex, with different collagens, laminins, and other structural barrier components showing differing basal expression levels and differential regulation between the two ages (Table 1). Both the overall gene expression of Col-IV, the principal collagen type in the neurovascular basal lamina, and the relative expression of individual Col-IV α chains, α1/α2/α5, was on average 10-fold higher in uninjured tissue in neonates and did not increase further in injured tissue, whereas its gene expression was induced after stroke in the adult. Interestingly, matrix metalloproteinase-9 (MMP-9), a proteinase critical for BBB disruption following stroke, was dramatically increased in the adult (63.2-fold) but not increased in the neonate. Similarly, VEGF receptor-2 (VEGFR-2) and angiopoietin 2, which are known to increase vascular permeability, also showed an increase in gene expression in the adult (5.3-fold) but not in the neonate. Several leukocyte adhesion molecules, including P-selectin, E-selectin, and Icam-1, were upregulated in both ages, but the extent and the dynamics of upregulation was different for individual adhesion molecules following neonatal and adult stroke (a 28.1-fold increase of P-selectin in the neonate and a 9.4-fold in the adult, whereas a 57.6-fold increase in E-selectin in the neonate and a 214.3-fold increase in the adult).
Table 1.
Expression of several groups of genes directly related to the BBB function
| Probe set identification | Gene symbol | Gene name | Neonatal |
Adult |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Contra | Ipsi | Ipsi/contra | Contra | Ipsi | Ipsi/contra | |||||
| Pan endothelial genes | ||||||||||
| 1393067_at | Tek | Endothelial-specific receptor tyrosine kinase (Tie2) | 11,647 | 11,985 | 1.0 | 12,776 | 14,525 | 1.1 | ||
| Tight junctions | ||||||||||
| 1374104_at | Cldn5 | Claudin 5 | 24,730 | 23,434 | 0.9 | 20,181 | 17,964 | 0.9 | ||
| 1368115_at | Cldn3 | Claudin 3 | 95 | 100 | 1.0 | 153 | 128 | 0.8 | ||
| 1373182_at | Cldn12 | Claudin 12 | 418 | 308 | 0.7 | 531 | 485 | 0.9 | ||
| 1378753_at | Ocln | Occludin | 9600 | 7132 | 0.7 | 8047 | 3034 | 0.4 | a | |
| 1372473_at | Tjp1 | Tight junction protein 1 (ZO-1) | 13,473 | 12,329 | 0.9 | 12,887 | 10,971 | 0.9 | ||
| 1370940_at | Tjp2 | Tight junction protein 1 (ZO-2) | 3237 | 3503 | 1.1 | 2664 | 4112 | 1.5 | ||
| 1384143_at | Cgn | Cingulin | 334 | 62 | 0.2 | a | 135 | 117 | 0.9 | |
| 1389107_at | Cgnl1 | Cingulin-like 1 | 11,857 | 8856 | 0.7 | 9625 | 9345 | 1.0 | ||
| Adhesion molecules | ||||||||||
| 1391946_at | Selp | P-selectin | 39 | 1094 | 28.1 | a | 43 | 400 | 9.4 | |
| 1388018_at | Sele | E-selectin | 22 | 1255 | 57.6 | b | 21 | 4414 | 214.3 | b |
| 1387202_at | Icam1 | Intracellular adhesion molecule 1 | 1471 | 5637 | 3.8 | 646 | 1892 | 2.9 | ||
| 1368474_at | Vcam1 | Vascular cell adhesion molecule 1 | 593 | 2058 | 3.5 | 747 | 807 | 1.1 | ||
| 1370043_at | Alcam | Activated leukocyte adhesion molecule | 962 | 489 | 0.5 | 890 | 620 | 0.7 | ||
| Extracellular matrix components | ||||||||||
| 1370927_at | Col12a1 | Collagen, type XII, α 1 | 60 | 423 | 7.0 | a | 65 | 112 | 1.7 | |
| 1388939_at | Col15a1 | Collagen, type XV, α 1 | 114 | 707 | 6.2 | a | 90 | 119 | 1.3 | |
| 1388459_at | Col18a1 | Collagen, type XVII, α 1 | 1712 | 5573 | 3.3 | 364 | 925 | 2.5 | ||
| 1387854_at | Col1a2 | Collagen, type I, α 2 | 273 | 295 | 1.1 | 156 | 235 | 1.5 | ||
| 1377631_at | Col9a3 | Collagen, type IX, α 3 | 8493 | 3315 | 0.4 | 707 | 181 | 0.3 | ||
| 1370895_at | Col5a2 | Collagen, type V, α 2 | 7215 | 6924 | 1.0 | 1572 | 1498 | 1.0 | ||
| 1374705_at | Col4a5 | Collagen, type IV, α 5 | 9183 | 7435 | 0.8 | 5664 | 3523 | 0.6 | ||
| 1388494_at | Col4a2 | Collagen, type IV, α 2 | 15,643 | 16,910 | 1.1 | 1478 | 6966 | 4.7 | a | |
| 1372439_at | Col4a1 | Collagen, type IV, α 1 | 15,622 | 19,599 | 1.3 | 3278 | 13,571 | 4.1 | a | |
| 1370959_at | Col3a1 | Collagen, type III, α 1 | 948 | 527 | 0.6 | 70 | 255 | 3.6 | a | |
| 1368380_at | Vtn | Vitronectin | 1324 | 246 | 0.2 | 2701 | 396 | 0.1 | ||
| 1370234_at | Fn1 | Fibronectin 1 | 19,206 | 19,778 | 1.0 | 14,529 | 9728 | 0.7 | ||
| 1390404_at | Lama2 | Laminin, α 2 | 1423 | 292 | 0.2 | 1066 | 495 | 0.5 | ||
| 1392926_at | Lama1 | Laminin, α 1 | 1215 | 611 | 0.5 | 142 | 84 | 0.6 | ||
| 1388932_at | Lama5 | Laminin, α 5 | 1079 | 1413 | 1.3 | 656 | 1618 | 2.5 | a | |
| 1398275_at | Mmp9 | Matrix metallopeptidase 9 | 187 | 88 | 0.5 | 42 | 2631 | 63.2 | a | |
| 1370301_at | Mmp2 | Matrix metallopeptidase 2 | 131 | 165 | 1.3 | 229 | 101 | 0.4 | ||
| Angiogenesis regulators | ||||||||||
| 1368463_at | Vegfc | Vascular endothelial growth factor C | 2221 | 2781 | 1.3 | 1120 | 1004 | 0.9 | ||
| 1380854_at | Vegfb | Vascular endothelial growth factor B | 324 | 249 | 0.8 | 345 | 202 | 0.6 | ||
| 1373807_at | Vegfa | Vascular endothelial growth factor A | 64 | 52 | 0.8 | 156 | 164 | 1.1 | ||
| 1367948_a_at | Kdr | VEGFR-2 | 14,626 | 12,974 | 0.9 | 4151 | 10,916 | 2.6 | a | |
| 1369216_a_at | Flt4 | VEGFR-3 | 95 | 57 | 0.6 | 37 | 40 | 1.1 | ||
| 1370492_a_at | Flt1 | VEGFR-1 | 3655 | 2348 | 0.6 | 1194 | 2417 | 2.0 | ||
| 1374207_at | Angpt2 | Angiopoietin 2 | 9569 | 6066 | 0.6 | 2761 | 14,586 | 5.3 | a | |
| 1381487_at | Angpt1 | Angiopoietin 1 | 14 | 14 | 1.0 | 16 | 13 | 0.8 | ||
| 1398358_a_at | Itgb5 | Integrin β 5 | 250 | 211 | 0.8 | 442 | 204 | 0.5 | ||
| 1368819_at | Itgb1 | Integrin β 1 | 10,566 | 11,530 | 1.1 | 8654 | 11,736 | 1.4 | ||
| 1370937_a_at | Itga7 | Integrin α 7 | 323 | 199 | 0.6 | 377 | 163 | 0.4 | ||
| 1385649_at | Itga5 | Integrin α 5 | 240 | 232 | 1.0 | 48 | 60 | 1.2 | ||
| 1387144_at | Itga1 | Integrin α 1 | 2723 | 1880 | 0.7 | 767 | 637 | 0.8 | ||
| Transporters | ||||||||||
| 1388750_at | Tfrc | Transferrin receptor | 20,171 | 6609 | 0.3 | 7354 | 3660 | 0.5 | ||
| 1370848_at | Slc2a1 | Glut-1 | 15,273 | 16,006 | 1.0 | 21,097 | 19,609 | 0.9 | ||
| 1386981_at | Slc16a1 | MCT1 | 18,730 | 9408 | 0.5 | 4735 | 4156 | 0.9 | ||
| 1387280_a_at | Slc7a5 | TA1 | 3950 | 735 | 0.2 | 678 | 313 | 0.5 | ||
| 1368391_at | Slc7a1 | CAT1 | 17,439 | 8603 | 0.5 | 11,275 | 5545 | 0.5 | ||
| 1370367_at | Slc1a1 | EEAC1 | 94 | 55 | 0.6 | 1475 | 2113 | 1.4 | ||
| 1368461_at | Slc22a8 | OAT3 | 7157 | 1153 | 0.2 | 1276 | 166 | 0.1 | ||
| 1387970_at | Slc38a5 | JM24 | 9584 | 8516 | 0.9 | 396 | 648 | 1.6 | ||
| 1370824_at | Slc38a3 | Snat3 | 6706 | 3092 | 0.5 | 1622 | 693 | 0.4 | ||
| 1368295_at | Slco2b1 | OATP-B | 2418 | 1320 | 0.5 | 1831 | 1017 | 0.6 | ||
| 1370205_at | Slco1c1 | OATP-F | 24,648 | 12,147 | 0.5 | 10,052 | 823 | 0.1 | ||
| 1387093_at | Slco1a4 | Oatp2 | 4272 | 1859 | 0.4 | 23,989 | 7497 | 0.3 | ||
| 1380577_at | Abcg2 | Bcrp | 10,466 | 5774 | 0.6 | 13,279 | 2549 | 0.2 | ||
| 1370583_s_at | Abcb1a, Abcb1b | P-glycoprotein | 2472 | 1078 | 0.4 | 1362 | 604 | 0.4 | ||
| 1370465_at | Abcb1a | P-glycoprotein | 4269 | 1965 | 0.5 | 4974 | 1941 | 0.4 | ||
| Wnt mediators | ||||||||||
| 1387184_at | Axin2 | Axin 2 | 312 | 69 | 0.2 | 70 | 42 | 0.6 | ||
| 1383785_at | Lef1 | Lymphoid enhancer binding factor 1 | 3283 | 3689 | 1.1 | 3071 | 2338 | 0.8 | ||
| 1388994_at | Fzd6 | Frizzled 6 | 5802 | 6912 | 1.2 | 1488 | 1836 | 1.2 | ||
| 1373067_at | Ctnnb1 | b-catenin | 4409 | 5293 | 1.2 | 8146 | 6466 | 0.8 | ||
aStatistical significance: only this age.
bStatistical significance: both ages.
Gene expression of TJ proteins was also unsynchronized. While only small changes in the expression level of claudin-5 and ZO-1 were observed in both ages after stroke, occludin was significantly downregulated in adults (2.7-fold) and, to a lesser extent, in neonates. Among other important groups of genes that affect BBB permeability, downregulation of several molecular transporters was observed, including P-glycoprotein (P-gp), breast cancer resistance-related protein (BCRP), organic anion transporter 3 (OAT3), and organic anion transporter F (OATRAF), in most cases in both neonates and adults. In contrast, other mediators of angiogenesis and genes involved in Wnt signaling remained largely unchanged.
Protein expression of TJ and basal membrane proteins differs in the normal developing and adult brain
Because the structure of the basement membrane at the time of the insult may affect BBB integrity, we first determined protein expression of several basal lamina and TJ components in brains of naive immature and adult rats.
Expression of Col-IV and laminin were significantly lower in the normal adult than in postnatal brain (Fig. 4A–C). Expression of the brain endothelial TJ protein claudin-5, which is critical for the endothelial–endothelial junction seal (Nitta et al., 2003), tended to be higher in the immature than in adult brain (p = 0.10) (Fig. 4A,E). Expression of another important TJ protein, occludin, the integral membrane protein, which is localized exclusively to TJ in endothelial cells (Hirase et al., 1997), was significantly higher in the immature than in adult brain (Fig. 4A,D). In contrast, PDGFRβ expression gradually increased from P7 to adulthood (Fig. 4A,F), consistent with previous findings on gradually increasing PDGFRβ expression from embryonic period to adulthood (Armulik et al., 2010). Together, these data demonstrate the vastly different and uncoordinated patterns of expression of several BBB proteins in the normal developing and adult brain.
Figure 4.

The effect of brain maturation on the expression of BBB proteins. A, Representative Western blots showing the expression of basal lamina proteins (Col-IV and laminin), TJ proteins (occludin and claudin-5), and the pericyte marker PDGFRβ in naive developing (P7–P17) and adult rats. B–F, Densitometric analysis of Western blots showing reduced expression of the basal lamina proteins laminin (B) and Col-IV (C) and the TJ proteins occludin (D) and claudin-5 (E) and increased expression of PDGFRβ (F) in the adult brain compared to the developing postnatal brain. Data are normalized for β-actin expression and expressed as fold-increase versus P7 brains.
The expression of TJ proteins is better preserved in acutely injured neonatal brain than in adult brain
We then tested the overall changes in protein expression of individual TJ proteins after stroke in both age groups. Compared to contralateral hemisphere, protein expression of claudin-5 was significantly increased in injured tissue of neonatal but not adult rats (Fig. 5A). Expression of occludin and ZO-1 was similar in both injured and contralateral tissue of the neonate, but was reduced in injured regions in adult rats (Fig. 5B,C), with variable extent of reduction in individual rats. Therefore, although the changes of protein expression of TJ proteins did not necessarily mirror changes in their gene expression, the overall protein expression for all tested TJ proteins was higher in injured regions of neonates than adults.
Figure 5.

Expression of occludin, ZO-1, and claudin-5 is better preserved in immature than in adult brain after acute focal stroke. A–C, Protein expression of claudin-5 (A), occludin (B), and ZO-1 (C) measured by Western blot in whole tissue lysates obtained from injured and matching contralateral brain regions. D–G. ZO-1 (red) is expressed as continuous strands around the vessel (green, IB4) in the contralateral hemisphere regardless of age (D, adult; E, neonate). Continuous strands of ZO-1 are seen in ischemic tissue in neonatal rat (G), while disrupted pattern of ZO-1 expression is observed in adult rat (F, arrows) 24 h after reperfusion. Note that both IB4 and ZO-1 expression are disrupted in adult ischemic brain (F). Images are 3D reconstructions from a z-stack of images captured with a Zeiss LSM confocal microscope using the 63× objective and a 0.5 μm step. IB4 (green) identifies vessels and DAPI shows nuclei morphology.
The spatial distribution and integrity of strands of ZO-1, an essential TJ accessory protein, was evaluated in IB4-positive vessels. As evident from the 3D reconstruction images of ZO-1/IB4 immunofluorescence (Fig. 5D–G), while these two labels did not completely overlay, continuous strands of both proteins within the same vessels were seen in contralateral regions of both adults (Fig. 5D) and neonates (Fig. 5E), in both larger and smaller vessels. The regularly spaced continuous ZO-1 strands were seen in cross sections of the vessels (data not shown). While ZO-1 strands were continuous along vessels (Fig. 5G) in injured tissue of neonatal rats, such strands in injured tissue of adult rats, though abundant, contained gaps (Fig. 5F, white arrows). Thus, the disappearance of ZO-1 in vessels of injured adult but not neonatal rats is consistent with the notion of a more preserved ZO-1 protein expression in acutely injured neonatal rats.
Basal lamina proteins are differentially affected by stroke in neonatal and adult rats
The intactness of the BBB greatly depends on adequate structural support from the adjacent basal lamina. Given that brain maturation markedly affected gene and protein expression of several basal lamina components in uninjured hemisphere, we first analyzed the spatial distribution of Col-IV and laminin in contralateral hemisphere in relation to the density of brain vessels. Density of RECA-1+ vessels was significantly higher in the contralateral caudate of adult rats compared to neonate rats (Fig. 6A vs Fig. 6C,E), showing a more extensive small vessel/capillary network in the adult (Fig. 6F vs Fig. 6G). In contrast, Col-IV coverage of RECA-1+ vessels, defined by the ratio of Col-IV and RECA-1 densities, was significantly higher in the contralateral caudate of neonates than adults (Fig. 6H vs Fig. 6J,L), in agreement with our observations of higher transcript levels of several Col-IV chains and higher Col-IV protein expression in noninjured neonates (Fig. 4). Brain injury did not affect the volume density of vessels in any of the two ages (Fig. 6A vs Fig. 6B; Fig. 6C vs Fig. 6D; Fig. 6E), but led to an increased Col-IV density in the adult (Fig. 6J vs Fig. 6K; Fig. 6L), which remained unchanged in neonates (Fig. 6H vs Fig. 6I; Fig. 6L). However, despite induced Col-IV expression in adults after injury, the coverage of brain vessels by Col-IV remained significantly higher in the injured caudate of neonates (Fig. 6L). To determine whether this increase in Col-IV coverage occurred in both large and small vessels, we analyzed the size distribution of Col-IV+ vessels in the contralateral and ipsilateral hemispheres and observed that stroke led to a redistribution of Col-IV in the adult, with preferential loss of coverage of smaller vessels (Fig. 6M), while coverage of small vessels by Col-IV was mostly unaffected in injured neonatal brain (Fig. 6N).
Figure 6.

Stroke differentially affects the expression of basal lamina proteins in the neonatal and adult brain. A–E, Immunofluorescence of the brain vasculature (RECA-1, green) showing higher vascular density in the contralateral caudate of adults compared to neonates (C vs A; E) 24 h after stroke. Vascular density was unaffected by stroke at both ages in the ipsilateral injured caudate (B vs A; D vs C; E). Data are expressed as percentage of the total sampled brain volume in three different Z-stacks per brain and region. F, G, The size distribution of RECA-1 vessels in the adult (F) and the neonate (G). H--L. Immunofluorescence of Col-IV (red) showing increased coverage of brain vessels (expressed as the ratio between Col-IV and RECA-1 volume densities) in the contralateral neonatal brain compared to the adult brain (H vs J; L). Coverage of brain vessels by Col-IV increased in the injured caudate of adults (K vs J; L), but remained unchanged in neonates (I vs H; L). M, N, The distribution of Col-IV/RECA-1 positive vessels in the adult (M) and the neonate (N). Note preferential loss of Col-IV coverage of smaller size vessels in the adult but mostly unaffected coverage in injured neonatal brain. *p < 0.05 for same size vessels in ipsilateral versus contralateral hemisphere. O, R, Immunofluorescence of the basal lamina protein laminin (red) showing increased coverage of brain vessels (expressed as the ratio between laminin and RECA-1 volume densities) in the contralateral neonatal brain compared to the adult brain (O vs Q; S). Coverage of brain vessels by laminin was not significantly affected after MCAO (P vs O; R vs Q; S).
The overall laminin coverage of RECA-1+ vessels, as defined by the ratio of laminin and RECA-1 densities, was also higher in the contralateral caudate in neonates (Fig. 6O vs Fig. 6Q,S), again in agreement with our data from the transcriptome analysis and Western blot analysis. Injury did not induce significant changes in the overall laminin coverage in the caudate of neonates and adults (Fig. 6O vs Fig. 6P; Fig. 6Q vs Fig. 6R,S), although coverage tended to be slightly higher in adults (Fig. 6S). Also, we observed a redistribution of laminin after injury, with preferential loss of coverage of small vessels and increased coverage of large vessels in adults but not in neonates (data not shown).
Neutrophils do not transmigrate into ischemic-reperfused tissue of neonatal rats but transmigrate in response to intracerebral crCINC-1 injection or peripheral administration of a neutralizing anti-CINC-1 antibody
Neutrophil infiltration, which occurs acutely after stroke in adult rodents (Garcia et al., 1994; Matsuo et al., 1995), is thought to contribute to BBB disruption. Since the BBB of neonatal rats remained largely impermeable to tracers of various sizes after MCAO, we then determined whether neutrophils were present in the injured tissue. No neutrophil infiltrates were observed in the injured tissue with H&E staining of brain sections at 0, 1, 4, 8, 24, and 72 h and at 7 d after reperfusion (n = 2–6 per time point), and only a few randomly distributed individual neutrophils were seen in brain parenchyma (data not shown). The use of a selective antigranulocyte antibody (His48) and an anti-PMN serum confirmed that, while a number of His48+ or PMN+ cells were seen in the meninges (data not shown) and within brain vessels after MCAO (Fig. 7A), in nonperfused brains in particular, essentially no such cells were seen in the parenchyma of the same animals (Fig. 7A). Neutrophil infiltration failed to occur in the injured neonates despite a 115-fold increase of the neutrophil chemoattractant protein CINC-1 in the brain 8 h after MCAO, a time when circulating CINC-1 concentration declines (Denker et al., 2007). In the adult, His48+ cells were seen in a subset of vessels (Fig. 7B, green) and in the parenchyma (Fig. 7B, arrowheads), as well as in the meninges (Fig. 7C) 4 h after reperfusion.
Figure 7.
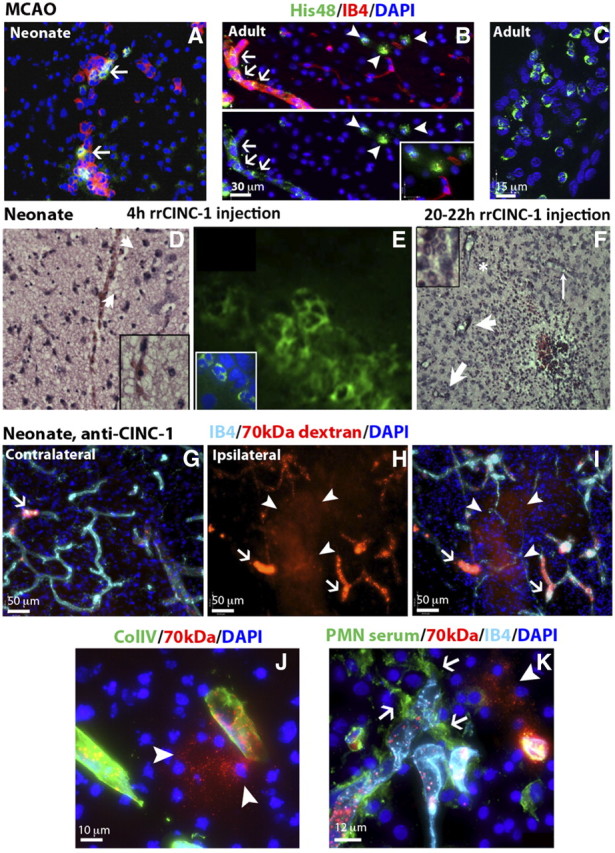
Neutrophil transmigration is limited after acute neonatal stroke. A, Neutrophil transmigration does not occur within 24 h after MCAO but His48+ cells are seen within vessels in animals without intracardiac perfusion (IB4, red; His48+ cells, green). B, C, Neutrophils (His48, green) are present in the brain parenchyma (B, arrowheads), meninges (C), and in a subpopulation of brain vessels (IB4, red) (B, arrows) in adult animals 4 h after reperfusion. D, E, Neutrophils are observed on H&E (D) and His48-immunofluorescence image (E) 4 h after intracerebral injection of rrCINC-1. Arrows in D point at neutrophils. His48+ cells are seen along the needle track and in parenchyma, but only a few His48+ neutrophils transmigrate following intracerebral rrCINC-1 injection (E). F, H&E staining showing infiltration of neutrophils in the brain 20–22 h after intracerebral injection of rrCINC-1. Neutrophils are observed in association with brain vessels (thin arrows) and in the brain parenchyma (thick arrows). Asterisk indicates the region magnified in the upper-left inset. G–I, Large areas of 70 kDa dextran (red) extravasation (H, I, arrowheads) were observed in the injured cortex of neonate rats injected with anti-CINC-1 15 min before MCAO. Increased accumulation of 70 kDa dextran (red) was also observed in brain vessels (arrows) in both the contralateral (G) and ipsilateral (H, I) cortex. (section thickness, 50 mm). J, Detail of a leaking vessel (70 kDa dextran, red, arrowheads) in a neonate rat injected with anti-CINC-1. K, Neutrophils (anti-PMN serum, green, arrows) associated with a leaking vessel (70 kDa dextran, red, arrowhead) in the injured cortex of a neonate rat injected with anti-CINC-1.
Because leukocyte transmigration is a multistep process that involves leukocyte rolling into, adhesion to, and migration through the endothelium (Engelhardt, 2003), we asked if low neutrophil transmigration in the presence of CINC-1 gradient in neonates is due to intrinsic properties of the immature BBB or due to the immaturity of neutrophils themselves. Intracortical injection of crCINC-1, which shares ∼92% amino acid sequence homology with rat CINC-1 (Nakagawa et al., 1994), induced neutrophil accumulation along the needle track at 4 h (n = 2), with only a limited number of neutrophils transmigrated into brain parenchyma adjacent to the injection site (Fig. 7D,E). No infiltration was observed at 18 h (n = 2). Infiltration varied from essentially none (n = 2) to massive (n = 3) 20–22 h after injection. Infiltrates were seen within and close to a subset of vessels in proximity of the injection site (Fig. 7F). No neutrophil infiltration was observed in vehicle-treated rats at any time point studied, demonstrating that mechanical disruption of the BBB at the site of injection without the chemoattractant gradient is insufficient for infiltration of these cells. The different patterns of P-selectin and E-selectin gene expression that we observed between neonatal and adult rats (Table 1) may also explain limited neutrophil transmigration into the tissue.
Given that neutrophils are capable of transmigrating into the neonatal brain in response to pharmacological doses of crCINC-1, but not after MCAO, we then tested if changing a balance between peripheral and cerebral levels of neutrophil chemoattractants affects neutrophil transmigration. Reduction of the peripheral CINC-1 levels by intravenous administration of a neutralizing anti-CINC-1 antibody adversely affected BBB integrity (Fig. 7G–I) and induced neutrophil adhesion and infiltration into the injured tissue (Fig. 7K) at 24 h after reperfusion. Compared to vehicle-treated rats, administration of a neutralizing anti-CINC-1 antibody also significantly increased injury volume from 51.6 ± 7.8% (n = 7) to 61.8 ± 11.6% (n = 9, p = 0.037) of ipsilateral hemisphere at this point. The pattern of 70 kDa dextran distribution showed that CINC-1 neutralization was associated with the increased number of vessels with dilated junctions in the contralateral hemisphere (Fig. 7G, arrow), and further increase in numbers of such abnormally looking vessel segments in injured regions (Fig. 7H,I, arrows). Furthermore, both large (Fig. 7G–I, arrowheads) and small (Fig. 7J, arrowheads) leakages of 70 kDa dextran occurred in injured regions. Vascular disruption occurred despite the presence of the basement membrane protein Col-IV (Fig. 7J). Importantly, neutrophil infiltration was also increased within injured regions of rats treated with an anti-CINC-1 antibody (Fig. 7K). Neutrophils were also seen within a subpopulation of vessels (Fig. 7K). Together, these results suggest that the lack of neutrophil infiltration into acutely injured regions preserves BBB integrity and protects the neonatal brain from injury.
Discussion
We show for the first time that the BBB is markedly more intact in injured neonatal brain than in adult brain after acute focal stroke. In contrast to the significantly increased albumin leakage into injured adult brain, leakage remained relatively low in injured neonatal brain within 2 to 24 h after reperfusion, and the BBB remained impermeable to large and small intravascular tracers. The endothelial transcriptome data showed the largely nonoverlapping patterns of upregulated and downregulated genes in injured regions of adults and neonates at 24 h, including the markedly different expression of the extracellular matrix and basal lamina proteins, and adhesion molecules. The protein expression of Col-IV and laminin and of several TJ proteins was higher in naive neonates and was better preserved in injured neonates compared to adults. Neutrophil extravasation was low in injured neonatal brain, but extravasation was increased and vascular integrity compromised following an intravenous anti-CINC-1 antibody injection. Therefore, multiple mechanisms contribute to the functional BBB integrity after acute neonatal stroke.
Albumin leakage was greatly increased in acutely injured tissue of the adults, consistent with previous reports (Belayev et al., 1996), but leakage remained low in the injured neonates. At 24 h in the neonate, leakage of 70 kDa dextran was low, a smaller intravascular tracer, 3 kDa dextran, also remained in the vessels, and the BBB was largely impermeable to an even smaller molecule, Gd-DTPA. The markedly lower BBB permeability in acutely injured neonatal brain was not due to lack of injury, because selection of all animals in both age groups was based on the presence of DWI injury during MCAO, which is predictive of brain injury 24 h later (Derugin et al., 2000). According to perfusion-sensitive MRI, the functional intactness of the BBB in neonates, but not in adults, was not due to better cerebral perfusion during MCAO or to better preserved perfusion upon reperfusion in neonates.
The BBB is complex. Its integrity, high electrical resistance, and ability to exclude the vast majority of molecules are controlled by a number of partially independent components (Bazzoni et al., 2000; Zlokovic, 2008) that are based, in part, on features of brain endothelial cells that are not present on nonbrain endothelial cells (Daneman et al., 2010b). The ability to maintain BBB integrity depends on adequate structural support from the basement membrane, and reduced Col-IV expression is associated with increased injury in the adult (McColl et al., 2008), as well as vascular abnormalities in the developing brain (Gould et al., 2005). Col-IV is not only critical for the mechanical stability of the BBB, through the formation of distinct networks via self-association of its α chains and interaction with other extracellular matrix components, including laminin and proteoglycans, but also for cell signaling via interaction with various receptors and adhesion molecules (Khoshnoodi et al., 2008). The transcript levels of individual Col-IVα and laminin-α chains were differentially affected by age in uninjured tissue. Furthermore, upregulation and downregulation of individual Col-IVα chains were unsynchronized and differed between injured neonates and adults.
Coverage of brain vessels by Col-IV and laminin was also markedly different between the mature and immature brain: coverage was significantly higher in uninjured neonatal brain and was largely unaffected by the injury, but was lost in small vessels and capillaries in injured adults. Gene expression of MMP-9 and several leukocyte adhesion molecules, including E-selectins and P-selectins, also differed greatly between age groups, whereas gene expression of efflux transporters, such as P-gp, changed similarly by MCAO in both groups. Gene expression of the regulators of vascular remodeling, VEGFR-2 and angiopoietin-2, increased only in injured adults, whereas several other regulators of angiogenesis remained unchanged. Thus, the neurovascular basement membrane is more prominent in neonatal brain than in adult brain, likely contributing to the relative impermeability of the neonatal BBB after stroke.
The TJs play a key role in the functional preservation of the BBB (Bazzoni et al., 2000), and the expression and localization of the TJ proteins directly correlate with BBB permeability and vasogenic edema (Nitta et al., 2003; Witt et al., 2003). We observed largely unaltered transcript levels of claudin-5 and ZO-1 in both age groups, but saw significantly reduced expression of occludin in adult stroke. The protein expression of claudin-5, occludin, and ZO-1 was higher in injured neonatal brain than in adult brain, and, consistently, strands of ZO-1 were highly organized in injured brain regions of the neonates but not adults. A decrease in occludin and claudin-5 expression, but unchanged ZO-1 expression, during normal development, together with the altered vascular regulation due to increasing PDGFRβ with age, may also explain the differential BBB response to injury in neonates and adults and through better preserved TJ integrity in injured neonates.
Both animal and human studies in the adult show that neutrophils contribute to BBB disruption after transient cerebral ischemia. Neutrophils are present early in ischemic tissue, contributing to the secondary perfusion deficits (del Zoppo et al., 1991; Kochanek and Hallenbeck, 1992), and producing ROS (Barone et al., 1991), and proteolytic enzymes (Gidday et al., 2005). These effects are reduced by neutropenia or treatments that prevent leukocyte adhesion (Matsuo et al., 1995; Yamasaki et al., 1997). In stark contrast to adults after MCAO, neutrophil infiltration in neonates was negligible 1–72 h after reperfusion, a finding consistent with a very limited (Hudome et al., 1997) or brief (Bona et al., 1999) neutrophil infiltration into injured neonatal rat brain reported after hypoxia-ischemia. Although the exact mechanisms that restrict neutrophil infiltration in injured neonatal brain are not yet understood, the differing patterns of adhesion molecule gene expression between rats of two ages may limit neutrophil extravasation in neonate. Alternatively, the transient increase of CINC-1 in the circulation before CINC-1 increase in the brain (Denker et al., 2007) is likely to diminish neutrophil infiltration, BBB disruption, and injury in the neonate despite the markedly increased CINC-1 levels after reperfusion. Mechanical damage (needle insertion) did not drive neutrophil infiltration, and, even following intracerebral rrCINC-1 injection, infiltration was spatially discrete, suggesting that intrinsic properties of immature and mature neutrophils might differ. Data on reduced atrophy and higher levels of adenine nucleotides in neutropenic pups subjected to hypoxia-ischemia when neutrophils are depleted before but not after injury (Palmer et al., 2004) suggest that these cells can exert multiple effects, which are not coordinated in time. Neutrophils exacerbate ischemic outcomes by systemic inflammation in the adult (McColl et al., 2008) and are the source of ROS (Kunz et al., 2008). They also mediate brain injury after intracranial cytokine injection in juvenile rats (Anthony et al., 1997). Although several types of cells, including endothelial cells and immune cells, can increase MMP-9 production, stroke studies in chimeric mice deficient in either peripheral or brain MMP-9 identified neutrophils as the major source of this proteolytic enzyme (Gidday et al., 2005). Striking differences in endothelial MMP-9 expression—unchanged transcript levels in the neonate but a 63-fold upregulation in the adult—may have major implications for both the structural and functional BBB integrity after neonatal stroke. Enzymatic degradation of TJs (Yang et al., 2007), vasogenic edema (Rosenberg and Yang, 2007), and initiation of the signaling cascade for neuronal cell death in the adult are MMP-9 dependent (Gu et al., 2005), while inhibition of genetic depletion of MMP-9 or other proteolytic enzymes in leukocytes reduce laminin degradation, neuronal apoptosis, and ischemic injury (Tonai et al., 2001; Gidday et al., 2005; Afshar-Kharghan and Thiagarajan, 2006). Therefore, it is possible that the differences in TJ protein levels are due to proteolysis by MMPs rather than to altered transcriptional levels.
Other components of the neurovascular unit may contribute to the low BBB permeability after acute neonatal stroke. Composition and phosphorylation of TJ complexes may affect placement and the dynamic nature of TJ function. The phenotypic endothelial cell heterogeneity, the more elaborate capillary network in the adult, regional size-dependent responsiveness of the vessels, and transcytosis, another major mechanism that regulates BBB intactness, may also contribute to the differential response of the immature and adult BBB. Furthermore, age-related differences in the astrocyte phenotypes and coverage (Cahoy et al., 2008; Zhang and Barres, 2010) and the gradually increasing pericyte vessel coverage with age (Armulik et al., 2010) may affect the associated hemodynamic regulation in many ways. Microglia are believed to contribute to the pathophysiology of adult stroke partly by producing superoxide (Cho et al., 2005), but we recently showed that microglia contribute to endogenous protection after neonatal stroke and that these cells do not produce superoxide nearly to the extent microglia do in adult stroke (Faustino et al., 2011), thereby likely protecting the endothelium. The relative impact of systemic versus local inflammation may also have a wide range of implications for BBB intactness (Vexler and Yenari, 2009; Denes et al., 2010; Stolp et al., 2011). While much is still to be learned about the underlying mechanisms of such strikingly different responses of the immature and mature BBB to stroke, it is vital to appropriately choose therapies given key role of largely intact BBB in limiting access of drugs into the neonatal brain.
Footnotes
This work was supported by the National Institutes of Health Research Project Grant RO1 NS55915 (Z.S.V.), National Institutes of Health Research Project Grant RO1 NS44025 (Z.S.V.), American Heart Association Grant-in-Aid 0855235F (Z.S.V.), the Ramón Areces Foundation (D.F.-L.), R01 NS045621 (Ben A. Barres), the Myelin Repair Foundation (Ben A. Barres, R.D.), and the Program for Breakthrough Biomedical Research (R.D.). We acknowledge Andra Dingman for her contribution to early stages of the study, Baomei Liu and Brianna Pickering for technical help, and Donna Ferriero, M.D., for useful discussions. We would also like to acknowledge the generosity of Dr. Ben A. Barres, as the genomic work was performed in his laboratory by Dr. Richard Daneman.
The authors declare no competing financial interests.
References
- Afshar-Kharghan V, Thiagarajan P. Leukocyte adhesion and thrombosis. Curr Opin Hematol. 2006;13:34–39. doi: 10.1097/01.moh.0000190107.54790.de. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Bolton SJ, Fearn S, Perry VH. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain. 1997;120(Pt 3):435–444. doi: 10.1093/brain/120.3.435. [DOI] [PubMed] [Google Scholar]
- Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol. 1998;8:923–926. doi: 10.1016/s0960-9822(07)00373-9. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Barone FC, Hillegass LM, Price WJ, White RF, Lee EV, Feuerstein GZ, Sarau HM, Clark RK, Griswold DE. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–345. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275:20520–20526. doi: 10.1074/jbc.M905251199. [DOI] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res. 1999;45:500–509. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010a;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010b;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Denker SP, Ji S, Lee SY, Dingman A, Derugin N, Wendland MF, Vexler ZS. Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J Neurochem. 2007;100:893–904. doi: 10.1111/j.1471-4159.2006.04162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derugin N, Ferriero DM, Vexler ZS. Neonatal reversible focal cerebral ischemia: a new model. Neurosci Res. 1998;32:349–353. doi: 10.1016/s0168-0102(98)00096-0. [DOI] [PubMed] [Google Scholar]
- Derugin N, Wendland M, Muramatsu K, Roberts TP, Gregory G, Ferriero DM, Vexler ZS. Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rat. Stroke. 2000;31:1752–1761. doi: 10.1161/01.str.31.7.1752. [DOI] [PubMed] [Google Scholar]
- Derugin N, Dingman A, Wendland MF, Fox C, Bollen A, Vexler ZS. Magnetic resonance imaging as a surrogate measure for histological sub-chronic endpoint in a neonatal rat stroke model. Brain Res. 2005;1066:49–56. doi: 10.1016/j.brainres.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Dzietko M, Faustino J, Derugin N, Wendland MF, Ferriero DM, Vexler ZS. MRI as a translational tool for the study of neonatal stroke. J Child Neurol. 2011;26:1145–1153. doi: 10.1177/0883073811408308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek CJ, Wong A, Liddelow SA, Johansson PA, Dziegielewska KM, Saunders NR. Efflux mechanisms at the developing brain barriers: ABC-transporters in the fetal and postnatal rat. Toxicol Lett. 2010;197:51–59. doi: 10.1016/j.toxlet.2010.04.025. [DOI] [PubMed] [Google Scholar]
- Engelhardt B. Development of the blood-brain barrier. Cell Tissue Res. 2003;314:119–129. doi: 10.1007/s00441-003-0751-z. [DOI] [PubMed] [Google Scholar]
- Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, Vexler ZS. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31:12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat) Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SW. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, Fujimoto K, Tsukita S, Rubin LL. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110(Pt 14):1603–1613. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- Hudome S, Palmer C, Roberts RL, Mauger D, Housman C, Towfighi J. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr Res. 1997;41:607–616. doi: 10.1203/00006450-199705000-00002. [DOI] [PubMed] [Google Scholar]
- Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech. 2008;71:357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniesel U, Risau W, Wolburg H. Development of blood-brain barrier tight junctions in the rat cortex. Brain Res Dev Brain Res. 1996;96:229–240. doi: 10.1016/0165-3806(96)00117-4. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- Kucharczyk J, Vexler ZS, Roberts TP, Asgari HS, Mintorovitch J, Derugin N, Watson AD, Moseley ME. Echo-planar perfusion-sensitive MR imaging of acute cerebral ischemia. Radiology. 1993;188:711–717. doi: 10.1148/radiology.188.3.8351338. [DOI] [PubMed] [Google Scholar]
- Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, Zhou P, Iadecola C. Nuclear factor-κB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lynch JK. Epidemiology and classification of perinatal stroke. Semin Fetal Neonatal Med. 2009;14:245–249. doi: 10.1016/j.siny.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Kihara T, Ikeda M, Ninomiya M, Onodera H, Kogure K. Role of neutrophils in radical production during ischemia and reperfusion of the rat brain: effect of neutrophil depletion on extracellular ascorbyl radical formation. J Cereb Blood Flow Metab. 1995;15:941–947. doi: 10.1038/jcbfm.1995.119. [DOI] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley ME, Wendland MF, Kucharczyk J. Magnetic resonance imaging of diffusion and perfusion. Top Magn Reson Imaging. 1991;3:50–67. [PubMed] [Google Scholar]
- Nakagawa H, Komorita N, Shibata F, Ikesue A, Konishi K, Fujioka M, Kato H. Identification of cytokine-induced neutrophil chemoattractants (CINC), rat GRO/CINC-2 alpha and CINC-2 beta, produced by granulation tissue in culture: purification, complete amino acid sequences and characterization. Biochem J. 1994;301(Pt 2):545–550. doi: 10.1042/bj3010545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Roberts RL, Young PI. Timing of neutrophil depletion influences long-term neuroprotection in neonatal rat hypoxic-ischemic brain injury. Pediatr Res. 2004;55:549–556. doi: 10.1203/01.PDR.0000113546.03897.FC. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007;22:E4. doi: 10.3171/foc.2007.22.5.5. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Habgood MD, Dziegielewska KM. Barrier mechanisms in the brain, II. Immature brain. Clin Exp Pharmacol Physiol. 1999;26:85–91. doi: 10.1046/j.1440-1681.1999.02987.x. [DOI] [PubMed] [Google Scholar]
- Stolp HB, Johansson PA, Habgood MD, Dziegielewska KM, Saunders NR, Ek CJ. Effects of neonatal systemic inflammation on blood-brain barrier permeability and behaviour in juvenile and adult rats. Cardiovasc Psychiatry Neurol. 2011;2011:469046. doi: 10.1155/2011/469046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonai T, Shiba K, Taketani Y, Ohmoto Y, Murata K, Muraguchi M, Ohsaki H, Takeda E, Nishisho T. A neutrophil elastase inhibitor (ONO-5046) reduces neurologic damage after spinal cord injury in rats. J Neurochem. 2001;78:1064–1072. doi: 10.1046/j.1471-4159.2001.00488.x. [DOI] [PubMed] [Google Scholar]
- Vexler ZS, Yenari MA. Does inflammation after stroke affect the developing brain differently than adult brain? Dev Neurosci. 2009;31:378–393. doi: 10.1159/000232556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285:H2820–H2831. doi: 10.1152/ajpheart.00589.2003. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuo Y, Zagorski J, Matsuura N, Onodera H, Itoyama Y, Kogure K. New therapeutic possibility of blocking cytokine-induced neutrophil chemoattractant on transient ischemic brain damage in rats. Brain Res. 1997;759:103–111. doi: 10.1016/s0006-8993(97)00251-5. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20:588–594. doi: 10.1016/j.conb.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]