

Abstract
Oligomerization of the amyloid β-protein (Aβ) is a seminal event in Alzheimer’s disease (AD). Aβ42, which is only two amino acids longer than Aβ40, is particularly pathogenic. Why this is so has not been elucidated fully. We report here results of computational and experimental studies revealing a C-terminal turn at Val36-Gly37 in Aβ42 that is not present in Aβ40. The dihedral angles of residues 36 and 37 in an Ile31–Ala42 peptide were consistent with β-turns, and a β-hairpin-like structure was indeed observed that was stabilized by hydrogen bonds and by hydrophobic interactions between residues 31–35 and residues 38–42. In contrast, Aβ(31–40) mainly existed as a statistical coil. To study the system experimentally, Aβ peptides containing amino acid substitutions designed to stabilize or destabilize the hairpin were chemically synthesized. The triple substitution Gly33Val–Val36Pro–Gly38Val (“VPV”) facilitated Aβ42 hexamer and nonamer formation, while inhibiting formation of classical amyloid-type fibrils. These assemblies were as toxic as were assemblies from wild type Aβ42. When substituted into Aβ40, the VPV substitution caused the peptide to oligomerize similarly to Aβ42. The modified Aβ40 was significantly more toxic than Aβ40. The double substitution D-Pro36-L-Pro37 abolished hexamer and dodecamer formation by Aβ42 and produced an oligomer size distribution similar to that of Aβ40. Our data suggest that the Val36-Gly37 turn could be the sine qua non of Aβ42. If true, this structure would be an exceptionally important therapeutic target.
Keywords: Amyloid β-protein, Alzheimer’s disease, β-hairpin
Introduction
Alzheimer’s disease (AD) is the most common cause of late-life dementia1. The predominant cerebral neuropathological features of AD are extracellular amyloid deposits formed by the amyloid β-protein (Aβ), intracellular neurofibrillary tangles formed by the protein tau, and neuron loss2. Aβ is a product of proteolytic cleavage of the amyloid β-protein precursor (AβPP)3. Two predominant species of Aβ exist in humans, Aβ40 and Aβ42, that are distinguished by the absence or presence, respectively, of an Ile–Ala dipeptide at the C-terminal end of an identical 40 amino acid peptide4. Aβ42 is the principal protein component of parenchymal plaques5–7. An increase in the absolute amount of Aβ42, or in the Aβ42/Aβ40 concentration ratio, is associated with familial forms of AD8,9. In humans, reduction of Aβ42 concentration correlates with a decreased risk for AD10. In vitro studies have shown that Aβ42 displays fibril nucleation and elongation rates that are significantly higher than those of Aβ4011 and that Aβ42 forms larger oligomers than does Aβ4012–16. Importantly, the assemblies formed by Aβ42 are more toxic than are those formed by Aβ4017.
To execute strategies for knowledge-based design of therapeutic agents, one must move from the regimes of morphology and kinetics to that of atomic structure and dynamics. In this way, specific atoms and their movements can be correlated with the biological consequences of peptide folding and assembly, providing critical information for drug targeting and design.
Previously, we used the method of photo-induced cross-linking of unmodified proteins (PICUP) to determine quantitatively the oligomer size frequency distribution 15,18. Aβ40 and Aβ42 oligomerized through distinct pathways. Aβ40 predominately assembled into dimeric, trimeric, and tetrameric species, whereas Aβ42 formed pentamer/hexamer units (paranuclei) that further assembled into larger oligomers (dodecamers, octadecamers)19. These results were confirmed and extended using ion mobility spectrometry–mass spectrometry14. Other dodecameric structures also have been described, including Aβ-derived diffusible ligands (ADDLs)20 and Aβ★5621. In addition, many other types of assemblies, ranging in size from dimer to μm-sized macrostructures (βamy balls22), have been reported (for a recent review, see Roychaudhuri et al.3).
To elucidate, at atomic resolution, the conformational dynamics of Aβ40 and Aβ42 that contribute to their distinct physical and biological behaviors, we previously performed simulations on the respective monomeric Aβ peptides23. We observed that both peptides were largely disordered but that frequent turn-like features were exhibited by residues 6–9 (Turn #1, “T1”), 14–16 (T2), and 23–27 (T3). All three regions exist both in Aβ40 and Aβ42, thus it is reasonable to speculate that these regions cannot alone contribute significantly to the idiotypic behavior of Aβ42. However, we did observe distinct behavior of the Aβ42 C-terminus (residues 31–42). This peptide segment tended to bend, resulting in the formation of a turn-like fold, involving residues 35–38 (T4), with a significantly larger number of intra-molecular contacts than observed in Aβ40. Computational and experimental studies have shown that both peptides display little regular structure, but that the Aβ42 C-terminus is more rigid than that of Aβ4024–26. Lazo et al. showed that the Aβ42 C-terminus is resistant to proteolytic digestion27. Taken together, these data suggest the existence of a folded structure at the Aβ42 C-terminus.
We discuss here the results of computational and experimental studies seeking to test the hypothesis that the C-terminal turn 1 element is the sine qua non of Aβ42, the structural feature that imparts on Aβ42 its unique assembly properties and biological activity relative to Aβ40.
Results
Simulation of Aβ C-terminal conformational dynamics
We used REMD simulations for a total of 3.2 μs to generate 20,000 conformations for Aβ(31–40/42) (see Supporting Information for details). To determine whether the simulation had converged, the conformational ensemble was divided into two equal parts. Each part was subjected to secondary structure analysis using the DSSP program28. The highly overlapped curves shown in (Fig. 1) suggest that the two conformational ensembles are similar, which in turn indicates convergence of the simulations. Demonstrating convergence was important because it showed that our simulation sampled sufficient volumes of the total conformational space to produce a representative subset of that space, from which meaningful data could be obtained.
Fig. 1.

The conformational ensemble collected from the REMD simulation of Aβ42 was divided into two equal populations of 10,000 conformers each. Each population then was subjected to secondary structure analysis using the DSSP program28 to determine the percentages of turn (upper panel) and anti-parallel β-sheet (lower panel). The highly overlapped curves suggest that the two conformational ensembles are similar, indicating convergence of the simulations.
We then clustered the collected conformations with an RMSD threshold of 2Å (Fig. 2). Though the Aβ42 C-terminus appears to be disordered overall, its most populated structure is a well-folded β-hairpin with residues 36 and 37 located at the i + 1 and i + 2 positions of the β-turn (Fig. 3, red arrows). This β-hairpin structure is stabilized by hydrogen bonds interactions between the following pairs of residues, Ile31:Ala42, Ile32:Ile41, Gly33:Val40, Leu34:Val39, and Met35:Gly38, and hydrophobic interactions between Ile31:Ile41, Leu34:Val39, Val40:Met35. The second most populated C-terminus structure is also compact and contains a well-defined β-turn at residues 35–38 (Fig. 4). The occurrence frequency for these two structures combined is ≈10 times greater than the third most populated structure (Fig. 1). For this reason, we do not discuss the third-most frequent conformational clusters nor clusters of even smaller occurrence frequency. We also calculated the dihedral angles of residues 36 and 37 to quantify β-turn content, as the propensity of residues 36 and 37 to exist in a β-turn is closely related to the stability of the β-hairpin. The turn type adopted by residues 36 and 37 is not unique, as type-I′, type-II, and βVIb turns were observed at frequencies of 10%, 7.5%, and 7%, respectively (Table 1). We refer to these turns collectively as “β-turns.” In contrast to these data from Aβ42, the most populated Aβ40 C-terminus conformer displayed no regular secondary structure, and β-turn population by residues 35–38 (8%) was that of Aβ42 (Table 1).
Fig. 2.

Occurrence frequencies (%) for each conformational cluster of each Aβ peptide.
Fig. 3.

Structures of the most populated conformers for each peptide. Yellow dotted lines indicate hydrogen bonds. C, N, O and S atoms are colored by green, blue, red and yellow, respectively. H atoms are not shown. Red arrows point toward the turn, residues 36 and 37. Each of the five conformers overlays a ribbon diagram of its own peptide backbone to help illustrate the position of the backbone and any secondary structure elements within it.
Fig. 4.

Structures of the second most populated conformers for each peptide. Yellow dotted lines indicate hydrogen bonds. C, N, O and S atoms are colored by green, blue, red and yellow, respectively. H atoms are not shown. Each of the five conformers overlays a ribbon diagram of its own peptide backbone to help illustrate the position of the backbone and any secondary structure elements within it.
Table 1. Amino acid substitutions engineered into the Aβ sequence.
The substituted positions are highlighted in bold italics. Lower case p signifies D-Pro.
| Aβ | Sequence | Structurea | β-turnb | Oligomers Formedc |
|---|---|---|---|---|
| Aβ42 | 31IIGLMVGGVVIA | β-hairpin | 25% | 1, 2, 3, 4, 5, 6 |
| [VPV]Aβ42 | 31IIVLMPGVVVIA | β-hairpin | 65% | 1, 5, 6, 12 |
| [pP]Aβ42 | 31IIGLMpPGVVIA | statistical coil | 50% | 1, 2, 3, 4 |
| Aβ40 | 31IIGLMVGGVV | statistical coil | 8% | 1, 2, 3, 4 |
| [VPV]Aβ40 | 31IIVLMPGVVV | statistical coil | 35% | 1, 3, 5 |
Structure of the predominant full-length conformer in the population.
Structure of residues 35–38 defined by dihedral angle.
Numbers indicate the assembly “order,” i.e., the number of monomers per oligomer, observed by SDS-PAGE. Monomers are signified by “1.”
Conformational dynamics of designed C-terminal peptide analogues
If the β-hairpin structure determined were a relevant structural feature of holo-Aβ, we hypothesized that it should be possible to design de novo Aβ analogues containing amino acid substitutions that would stabilize the β-hairpin. To test this hypothesis, we first used computer simulations to determine whether specific amino acids would indeed stabilize the β-hairpin (Table 1). The first Aβ(31–42) peptide we designed contained D-Pro36–L-Pro37, as this sequence has been shown to stabilize β-hairpin structure significantly29. This peptide is designated [pP]Aβ42. Unexpectedly, the simulation revealed that though significantly more (50%) β-turn structure was observed for residues 35–38, the most populated structure was actually a statistical coil and the overall conformational diversity was higher that that of wild type Aβ(31–42) (Fig. 3 & Fig. 1). This peptide thus was studied to determine how destabilizing substitutions affected peptide dynamics.
We then designed a second Aβ42 analogue, but with an L-Pro36–L-Gly37 sequence that was reported to stabilize β-hairpin structure30,31. In this peptide, we also replaced Gly33 and Gly38 with Val to reduce the flexibility of the peptide backbone and to strengthen putative hydrophobic interactions between the two predicted β-strands. We designate this peptide [VPV]Aβ42. With these modifications, β-hairpin content increased from 5.5% to 12.5%, and the β-turn population increased to 65%, as revealed by MD simulation (Fig. 3 & Fig. 1).
Because the [pP] substitution in Aβ42 did not stabilize its turn, we did not incorporate it into Aβ40 (see Fig. 3 for wild type Aβ40 conformers). Instead, we focused on [VPV]Aβ40. We observed that this substitution did not produce a β-hairpin structure (Fig. 3 & Fig. 1), though higher β-turn content (35%) was observed for residues 35–38 (Table 1).
Peptide secondary structure dynamics
To determine the temporal dynamics of peptide secondary structure, we monitored peptide assembly using CD (Fig. 5 and Fig. 6). Wild-type Aβ42 and Aβ40 initially displayed statistical coil (SC) structures (Fig. 5, A and B, respectively), which underwent rapid SC→β-sheet transitions to produce maximal β-sheet levels of ≈40–45% by days 4 and 5, respectively (Fig. 6). Relative to Aβ42, and to all the other peptides, [VPV]Aβ42 displayed significantly more β-structure initially (≈30%) and showed maximal β-structure at day 5 (Fig. 6 & Fig. 5C). [VPV]Aβ40 displayed slower kinetics, not displaying maximal β-sheet structure until day 9 (Fig. 6 & Fig. 5D). In contrast to the structural transitions observed for the other peptides, [pP]Aβ42 remained as a statistical coil throughout the experiment (Fig. 6 and Fig. 5E).
Fig. 5.

Circular dichroism (CD) spectroscopy. LMW Aβ solutions were prepared at concentrations of 60–80 μM and then incubated at 37° C with slow inversion. Aliquots were removed periodically for CD analysis. (A) Aβ42, (B) Aβ40, (C) [VPV]Aβ42, (D) [VPV]Aβ40, (E) [pP]Aβ42. The spectra are representative of those obtained in each of three independent experiments. Spectra from different days that were essentially superimposable are represented by a single spectrum, for clarity of viewing.
Fig. 6.

Kinetics of β-strand formation. Data from the experiments shown in Fig. 5 were deconvolved using Dichroweb64 to yield percent β-strand content for each peptide on each day of observation. These data then were combined to produce the figure shown.
Time evolution of β-sheet structure
ThT fluorescence was used to monitor the time dependence of β-sheet formation during Aβ incubation (Fig. 7)32. Aβ40 and Aβ42 displayed rapid rises in fluorescence that peaked at days 4 and ≈2, respectively. These peaks were followed by declines, an observation that is typical for Aβ assembly33. We did not observe a lag phase because relatively high peptide concentrations were used (≈35–40 μM). [VPV]Aβ40 displayed a monotonic increase in fluorescence that started at day 1 and peaked at day 6 at a level somewhat higher than that produced by Aβ40. [VPV]Aβ42, in contrast, produced substantial fluorescence immediately. The fluorescence intensity was ≈40% that of the maximal level displayed by Aβ42. The fluorescence remained relatively constant, or trended slightly downward, during the observation period. [pP]Aβ42 showed a very modest monotonic increase in fluorescence over time, producing a final fluorescence intensity that was <5% of the maximum levels of Aβ40 or Aβ42.
Fig. 7.

Kinetics of β-sheet formation. Peptides were incubated in 10 mM phosphate buffer, pH 7.4, for 7 d at 37° C with slow inversion. Aliquots were removed every 24 h to determine the level of ThT fluorescence. Error bars show S.E., which is some cases are smaller than the figure symbols.
Peptide oligomerization
To determine the effects of the designed amino acid substitutions on peptide oligomerization, we used the technique of photo-induced cross-linking of unmodified proteins (PICUP)34. PICUP enables quantitative determination of the oligomer size frequency distribution. Cross-linking Aβ40 and Aβ42 produced typical distributions15, namely Aβ40 formed oligomers predominately of orders 2 2–4 (Fig. 8, lane 5) and Aβ42 formed oligomers of orders 2–6 (Fig. 8, lane 3). Un-cross-linked Aβ40 displayed only a monomer band, whereas un-cross-linked Aβ42 displayed monomer and trimer bands (results not shown), as has been reported previously 15. The [VPV]Aβ42 peptide oligomerized distinctly from its wild type homologue (Fig. 8, lane 2). Prominent bands were observed with molecular masses of ≈4.5 kDa, ≈23 kDa and ≈28 kDa, corresponding to monomer, pentamer and hexamer, respectively. Relatively faint bands with molecular masses of ≈9 kDa and ≈18 kDa, corresponding to dimer and tetramer, were observed. No trimer band was observed. Bands of molecular mass ≈42–56 kDa also were seen. These bands may correspond to nonamer–dodecamer. In contrast, the oligomer distribution of [pP]Aβ42 (Fig. 8, lane 1) was very similar to that of Aβ40, with the exception that the apparent molecular masses of each band were slightly higher due to the increased mass of this substituted Aβ42 peptide. The oligomer distribution of [VPV]Aβ40 (Fig. 8, lane 4) was distinct from that of wild type Aβ40 (Fig. 8, lane 5). The [VPV]Aβ40 distribution was characterized by four prominent bands, monomer, dimer, a band between trimer and tetramer, and a band between tetramer and pentamer. This distribution displayed similarities to the distribution of wild type Aβ42 in its relative paucity of trimer and greater abundance of higher order oligomers.
Fig. 8.

Analysis of Aβ oligomerization. Each peptide was solvated freshly from the lyophilized state and then immediately subjected to PICUP, followed by SDS-PAGE and silver staining. [pP]Aβ42 (lane 1), [VPV]Aβ42 (lane 2), Aβ42 (lane 3), [VPV]Aβ40 (lane 4) and Aβ40 (lane 5). “MWM” is molecular weight marker. The data in the experiment shown are essentially identical to those observed in two other independent experiments done on different days.
Temporal changes in peptide assembly size
Dynamic light scattering (DLS) was used to monitor time-dependent changes in the distribution of oligomer sizes (Fig. S1). No significant time-dependent changes in the oligomer distributions of Aβ40 or [VPV]Aβ40 peptides were observed over a time period of one month. Both peptides formed small oligomers (RH≈2 nm) and a broad distribution of larger assemblies. In Aβ40, assemblies of RH≈10 nm, and large aggregates with RH≈60–80 nm, were observed. In addition, occasional contributions to the scattering intensity from very large (many hundreds of nm) were observed. These contributions increased over time, as reflected by the decreasing scattering intensity noted for the shaded oligomer peaks. Additionally, numerous intensity spikes appeared after a few days (data not shown). Such intensity spikes indicate formation of very large aggregates that drift in and out of the scattering volume.
In comparison to the Aβ40 system, Aβ42 and [VPV]Aβ42 displayed more prominent contributions from oligomers. This means that many fewer 60–80 nm aggregates were present. The oligomer fraction remained stable over a month of observation. In addition, oligomers of [VPV]Aβ42 had RH≈8 nm. This size was less than the 10 nm size typically observed in Aβ42 experiments19,35 and may reflect a difference in oligomer structure of Aβ42 and [VPV]Aβ42. Interestingly, [pP]Aβ42 behaved much more like Aβ40. It predominantly formed small oligomers with RH≈2 nm, and no significant increase in size occurred. Some larger aggregates were present that had RH≈20–30 nm. These aggregates were much smaller than those in the Aβ40 samples and scattered much less light. As a consequence, the relative contribution of the 2 nm oligomer fraction in [pP]Aβ42 was very prominent.
Aβ assembly morphology
To determine if assembly stage-specific differences in morphology existed among the different Aβ peptides, aliquots of the assembly reactions were examined using electron microscopy (EM). The kinetics of assembly differs among the peptides. For this reason, examination of different peptide samples at the same times would not allow morphologic comparisons of each peptide at the same stage of assembly. To control for this variance, we used a temporal normalization procedure. In an independent set of experiments, we monitored the time-dependent evolution of β-sheet structure. We determined β-sheet content at the initiation of peptide incubation (tβ=0%) and at the time at which β-sheet content was maximal (tβ=100%). We then determined the half-time (tβ=50%) for this process. Within experimental error, this kinetics was reproducible, which allowed us in subsequent experiments to remove aliquots of each peptide for EM analysis at equivalent assembly stages. However, in addition, CD monitoring was done on the actual samples used for EM to ensure that aliquot removal was done at equivalent stages. Each aliquot was frozen in liquid nitrogen and stored at −80° C until analysis. Importantly, the thawed samples were used concurrently for EM and cytotoxicity assays (see below) to ensure that rigorous structure–activity correlations could be accomplished.
Initially, small (10–30 nm diameter) circular or irregular structures were observed in the Aβ40 (Fig. 9, panel tβ=0) and Aβ42 (Fig. 9, panel tβ=0) samples. [VPV]Aβ40 formed structures that were larger than those of Aβ40, ≈20–30 nm in diameter compared with ≈10–20 nm. Each of the [VPV]Aβ mutants produced structures that were larger in size than their wild type Aβ42 counterparts and often were found clumped into larger superstructures. [VPV]Aβ42 formed a mixture of spherical oligomers ranging in size from 13–20 nm and worm-like aggregates that were ≥100 nm in size. [pP]Aβ42 formed comparatively smaller structures than did [VPV]Aβ42.
Fig. 9.

Morphology of peptide assemblies. Electron micrographs of Aβ40, Aβ42, and their analogues at tβ=0%, 50% and 100% (0, 50, and 100% points in the process of β-sheet formation: see text for explanation). Red arrows point to structures discussed in the text. In the plate of [VPV]Aβ42 at tβ= 100, a yellow arrow shows a thin filament, a thick white arrow points to the section of the micrograph expanded in the inset, in which a smaller white arrow points to a railroad track-like structure. Magnification is 29,000. Scale bar is 100 nm.
At tβ=50% point in assembly, Aβ40 and [VPV]]Aβ40 formed aggregates containing globular units of 5–10 nm diameter and 10–40 nm diameter, respectively. Aβ42 formed globular species with diameter ranging from 20–30 nm. [VPV]Aβ42 formed globular structures ranging in diameter from 50–100 nm. Small numbers of fibrils, with diameters of 10–20 nm, also were observed. [pP]Aβ42 formed comparatively smaller structures than did [VPV]Aβ42. Some irregular structures had diameters of 20 nm. Others appeared to cluster in aggregates with sizes ranging from 40–100 nm. Each respective Aβ assembly formed at tβ=50% was larger than that observed at tβ=0%.
Cursory examination of the [VPV]Aβ42 assemblies suggested that the distribution of sizes might not be continuous. For this reason, we determined quantitatively the size frequency distribution of the assemblies (Fig. 10). The distribution showed that the predominant assembly diameter was 50 nm. Substantial numbers of structures with diameters of 36 nm, 43nm and 57 nm also were observed.
Fig. 10.
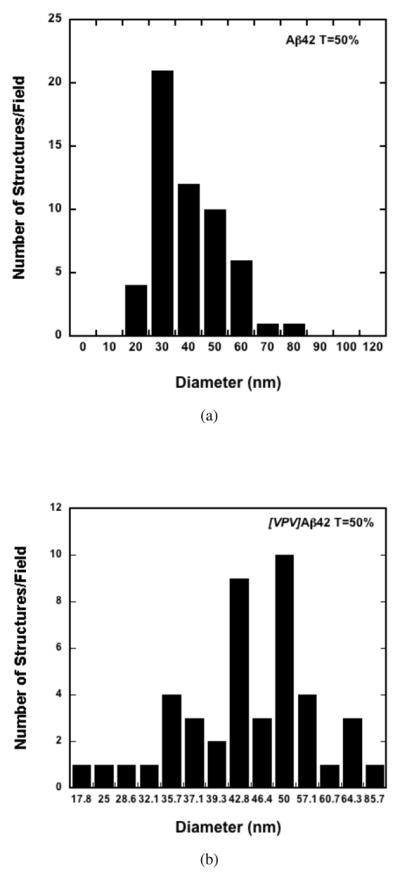
Histograms of assembly diameters observed by EM of Aβ42 (top panel) and [VPV]Aβ42 (bottom panel) at tβ=50%. The total number of structures examined were 56 and 44, respectively.
At tβ=100%, Aβ40 and [VPV]Aβ40 formed fibrils that ranged in diameter from 5–10 nm and 8–10 nm, respectively. Aβ42 produced a dense meshwork of fibrils with diameters of 10–15 nm. Many of the fibrils appeared helical with a pitch of ≈40 nm. [VPV]Aβ42 displayed quasicrystalline structures (Fig. 9, white arrow), along with fibrils. The quasicrystalline structures were 20–60 nm in length and 40–80 nm in diameter and resembled railroad tracks and ties (Fig. 9, inset). Needle-like fibrils also were observed (Fig. 9, yellow arrow), and these had diameters of 5 nm, thinner than those of Aβ42. In contrast to the other four peptides, [pP]Aβ42 did not form fibrils but rather remained in a relatively amorphous state characterized by masses of assemblies dispersed throughout the grid.
Cytotoxicity assays
To establish structure-activity relationships, two types of cytotoxicity assays were performed, MTS and LDH. The MTS assay was employed to evaluate the effects of the assemblies on cellular metabolism, including MTS reduction and exocytosis, and the LDH assay was performed to evaluate cell viability (plasma membrane integrity)36. Aβ samples were prepared as they were for EM studies and then the samples were added to rat primary hippocampal and cortical neurons.
Samples assayed immediately after preparation (tβ=0%) had no significant effect on MTS metabolism (Fig. 12). However, at tβ=50%, all the peptides except Aβ40 were toxic (p < 0.01). The toxicity of [VPV]Aβ40 trended lower than those of the Aβ42 peptides, but this difference was not statistically significant. At tβ=100%, [pP]Aβ42, [VPV]Aβ42, and [VPV]Aβ40 remained as toxic as they were at tβ=50%. The toxicity of Aβ42 trended toward greater toxicity than the control, but the difference was insignificant statistically. Aβ40 remained non-toxic.
Fig. 12.
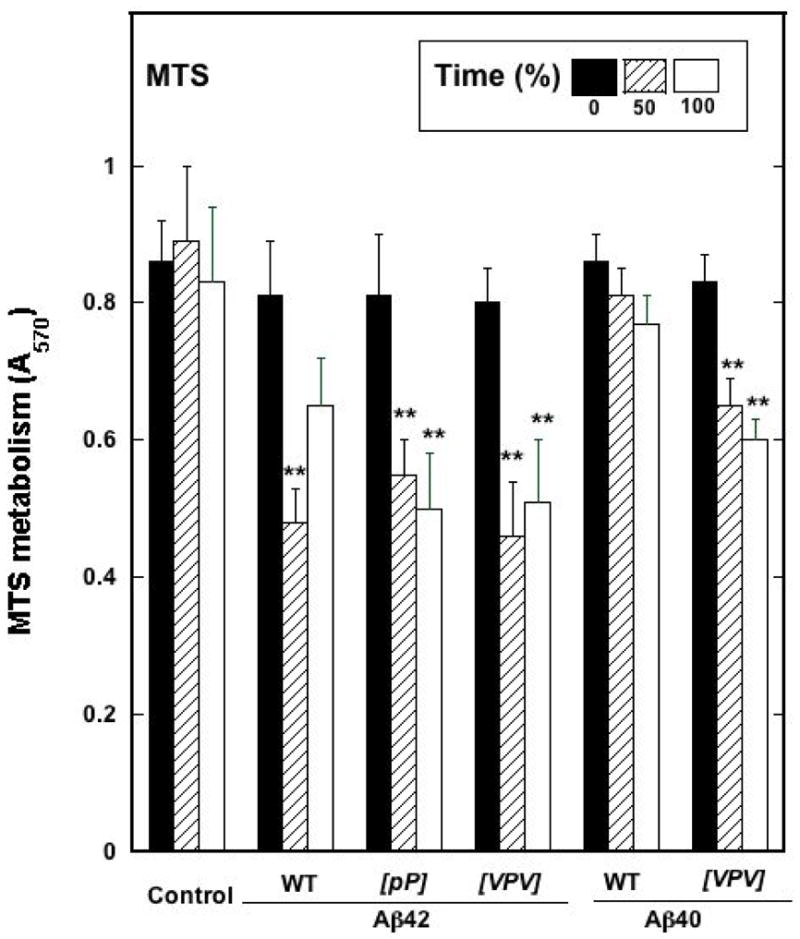
MTS activity as measured in rat primary hippocampal and cortical neurons for the different Aβ peptides and their substitutions at tβ=0%, 50%, and 100%. Absorbance was measured at 570 nm.
Results for the LDH assay were consistent with those of the MTS assay (Fig. 11). At tβ=0%, no significant toxicity was observed for any of the peptides. At tβ=50%, large, statistically significant (p < 0.01) increases in LDH activity were seen for all the Aβ42 peptides and for [VPV]Aβ40. Aβ40 toxicity was significantly higher than that of the control (p < 0.05), but the absolute increase was small. The absolute toxicity levels of the Aβ42 assemblies were higher than those of the Aβ40 assemblies. At tβ=100%, Aβ42 peptide toxicities remained approximately equal to, or were lower than, those observed for the same peptides at tβ=50%. However, in all cases, significant (p < 0.01) toxicity was observed (versus controls). Aβ40 was not toxic at tβ=100%, whereas the toxicity of [VPV]Aβ40 remained identical, within experimental error, to that observed at tβ=50%.
Fig. 11.
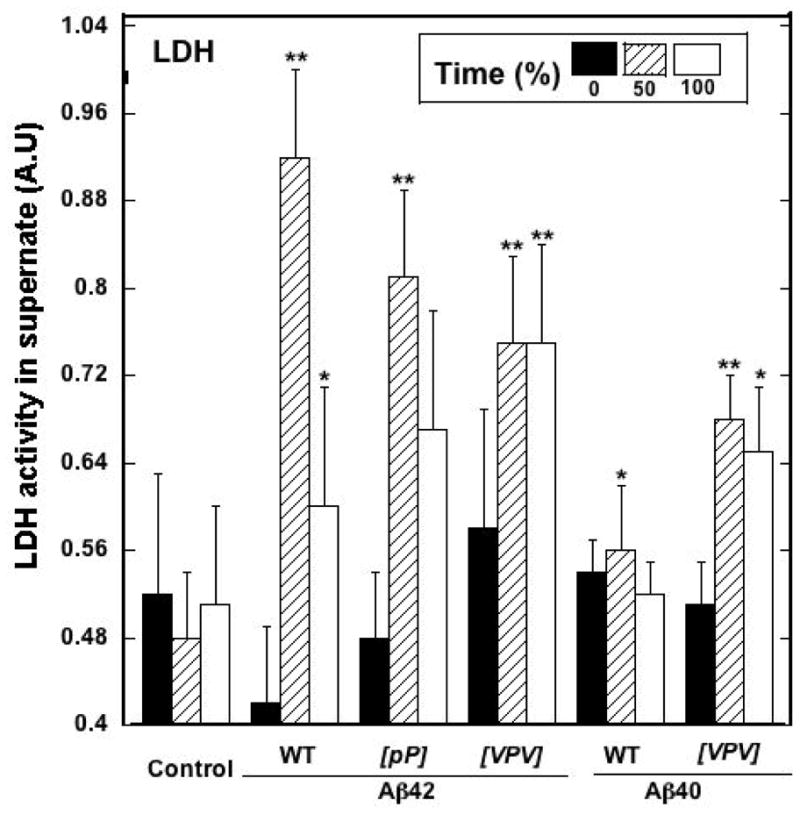
Lactate dehydrogenase (LDH) toxicity assays of Aβ assemblies. Aliquots removed from the samples used for CD experiments were used to treat rat primary hippocampal and cortical neurons. LDH activity released into the medium was used to assess assembly toxicity. **p<0.01 compared to control and *p<0.05 compared to control. Error bars are ±SD.
Discussion
Aβ40 and Aβ42 have been found to oligomerize in two distinct manners. Aβ40 forms primarily dimers, trimers, and tetramers, whereas Aβ42 assembles into pentamer/hexamer units (paranuclei) that can self-associate to produce dodecamers and hexadecamers14,19. Interestingly, experimental and in silico studies suggest that the overall conformational dynamics of the two peptides are similar, with the exception of their C-termini24–26. The C-terminus of Aβ42 is more rigid, an observation likely due to the more frequent intramolecular contacts within this segment23. In this study, we first used REMD simulations to extensively sample and then compare the conformational spaces of Aβ(31–42) and Aβ(31–40) foldons. We discovered that the most populated conformational cluster of Aβ(31–42) was a β-hairpin-like structure with a β-turn centered at V36–G37, whereas Aβ(31–40) existed predominately as a statistical coil (Fig. 3 & Fig. 1).
We hypothesized that if this β-hairpin conformation was the key structural element responsible for the distinct oligomerization behavior of Aβ40 and Aβ42, then we could perturb oligomerization by engineering stabilizing or destabilizing amino acid substitutions in this region, a study that would have important implications for targeting therapeutic agents. To test our hypothesis, we first engineered [pP]Aβ42, in which V36 and G37 were replaced by D-Pro and L-Pro, respectively. The D-Pro-L-Pro dipeptide is know to effectively constrain the backbone dihedral angles in a region favoring β-turn structure29. Surprisingly, simulations using [pP]Aβ(31–42) revealed that this peptide segment mostly existed as statistical coil. We interpret this result as an effect of the positive entropic contribution of the two Gly residues at 33 and 38, which favors a flexible conformer.
We next engineered [VPV]Aβ42, in which G33, V36, and G38 were replaced by Val, L-Pro, and Val, respectively. The G33→V and G38→V replacements should increase the rigidity of the backbone, as well as the hydrophobic interaction between the two β-strands. The L-Pro-Gly sequence is known to constrain the β-turn conformation. As predicted, the β-hairpin population of [VPV]Aβ(31–42) nearly doubled compared with Aβ(31–42) (Fig. 1). The increased stability of β-hairpin was reflected by the fact that [VPV]Aβ42 displayed high β-content immediately after dissolution, whereas Aβ42 existed in SC form after 5 days of incubation. ThT fluorescence results were consistent with these observations. Wild type Aβ40 and Aβ42 exhibited progressive increases in ThT binding as fibril formation proceeded. In contrast, [VPV]Aβ42 produced substantial ThT fluorescence immediately upon solvation, and the fluorescence intensity remained nearly constant during the duration of the experiment. These results suggest that substantial and increased β-sheet formation, relative to that found in wild-type Aβ42, occurs in [VPV]Aβ42 and that this β-sheet structure is stable. FTIR experiments have suggested that anti-parallel β-sheet is a structural signature of Aβ42 oligomers37, and this feature is consistent with our model of the Aβ42 C-terminus as a β-hairpin. DLS experiments also revealed that [VPV]Aβ42 formed oligomers that were more stable than those of Aβ42 (Figure S1).
To investigate how the stabilized and destabilized β-turn affected oligomerization, we used PICUP and SDS-PAGE (Fig. 8). Stabilization of the β turn, in the form of the [VPV]Aβ42 peptide, produced a tri-nodal distribution involving primarily monomer, pentamer/hexamer, and nonamer. The decreased dispersity of this distribution combined with the appearance of higher-order oligomers is consistent with the behavior of a peptide that could be characterized as a “super Aβ42.” Such a peptide explores a much more restricted volume of conformational space than does its wild type homologue, a volume comprising oligomeric conformational states of lower overall free energies or relatively high transitional activation energies. In the mirror-image experiment involving turn destabilization, the oligomer distribution of [pP]Aβ42 was indistinguishable, within experimental error, from that of Aβ40. This demonstrates that destabilization of the C terminal β-turn converts Aβ42 into Aβ40.
Unlike wild type or [VPV]Aβ42, [VPV]Aβ40 formed β-rich fibrillar structures, though at slower pace than Aβ42 and Aβ40. This was not surprising considering the decreased number of hydrogen bond donors/acceptors and the decreased hydrophobic interaction potential at the C-terminus of the Aβ40 peptides compared with the Aβ42 peptides. Only three residues exist after the turn proper in the Aβ40 system, as opposed to five in the Aβ42 system. This means that although the VPV substitution enables formation of a relatively stable turn, the overall stability is lower due to the lack of the other two amino acids. Nevertheless, the VPV substitutions alone are sufficient to support β-turn formation at residues 36 and 37 of Aβ40 and thus produce a C-terminal structure engendering Aβ42-like behavior in [VPV]Aβ40.
Aβ42 formed assemblies resembling strings of spherical oligomers at tβ=50% (Fig. 9), the midpoint of the assembly process. These structures also were observed with [VPV]Aβ42, but they remained dispersed and did not coalesce into protofibrils, suggesting that the [VPV]Aβ42 oligomers were more stable than those formed by Aβ42. Assemblies formed by both peptides exhibited significant and similar levels of neurotoxic activity (Fig. 11). After 5 d of incubation, Aβ42 formed amyloid fibrils and [VPV]Aβ42 formed quasicrystalline structures. Interestingly, at this time point, Aβ42 toxicity declined. This might have been due to the formation of macro-molecular aggregates that had decreased intrinsic toxicity or decreased ability to diffuse to and interact with cell membranes. In contrast, the toxicity of [VPV]Aβ42, which remained in oligomeric form, remained undiminished. [pP]Aβ42 also was toxic and its toxicity remained high both at tβ=50% and at tβ=100%. For each mutant Aβ42 peptide, substantially less higher-order assembly was observed relative to the wild type peptide form, consistent with an enhanced toxic potency of the oligomeric assemblies16,17. Consistent with a potential relationship between oligomer content and toxicity, [VPV]Aβ40 was quite toxic in MTS and LDH assays, unlike its wild type homologue. In addition, after incubation for 5 d, [VPV]Aβ40 toxicity was equivalent to that of wild type Aβ42 (Fig. 11), as would be predicted for an Aβ42-like peptide.
In previous experiments, we produced pure, stable dimers, trimers, and tetramers of Aβ40 and showed that each species seeded growth of amyloid fibrils16. Seeding capacity depended directly on the extent of structural order within each oligomer population, as determined by CD and ThT analyses. This suggested that these oligomers shared at least some structural features with fibrils. Pentamers and hexamers were not studied. Fibril models have suggested that the C-termini of Aβ40 and Aβ42 form parallel, in-register β-strands38,39. However, structural diversity exists among fibril populations because differences in fibril preparation method produce fibrils of differing morphology40. Our results provide one mechanistic interpretation for these results, namely that differences in monomer C-terminal structure drive assembly down different pathways (Fig. 13). In the simplest case, low-order oligomers (dimers, trimers, tetramers, and certain types of pentamer or hexamer) possess C-termini that do not form β-hairpins. This state exists in the Aβ40 system in particular14, but also in the Aβ42 system, and gives rise to classical amyloid-type fibrils (Fig. 13A). When stable C-terminal β-hairpins do exist, pentamers and hexamers (paranuclei) are stabilized, which simultaneously hinders the formation of dimers, trimers, and tetramers (Fig. 13B). One mechanism for this stabilization may be the increase in hydrophobic surface created by turn formation at residues 35–38, which facilitates inter- and intra-peptide interactions leading to and stabilizing oligomers. This is especially evident in the case of [VPV]Aβ42. The result of this stabilization is subsequent formation of distinct fibrillar structures with relatively small aspect ratios and a unique (“railroad tracks and ties”) morphology. In contrast, [pP]Aβ42 cannot form paranucleus because its C-terminus cannot fold into the necessary β-hairpin structure.
Fig. 13.
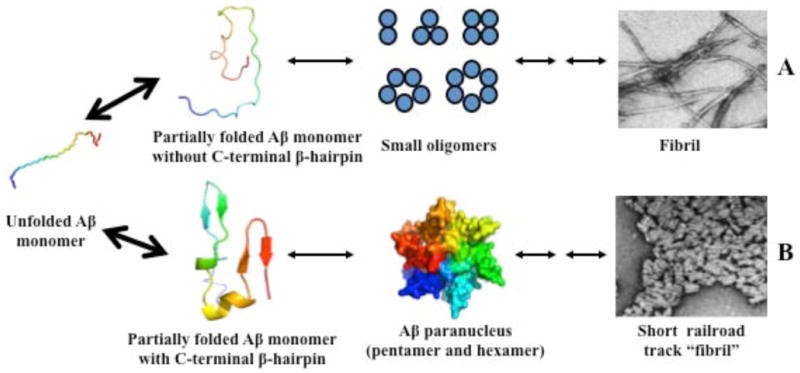
A proposed mechanism for Aβ assembly. Aβ monomer can adopt two different types of conformation, one without C-terminal β-hairpin, the other with C-terminal β-hairpin. The former may form small oligomers which eventually deposit as amyloid fibrils, and the later may form paranuclei and ultimately form “railroad track-like fibrils”. Big arrows indicate there are rapid equilibrium among unfolded Aβ monomer and partially folded monomers. The Aβ monomeric structure is colored from blue (N-terminus) to red (C-terminus). Pentamer and hexamer on the two different pathways are colored as blue and red, respectively, which indicates they may have different structures.
Other C-terminal turns may exist. Ahmed et al., using low temperature and low salt conditions to produce Aβ42 pentamers, reported that residues 37 and 38 underwent hydrogen-deuterium exchange, whereas flanking residues did not, suggesting that these two residues adopted a turn-like conformation41. This turn position previously had been proposed in in silico modeling studies42,43. More recently, Rajadas et al. replaced Gly37–Gly38 with Pro-Gly and found that the substitutions caused Aβ42 to form more stable oligomers, but these oligomers were relatively disordered44. It is noteworthy that a recent study suggested that β-hairpins involving Gly–Gly are relatively unstable29. Murakami et al. suggested that a turn is centered at residues 38–39 and that this turn may be responsible for bringing the C-terminal carboxylate anion close to an S-oxidized radical cation of Met35, thus stabilizing it45. These other turn positions are different from that reported here, but taken together emphasize the importance of C-terminal turn formation in controlling Aβ oligomerization and higher-order assembly.
Recently, a very interesting new structure, the “cylindrin,” was described46. This hexamer of peptide undecamers forms a cylindrical structure that has secondary structure, immunological (A11+), and toxicity characteristics similar to Aβ paranuclei. It is possible that one or more short Aβ peptide segments could form a cylindrical core that organizes paranucleus formation. No evidence yet exists for this possibility, but the question currently is under active study.
In summary, our data suggest that the C-terminal Val36–Gly37 turn is the sine qua non of Aβ42. Facilitating its formation in Aβ40 creates a more Aβ42-like peptide. Stabilizing the turn in Aβ42 creates a “super Aβ42.” The VPV substitutions stabilized the β-hairpin and facilitated Aβ42 paranuclei formation. [VPV]Aβ42 assemblies were neurotoxic and comprised a population with few classical amyloid-type fibrils but with substantial numbers of unusual, short, quasi-crystalline structures resembling railroad tracks and ties. Destabilizing the turn in Aβ42 makes this peptide “Aβ40-like.” This makes the turn a particularly attractive and important target for therapeutic agents. In addition, our engineered mutants should be useful tools for mechanistic studies of Aβ neurotoxicity because of the relatively high stability of the oligomers formed.
Materials and methods
Molecular dynamics simulation
We previously used the Generalized Born (GB) implicit solvent model47 and replica-exchange molecular dynamics (MD) for our simulations, obtaining a qualitative picture of the conformational dynamics of full-length Aβ40 and Aβ42 23. However, modeling solvent implicitly may preclude the definition of the high-resolution structure of Aβ because these models do not represent the explicit atomic interactions between water and protein molecules and they may underestimate the frictional effects of water molecules surrounding the protein 47. As a result, peptide populations may appear to possess higher conformational freedom and lower structural stability, when in fact they do not.
Simulation using full-length Aβ in explicit water remains impractical, as it requires enormous computational resources 23. For this reason, we study the representative C-terminal folding units of Aβ40 and Aβ42, Aβ(31–40) and Aβ(31–42), respectively. Aβ23, and many other proteins 48, comprise autonomous or semi-autonomous folding units (“foldons”)49. The study of the conformational dynamics of these foldons can provide information relevant to the segmental folding of the holoprotein48,49. For Aβ, a large body of computational work has been done successfully on the Aβ(21–30) segment that comprises a peptide monomer folding nucleus27,50–53. These computational studies confirmed and extended prior experimental studies of the decapeptide and of the full-length Aβ peptide27,54.
Simulations were performed with the SANDER module of the Amber simulation package (version 10)55. The peptides were modeled by PARM99SB, a recently improved all-atom force field56. An extended copy of the peptide was heated to 300 K and subjected to a 20 ps MD run. The final conformation then was used as the starting conformation for the production runs. The starting conformers were desolvated in an octahedral TIP3P water box 57. The minimum distance of a protein atom to the edge of the box was 12 Å. A single Na+ ion was added to the system to maintain system neutrality. This system models a very dilute aqueous peptide solution at neutral pH. The system was minimized by 1000 steps of energy minimization to release geometry collision before being subjected to 500 ps of equilibration at NTP (1 bar and 298 K). Replica exchange molecular dynamics (REMD) simulations then were performed. Sixty-four replicas that exponentially spanned the temperature range 270–600 K were created. The temperature of the system was regulated using the Langevin dynamics algorithm 58 with a collision frequency of 3.0 ps−1. The particle mesh Ewald summation method 59 was used to treat the long-range electrostatic interaction. During the simulation, hydrogen atoms were constrained using the SHAKE algorithm 60. The integration time step was 2 fs. Exchange between replicas was attempted every 2 ps. Other relevant parameters were set by default. For each replica, the simulation length was 50 ns and 50,000 conformations were collected. The first 30 ns was treated as equilibration and the last 20 ns were used for data analysis.
In our studies here, each peptide was subjected to 50 ns of replica-exchange molecular dynamics (REMD) simulations at 298 K. The first 30 ns was used to equilibrate the system. The production run comprised 20,000 conformations collected from the last 20 ns. To determine if the simulation had converged, we divided the last 20 ns of data into two equal parts and then subjected each to secondary structure analysis using DSSP 28. The extent of overlap of the curves suggests that the two conformational ensembles are highly similar, indicating convergence (Fig. S1).
Peptide Synthesis
Aβ40, Aβ42, and their analogues were synthesized using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry and purified by reverse phase high performance liquid chromatography (RP-HPLC), essentially as described61. The identity and purity (usually >97%) of the peptides were confirmed by amino acid analysis followed by mass spectrometry and reverse phase high performance liquid chromatography (RP-HPLC).
Preparation of low molecular weight (LMW) Aβ42
Two hundred μg of each peptide lyophilizate was dissolved in 10% (v/v) 60 mM NaOH, followed by 45% (v/v) MilliQ water. The pH was adjusted to 7.5 by addition of 45% (v/v) 10 mM sodium phosphate, pH 7.5, yielding final nominal concentrations of 25–80 μM (depending on the experiment) in 4.5 mM phosphate buffer, pH 7.5. The peptide solution then was sonicated for 1 min in a Branson ultrasonic water bath (Branson Ultrasonics Corp, Danbury CT) and then centrifuged at 16,000 × g at room temperature (RT; usually 22° C) for 10 min. The supernatant fluid was filtered using a 0.2 μm Anotop filter and placed on ice. The filtrate is defined as “low molecular weight” Aβ and comprises an equilibrium mixture of monomer and low-order oligomers 62. Protein concentrations of these and other preparations were determined by quantitative amino acid analysis, unless otherwise indicated.
Photo-induced chemical cross-linking of Aβ
Aβ oligomerization was studied using Photo-Induced Cross-linking of Unmodified Proteins (PICUP)18, essentially as described63. Briefly, LMW Aβ was prepared at a concentration of 25–35 μM in 4.5 mM sodium phosphate, pH 7.5, at RT. Cross-linking was performed by adding 18 μL of sample to a 0.2 ml volume PCR tube. One μL of 2 mM Tris (2,2′-bipyridyl)dichlororuthenium (II)hexahydrate (Ru (bpy)) and 1μL of 40 mM ammonium persulfate (APS) then were added, after which the tube was irradiated for 1s with visible light. The reaction was quenched immediately with 1μL of 1 M dithiothreitol (DTT) and the sample then was placed on ice. An equal volume of 2× Tris-Tricine SDS sample buffer was added to each sample. The samples were boiled in a 100° C water bath for 10 min, centrifuged for 5–10 s at 16,000 × g, and then electrophoresed on a 10–20% T 1 mm thick Tris-Tricine SDS gel. The gel was silver stained using an Invitrogen X-press silver staining kit.
Circular Dichroism Spectroscopy
LMW Aβ solutions were prepared at a concentration of 60–80 μM. After sonication, the peptide samples were incubated at 37° C with slow inversion on a MiniLabroller (Edison, NJ). CD spectroscopy then was performed every 24 h using a JASCO J-810 spectropolarimeter (Tokyo, Japan). The CD parameters were wavelength range of 190–260 nm, data pitch of 0.2 nm, continuous scan mode, scan speed of 100 nm/min, 1 sec response, band width of 2 nm, and an accumulation of 10 scans per sample. The spectra were smoothed using the different adaptive smoothing parameters within the data acquisition software (Spectra Manager). The data subsequently were deconvoluted using DichroWeb 64.
Thioflavin T (ThT) Fluorescence
ThT is a fluorescent dye which has been used to measure the time-dependent acquisition of β-sheet structure associated with fibrillar assemblies. ThT fluorescence does not measure fibril concentration per se (some fibrils do not possess the β-sheet structures to which ThT binds), but fluorescence intensities do correlate with Aβ fibril content 32. LMW Aβ peptides were prepared at nominal concentrations of 35–40 μM. The samples were incubated with slow end-over-end mixing (inversion) on a MiniLabroller (Edison, NJ). At 24 h intervals, 10 μL of each sample were removed and added to 190 μL of 20 μM ThT dissolved in the same buffer. The solution was vortexed gently, incubated for 5 min at RT, and then fluorescence was determined using a Hitachi 4500 fluorimeter (Tokyo, Japan). Readings were obtained at an excitation wavelength of 450 nm and an emission wavelength of 482 nm. The slit widths were 5 nm and 10 nm, respectively. The readings were repeated three times at intervals of 30 seconds and the mean of the blank-corrected three readings was calculated. “Blanks” contained 20 μM ThT in buffer.
Quasielastic Light Scattering Spectroscopy (QLS)
DLS complements PICUP35. It requires no chemical stabilization of oligomers and its sensitivity increases with increasing oligomer molecular weight. PICUP, in contradistinction, is particularly useful for quantitation of low-order oligomer frequency distributions, but because cross-linking efficiency is <100%, it becomes increasingly inaccurate as molecular weight rises. Fig. S1 shows the temporal evolution of the size distributions of the wild type and modified Aβ peptides. Aβ40, Aβ42, and their respective mutants were dissolved at a concentration of 0.5 mg/ml in 20 mM sodium phosphate buffer, pH 7.5, briefly vortexed, sonicated for 20 s, and filtered using a 20 nm Anotop filter. Samples were subjected to QLS at RT for 7–10 days. Measurements were done using a custom optical setup comprising a 40 mW He-Ne laser (λ = 633 nm) (Coherent, Santa Clara, CA) and a PD2000DLS detector/correlator unit (Precision Detectors, Bellingham, MA). Light scattering was measured at an angle of 90°. The intensity correlation function and the diffusion constant (D) frequency distribution were determined using Precision Deconvolve software (Precision Detectors, Bellingham, MA). Hydrodynamic radius (RH) values were obtained from those for (D) using the Stokes-Einstein relationship 11 D = kTB=6πηRH, allowing inferences to be made about the distribution of scatterer sizes.
Electron Microscopy
Formvar 400 mesh grids were glow-discharged on a MED 010 EM glow discharge apparatus containing a cylindrical discharge compartment and an adjacent discharge control and timer unit. Peptide samples were mixed thoroughly and 8 μL of sample were layered carefully on the grid. The grid was incubated for 20 min under cover to prevent dust accumulation. After incubation, the solution was carefully drained using a filter paper wick by gently touching the tip of the filter paper to the edge of the grid. Five μL of 2.5% (v/v) glutaraldehyde (GTA) was added to the grid, which then was incubated for 3 min in the absence of light. The GTA solution was removed after incubation using a filter paper wick. Five μL of 1% (w/v) uranyl acetate was applied to the grid, which was incubated for 3 min in the dark. The solution was blotted away and the grids were air dried and examined on a JEOL 1200 EX transmission electron microscope.
Primary Neuronal Cultures
Rat cortical cultures were established from embryonic day 17 fetuses, as described previously 65. Briefly, the brain tissue was dissociated into a single-cell suspension by incubation with 0.25% trypsin/PBS at 37° C for 30 min and mechanical dissociation using a fire-polished glass Pasteur pipette. Cells were plated at a density of 20,000 cells/cm2 on glass coverslips in 35- and 100-mm culture dishes. Two h after plating, the medium was changed to Neurobasal plus N2 and B27 supplements (Invitrogen, Grand Island, NY). Cells were maintained at 37° C and 5% CO2 with 50% of the medium changed every five days. Cells were treated with various preparations of Aβ at 14 d in vitro for 12 and 24 h.
Neurotoxicity Assays
Cell death was assessed by quantifying lactate dehydrogenase (LDH) release using the CytoTox 96 kit (Promega, Madison, WI) 66. Cells were treated with Aβ peptides removed at different time points from the CD reaction mixtures. Each aliquot was snap frozen in liquid nitrogen and then stored at −85° C until assay. LDH released into the culture supernate due to Aβ-induced cell lysis was measured with a 30 min coupled enzymatic assay that resulted in the conversion of a tetrazolium salt (INT) into a red formazan product. The amount of color formed is proportional to the number of lysed cells.
Mitochondrial oxidoreductase activity was determined by analyzing the conversion of a tetrazolium compound to formazan. The reagent 3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium (MTS) is reduced to formazan by mitochondrial succinate dehydrogenase in complex II (succinate/ubiquinone oxidoreductase complex) and possibly other complexes of the electron transport chain (CellTiter 96 AQueous; Promega) 66. The quantity of formazan product measured by A490 is directly proportional to the number of living cells in culture.
Protein concentrations were determined using the BCA protein assay kit (Thermo Scientific, CA), using the microtiter plate protocol. The concentrations of the Aβ40 and Aβ42 peptides were adjusted with 10 mM sodium phosphate, pH 7.5, to maintain uniformity. The final peptide concentration used in both assays was 2.5 μM.
Supplementary Material
Highlights.
A structural basis for the enhanced amyloidogenicity of Aβ42 has been determined.
A β-turn exists at the C-terminus of Aβ42 but not Aβ40.
Amino acid substitutions within the turn can interconvert Aβ40 and Aβ42.
The turn could be a useful therapeutic target.
Acknowledgments
We thank Margaret Condron for synthesizing all the peptides used here. We gratefully acknowledge grants from the State of California Alzheimer’s Disease Research Fund (No. 07-65806), a UCLA Faculty Research Grant, the National Institutes of Health (AG027818, NS038328), and the Jim Easton Consortium for Alzheimer’s Drug Discovery and Biomarkers.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid β-protein
- CD
circular dichroism spectroscopy
- EM
electron microscopy
- LMW
low molecular weight
- REMD
Replica exchange molecular dynamics simulations
- ThT
Thioflavin T
- RH
hydrodynamic radius
Footnotes
Throughout this publication we refer to both the “turn” and the “hairpin.” The former term is restricted to Val36 and Gly37 and its neighboring residues, Met35 and Gly38, that are involved in H bond formation that stabilizes the turn. The term “hairpin” refers to the global structure of the Aβ(31–42) peptide, a structure that must possess a turn to exist.
In the context of oligomerization experiments, the term “order” refers to the number of Aβ monomers comprising the assembly.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer disease. J Biol Chem. 2009;284(8):4749–4753. doi: 10.1074/jbc.R800036200. URL http://dx.doi.org/10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glenner GG, Wong CW. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 5.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Younkin LH, Suzuki N, Younkin SG. Amyloid β-protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ 42(43) J Biol Chem. 1995;270(13):7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 6.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13(1):45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid β-protein secreted by familial amyloid β-protein precursor (β APP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 8.Golde TE, Eckman CB, Younkin SG. Biochemical detection of Aβ isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502(1):172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 9.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 10.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–216. doi: 10.1038/35102591. URL http://dx.doi.org/10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 11.Lomakin A, Teplow DB, Benedek GB. Quasielastic light scattering for protein assembly studies. Methods Mol Biol. 2005;299:153–174. doi: 10.1385/1-59259-874-9:153. [DOI] [PubMed] [Google Scholar]
- 12.Jarrett JT, Berger EP, Lansbury PT. The carboxy terminus of the β-amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 13.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA. 1996;93(3):1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea J-E, Ruotolo BT, Robinson CV, Bowers MT. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem. 2009;1(4):326–331. doi: 10.1038/nchem.247. URL http://dx.doi.org/10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bitan G, Vollers SS, Teplow DB. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J Biol Chem. 2003;278(37):34882–34889. doi: 10.1074/jbc.M300825200. URL http://dx.doi.org/10.1074/jbc.M300825200. [DOI] [PubMed] [Google Scholar]
- 16.Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc Natl Acad Sci USA. 2009;106(35):14745–14750. doi: 10.1073/pnas.0905127106. URL http://dx.doi.org/10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahlgren KN, Manelli AM, Stine WB, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem. 2002;277(35):32046–32053. doi: 10.1074/jbc.M201750200. URL http://dx.doi.org/10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 18.Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci U S A. 1999;96(11):6020–6024. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003;100(1):330–335. doi: 10.1073/pnas.222681699. URL http://dx.doi.org/10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. URL http://dx.doi.org/10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 22.Westlind-Danielsson A, Arnerup G. Spontaneous in vitro formation of supramolecular β-amyloid structures, “βamy balls”, by β-amyloid 1-40 peptide. Biochemistry. 2001;40(49):14736–14743. doi: 10.1021/bi010375c. [DOI] [PubMed] [Google Scholar]
- 23.Yang M, Teplow DB. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J Mol Biol. 2008;384(2):450–464. doi: 10.1016/j.jmb.2008.09.039. URL http://dx.doi.org/10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J Mol Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. URL http://dx.doi.org/10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon I-JL, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the Aβ(1-40) and Aβ(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126(7):1992–2005. doi: 10.1021/ja036813f. URL http://dx.doi.org/10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 26.Yan Y, Wang C. Aβ42 is more rigid than Aβ40 at the C terminus: Implications for Aβ aggregation and toxicity. J Mol Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. URL http://dx.doi.org/10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 27.Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14(6):1581–1596. doi: 10.1110/ps.041292205. URL http://dx.doi.org/10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. URL http://dx.doi.org/10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 29.Gibbs AC, Bjorndahl TC, Hodges RS, Wishart DS. Probing the structural determinants of type II′ β-turn formation in peptides and proteins. J Am Chem Soc. 2002;124(7):1203–1213. doi: 10.1021/ja011005e. [DOI] [PubMed] [Google Scholar]
- 30.Budesinsky M, Sebestik J, Bednarova L, Baumruk V, Safarik M, Bour P. Conformational properties of the Pro-Gly motif in the D-Ala-l-Pro-Gly-D-Ala model peptide explored by a statistical analysis of the NMR, Raman, and Raman optical activity spectra. J Org Chem. 2008;73(4):1481–1489. doi: 10.1021/jo702297y. URL http://dx.doi.org/10.1021/jo702297y. [DOI] [PubMed] [Google Scholar]
- 31.Rao MHVR, Kumar SK, Kunwar AC. Formation of β-hairpins in L-Pro-Gly containing peptides facilitated by 3-amino benzoic acid. Tetrahedron Lett. 2003;44:736965027372. [Google Scholar]
- 32.Groenning M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J Chem Biol. 2009;3:1–18. doi: 10.1007/s12154-009-0027-5. URL http://dx.doi.org/10.1007/s12154-009-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiang W, Yau W-M, Tycko R. Structural evolution of Iowa mutant β-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J Am Chem Soc. 2011;133(11):4018–4029. doi: 10.1021/ja109679q. URL http://dx.doi.org/10.1021/ja109679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bitan G, Teplow DB. Rapid photochemical cross-linking–a new tool for studies of metastable, amyloidogenic protein assemblies. Acc Chem Res. 2004;37(6):357–364. doi: 10.1021/ar000214l. URL http://dx.doi.org/10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 35.Lomakin A, Teplow DB. Quasielastic light scattering study of amyloid β-protein fibril formation. Protein Pept Lett. 2006;13(3):247–254. doi: 10.2174/092986606775338515. [DOI] [PubMed] [Google Scholar]
- 36.Lobner D. Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis? J Neurosci Methods. 2000;96(2):147–152. doi: 10.1016/s0165-0270(99)00193-4. [DOI] [PubMed] [Google Scholar]
- 37.Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufr64ne Y, Narayanaswami V, Goormaghtigh E, Ruysschaert J-M, Raussens V. Anti-parallel β-sheet - a signature structure of the oligomeric amyloid-β peptide. Biochem J. 2009;421:415–423. doi: 10.1042/BJ20090379. URL http://dx.doi.org/10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 38.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99(26):16742–16747. doi: 10.1073/pnas.262663499. URL http://dx.doi.org/10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-β(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science. 2005;307(5707):262–265. doi: 10.1126/science.1105850. URL http://dx.doi.org/10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Nostrand WEV, Smith SO. Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17(5):561–567. doi: 10.1038/nsmb.1799. URL http://dx.doi.org/10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In silico study of amyloid β-protein folding and oligomerization. Proc Natl Acad Sci USA. 2004;101(50):17345–17350. doi: 10.1073/pnas.0408153101. URL http://dx.doi.org/10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelley NW, Vishal V, Krafft GA, Pande VS. Simulating oligomerization at experimental concentrations and long timescales: A Markov state model approach. J Chem Phys. 2008;129(21):214707. doi: 10.1063/1.3010881. URL http://dx.doi.org/10.1063/1.3010881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajadas J, Liu CW, Novick P, Kelley NW, Inayathullah M, Lemieux MC, Pande VS. Rationally designed turn promoting mutation in the amyloid-β peptide sequence stabilizes oligomers in solution. PLoS One. 2011;6(7):e21776. doi: 10.1371/journal.pone.0021776. URL http://dx.doi.org/10.1371/journal.pone.0021776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, Shirasawa T. Formation and stabilization model of the 42-mer Aβ radical: implications for the long-lasting oxidative stress in Alzheimer’s disease. J Am Chem Soc. 2005;127(43):15168–15174. doi: 10.1021/ja054041c. URL http://dx.doi.org/10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- 46.Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, Eisenberg D. Atomic view of a toxic amyloid small oligomer. Science. 2012;335(6073):1228–1231. doi: 10.1126/science.1213151. URL http://dx.doi.org/10.1126/science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bashford D, Case DA. Generalized Born models of macromolecular solvation effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. URL http://dx.doi.org/10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 48.Maity H, Maity M, Krishna MMG, Mayne L, Englander SW. Protein folding: the stepwise assembly of foldon units. Proc Natl Acad Sci USA. 2005;102(13):4741–4746. doi: 10.1073/pnas.0501043102. URL http://dx.doi.org/10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panchenko AR, Luthey-Schulten Z, Wolynes PG. Foldons, protein structural modules, and exons. Proc Natl Acad Sci U S A. 1996;93(5):2008–2013. doi: 10.1073/pnas.93.5.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cruz L, Urbanc B, Borreguero JM, Lazo ND, Teplow DB, Stanley HE. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc Natl Acad Sci USA. 2005;102(51):18258–18263. doi: 10.1073/pnas.0509276102. URL http://dx.doi.org/10.1073/pnas.0509276102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea J-E. Structure of the 21–30 fragment of amyloid β-protein. Protein Sci. 2006;15(6):1239–1247. doi: 10.1110/ps.062076806. URL http://dx.doi.org/10.1110/ps.062076806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen W, Mousseau N, Derreumaux P. The conformations of the amyloid-β (21-30) fragment can be described by three families in solution. J Chem Phys. 2006;125(8):084911. doi: 10.1063/1.2337628. URL http://dx.doi.org/10.1063/1.2337628. [DOI] [PubMed] [Google Scholar]
- 53.Fawzi NL, Phillips A, Ruscio J, Doucleff M, Wemmer D, Head-Gordon T. Structure and dynamics of the Aβ 21–30 peptide from the interplay of NMR experiments and molecular simulations. J Am Chem Soc. 2008;130:614565026158. doi: 10.1021/ja710366c. URL http://dx.doi.org/10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, Rigby AC, Teplow DB. Familial Alzheimer’s disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc Natl Acad Sci U S A. 2007;104(42):16522–16527. doi: 10.1073/pnas.0705197104. URL http://dx.doi.org/10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Case D, Darden T, Cheatham T, CS, Wang J, Duke R, Luo R, Merz K, Wang B, Pearlman D, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell J, Ross W, Kollman P. Amber 8. University of California; San Francisco: 2004. [Google Scholar]
- 56.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–725. doi: 10.1002/prot.21123. URL http://dx.doi.org/10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 58.Lamm G, Szabo A. Langevin modes of macromolecules. J Chem Phys. 1986;85(12):7334–7348. [Google Scholar]
- 59.Darden T, York D, Pedersen L. Particle mesh Ewald: a N-log(n) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 60.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of nalkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 61.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272(35):22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 62.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274(36):25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 63.Bitan G, Lomakin A, Teplow DB. Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem. 2001;276(37):35176–35184. doi: 10.1074/jbc.M102223200. URL http://dx.doi.org/10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 64.Whitmore L, Wallace BA. Dichroweb, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue):W668–W673. doi: 10.1093/nar/gkh371. URL http://dx.doi.org/10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma Q-L, Yang F, Calon F, Ubeda OJ, Hansen JE, Weisbart RH, Beech W, Frautschy SA, Cole GM. p21-activated kinase-aberrant activation and translocation in alzheimer disease pathogenesis. J Biol Chem. 2008;283(20):14132–14143. doi: 10.1074/jbc.M708034200. URL http://dx.doi.org/10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid-β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26(22):6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. URL http://dx.doi.org/10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.