

Abstract
Introduction
Impaired inhibition of fear in the presence of safety cues and a deficiency in the extinction of fear cues are increasingly thought to be important biological markers of Posttraumatic Stress Disorder (PTSD). Other studies have suggested that there may be altered neural activation during behavioral inhibition tasks in subjects with PTSD. The current study aimed to see whether neural activation during inhibition was reduced in a highly traumatized civilian population, and whether atypical activation was associated with impaired fear inhibition.
Methods
The participants were 41 traumatized women (20 PTSD+, 21 PTSD−) recruited from Grady Memorial Hospital in Atlanta, GA. We used a Go/NoGo procedure with functional magnetic resonance imaging (fMRI) in a high-resolution 3T scanner. Participants were instructed to press a button whenever an “X” or “O” appeared on the screen, but not if a red square appeared behind the letter. Participants were assessed for trauma history and PTSD diagnosis, and completed a fear-potentiated startle and extinction paradigm.
Results
We found stronger activation in the ventromedial prefrontal cortex (vmPFC) in traumatized subjects without PTSD compared to those with PTSD in the NoGo greater than Go contrast condition. Activation in the vmPFC was negatively correlated with fear-potentiated startle responses during safety signal learning (p=.02) and fear extinction (p=.0002).
Conclusions
These results contribute to understanding of how the neural circuitry involved in inhibitory processes may be deficient in PTSD. Furthermore, the same circuits involved in behavioral inhibition appear to be involved in fear inhibition processes during differential fear conditioning and extinction.
1. Introduction
Posttraumatic stress disorder (PTSD) can develop in some individuals after exposure to an event that causes extreme fear, horror, or helplessness (APA, 1994). PTSD is characterized by three primary symptom clusters following a traumatic experience: a) the first cluster of symptoms includes re-experiencing of the traumatic event through intrusive thoughts, nightmares, flashbacks, and related phenomena that are often produced by reminders of the traumatic event; b) the second cluster is characterized by avoidance symptoms including loss of interest in social situations and emotional detachment; and c) the third cluster includes psychophysiological reactivity in response to trauma-related stimuli including exaggerated startle, hypervigilance, elevated perspiration, and shortness of breath (APA, 1994). Dysregulation of the fear processing system appears to be central to many of these symptoms of PTSD. Studies with combat and civilian trauma populations have shown that inhibition of fear-potentiated startle is impaired in PTSD compared to controls (Jovanovic et al., 2011). Inhibition of fear responses involves learning to discriminate between danger and safety cues and to suppress fear responses in the presence of safety cues (Jovanovic and Norrholm, 2011), using a Pavlovian conditioning model in which a neutral stimulus (CS+) is paired with an aversive unconditioned stimulus (US). After several pairings, the association is formed so that the CS+ alone elicits the conditioned response (CR) (Pavlov, 1927). In differential conditioning, a separate cue that is never paired with the US (CS−, safety signal) does not elicit the CR if the fear response is appropriately inhibited. An additional paradigm used to investigate fear inhibition is extinction, in which the previously fear-conditioned CS+ is repeatedly presented without the US, until the subject learns that it no longer predicts danger.
There are several lines of evidence that implicate the prefrontal cortex (PFC) as an anatomical substrate for fear inhibition (Jovanovic and Norrholm, 2011). For example, functional MRI data indicate increased activation of the ventromedial (vm)PFC during an extinction recall task that is presented after extinction learning has occurred (Phelps et al., 2004; Milad et al., 2007). Furthermore, morphometric MRI analyses suggest that the thickness of vmPFC cortical tissue is correlated with extinction retention (Milad et al., 2005; Hartley et al., 2011). The PFC is also activated during response inhibition tasks in the absence of fearful stimuli. In such tasks, the participant is presented with a “Go” signal indicating that a response is required, for example, to press a button when a letter appears on the monitor. On a fraction of trials, however, the participant is required to withhold a response during a “NoGo” signal (the Go/NoGo task) (Eagle et al., 2008; Hester et al., 2004). Go/NoGo tasks used in subjects with PTSD with functional magnetic resonance imaging (fMRI) (Carrion et al., 2008; Falconer et al., 2008) have found decreased activation in the mPFC in PTSD subjects compared to controls.
A hallmark of PTSD neurobiology is exaggerated amygdala activity during fearful stimulation coupled with reduced top-down control of the amygdala by the PFC, indicating dyregulation of this inhibitory neurocircuit (Rauch et al., 2000; Rauch et al., 2006; Shin et al., 2004; Liberzon and Martis, 2006; Etkin et al., 2006). A recent meta-analysis of imaging studies during emotion processing in PTSD, social anxiety, and specific phobia indicated that the vmPFC (including the rostral anterior cingulate cortex, rACC) is less active in PTSD patients relative to controls (Etkin and Wager, 2007). Additionally, a recent fMRI study of extinction recall demonstrated decreased activation of the vmPFC in PTSD patients (Rougemont-Bücking et al., 2010). Finally, structural MRI data indicate that greater rACC volume predicts positive treatment outcomes in PTSD patients (Bryant et al., 2008). This area has been found to differ in PTSD patients compared to controls in shape and size (Corbo et al., 2005).
Differential fear conditioning and extinction paradigms in a highly traumatized civilian population (Jovanovic et al., 2010b; Jovanovic et al., 2010a; Glover et al., 2011; Norrholm et al., 2011) suggest that participants with PTSD show higher fear-potentiated startle to the CS+ (danger signal) and CS− (safety signal) than trauma controls (Glover et al., 2011). Data from our study on extinction suggest that a high degree of fear during late extinction is related to impaired inhibition, as it is best predicted by higher fear responses to the safety signal at the end of conditioning (Norrholm et al., 2011). In the current study, we investigated the neurocircuitry of response inhibition using an fMRI Go/NoGo task in a sample of traumatized women from inner-city Atlanta with and without PTSD. We hypothesized that women with PTSD would have less activation of the vmPFC/rACC during the inhibition condition compared to trauma controls. Furthermore, we examined inhibition of fear-potentiated startle in relation to neural activation to the response inhibition task. We hypothesized that impaired inhibition of fear would be associated with decreased activation in the vmPFC during the NoGo condition.
2. Methods
2.1 Participants
A total of 53 African American females aged 20–62 years were recruited through an ongoing study of risk factors for PTSD from the primary care medical clinics of a publicly funded hospital that serves a low-income minority population in inner-city Atlanta (Bradley et al., 2008; Schwartz et al., 2005). After complete description of the study to the subjects, written informed consent was obtained. Study procedures were approved by the institutional review boards of Emory University and Grady Memorial Hospital.
Women were considered eligible for participation if they were able and willing to give informed consent. Participants were screened with a short questionnaire to assess for the presence of these exclusion criteria: current psychotropic medication use, medical or physical conditions that preclude MRI scanning (e.g., metal implants), a history of schizophrenia or other psychotic disorder, history of head injury or loss of consciousness for longer than 5 minutes, or a history of neurological illness. Participants were also screened for pregnancy using a urine test. Of the 53 women recruited, data from 12 participants were excluded from the analyses due to a) motion artifacts (n=3), b) neurological abnormalities on the structural scan (n=4), c) drug use (n=2), d) computer error (n=2), and e) HIV+ (n=1). The final sample included 41 women (20 PTSD+ and 21 PTSD−).
2.2 Psychological assessment
The following measures were used to index PTSD symptoms, childhood maltreatment and lifetime trauma history, respectively: Modified PTSD Symptom Scale (PSS) (Foa and Tolin, 2000), Childhood Trauma Questionnaire (CTQ) (Bernstein and Fink, 1998), and the Traumatic Events Inventory (TEI). These measures have all been used previously in our work with this population (Binder et al., 2008; Schwartz et al., 2005). The categorical definition of PTSD+ vs. PTSD− was determined from responses to the DSM-IV-based criteria in the PSS. Immediately prior to the MRI scan, the participants filled out the state and trait forms of the State-Trait Anxiety Inventory (STAI (Speilberger and Vagg, 1984)).
2.3 MRI procedures
Scanning took place in a Siemens 3-Tesla scanner at Emory University Hospital. Participants viewed task stimuli via an adjustable mirror affixed to the radiofrequency coil, which reflected a computer screen located at the end of the MRI aperture.
Following a short calibration scan, a high-resolution T1-weighted structural scan was acquired using an MPRAGE sequence (176 slices, field of view=256 mm cubic voxels; 1×1×1 mm slice; TR= 2600ms; TE= 3.02ms; TI= 900ms; flip angle= 8 degrees). During task administration, a total of 26 contiguous echo-planar, T2-weighted images parallel to the anterior-posterior commissure line were acquired (TR=2530m sec; TE=30m sec; field of view=240 mm; 64×64 matrix; 3.75×3.75×4.0 mm voxel). fMRI images were acquired using the Z-saga pulse sequence (Heberlein and Hu, 2004) to minimize susceptibility signal loss. Statistical Parametric Mapping, version 5 (SPM5, Wellcome Trust Centre for Neuroimaging, London, UK: http://www.fil.ion.ucl.ac.uk/spm/) was used for file conversion, image pre-processing and statistical analyses. Functional images were slice-time corrected with a high-pass filter applied, realigned to the first image in the session to correct for motion. The structural T1 volume was then co-registered to the mean of the realigned functional images and spatially normalized to standardized Montreal Neurological Institute (MNI) space using the VBM toolbox (Christian Gaser; University of Jena, Department of Psychiatry). The normalization parameters were then applied to the functional volumes and the images smoothed with an 8mm FWHM Gaussian kernel. MNI coordinates were transformed into Talairach coordinates using http://imaging.mrccbu.cam.ac.uk/imaging/MniTalairach.
The Go/NoGo task was modified from previous work published by Leibenluft and colleagues (Leibenluft et al., 2007). On all trials, a white fixation cross appeared on a black background for 500ms; it was replaced by an X or an O “Go” signal for 1000ms and followed by 750ms of blank screen. On a response pad, the subjects pressed 1 for X and 2 for O. The subjects were instructed to respond to each trial as fast as they could unless the “NoGo” signal appeared (i.e., the background changed to red), in which case they should not press either button. The task comprised four runs separated by three 20 sec rest periods. Each run contained 26 “Go”, 13 “NoGo”, and 14 blank trials distributed randomly.
2.4 Fear-potentiated startle assessment
The startle and MRI sessions occurred at separate visits. Startle response data were acquired at a 1000 Hz sampling frequency using the electromyography (EMG) module of the BIOPAC MP150 for Windows (Biopac Systems, Inc., Aero Camino, CA). The acquired data were filtered, rectified, and smoothed using the MindWare software suite (MindWare Technologies, Ltd., Gahanna, OH) and exported for statistical analyses. The EMG signal was filtered with low- and high-frequency cutoffs at 28 and 500 Hz, respectively. The maximum amplitude of the eyeblink muscle contraction 20–200ms after presentation of the startle probe was used as a measure of the acoustic startle response.
The eyeblink component of the acoustic startle response was measured by EMG recordings of the right orbicularis oculi muscle using two 5-mm Ag/AgCl electrodes filled with electrolyte gel. One electrode was positioned 1 cm below the pupil of the right eye and the other was placed 1 cm below the lateral canthus. Impedance levels were less than 6 kilo-ohms for each participant. The startle probe was a 108-dB (A) SPL, 40-ms burst of broadband noise with near instantaneous rise time, delivered binaurally through headphones.
The fear-potentiated startle protocol consisted of two phases: Fear Acquisition and Fear Extinction. The Fear Acquisition phase consisted of three blocks with four trials of each type (a reinforced conditioned stimulus, CS+; a nonreinforced conditioned stimulus, CS−; and the 40 ms, 108 dB noise probe alone, NA). Both CSs were colored shapes presented on a computer monitor for 6sec. The US was a 250ms air blast with an intensity of 140 p.s.i. directed at the larynx. This US has been used in several of our previous studies and consistently produces robust fear-potentiated startle (Norrholm et al., 2011; Jovanovic et al., 2010b). Ten minutes after the conclusion of the Fear Acquisition phase, participants underwent the Fear Extinction phase that consisted of 6 blocks with four trials of each type (the previously reinforced CS+, CS, and NA). None of the CS presentations during Extinction was reinforced with an airblast US. In all phases of the experiment, the inter-trial intervals were randomized to be 9–22 sec in duration.
2.5 Statistical analyses
Functional imaging data were analyzed using SPM software (Wellcome Trust Centre for Neuroimaging, University College London, U.K.). After pre-processing, the images were entered into a two-level general linear model (GLM) statistical analysis (Friston et al., 1995).
The first level was an event-related model fitting subject-specific parameters. To examine blood-oxygen-level dependent (BOLD) signal change to task conditions, fixed-effects analysis was conducted by creating vectors for onset time of each condition, including NoGo correct and incorrect trials and Go correct and incorrect trials, derived from the behavioral data on the response pad during the task. The primary t-contrast was for examining BOLD signal change corresponding to NoGo greater than Go (NoGo>Go). The resulting individual contrast images were then entered into second-level random-effects GLM to obtain group estimates and correlations with variables of interest. A 2-sample t-test with PTSD+ and PTSD− groups was used to compare activation between groups. Additionally, startle data were entered into a regression model to predict BOLD signal change to the NoGo>Go contrast condition. An initial exploratory statistical threshold of p<.005 (uncorrected) and an extent threshold of ≥ 5 voxels per cluster was used to determine significant activations in the whole-brain analysis; secondarily, significant activations were thresholded with an false discovery rate (FDR) corrected p<.05 for small volume (6mm sphere) in specific ROIs derived from the literature, i.e. vmPFC/rACC (Etkin and Wager, 2007; Falconer et al., 2008).
The presence of fear-potentiated startle was assessed by comparing startle magnitude on the CS+ trials to startle magnitude to the noise alone (NA) trials. Differential conditioning and extinction was assessed by calculating a difference score obtained by subtracting startle magnitude to the NA trials from the startle magnitude on CS+ trials and CS− trials for each conditioning block. To examine differences in fear conditioning within each group, a repeated-measures ANOVA was conducted with trial type (2 levels: CS+, CS−) as the within-subjects variable, during late acquisition (i.e., blocks 2 and 3, when the discrimination is maximal). Extinction to the previously reinforced CS+ was divided into early (blocks 1 and 2), mid (blocks 3 and 4), and late (blocks 5 and 6) phases. Fear-potentiated startle during extinction was entered as a within-groups variable of phase (3 levels) in a repeated-measures ANOVA, within each group. Fear conditioning data were available on 29 (16 PTSD−, 13 PTSD+), and extinction data on 24 (13 PTSD−, 11 PTSD+) participants. Missing data were due to either: noisy psychophysiological data, computer or experimenter error, or participant drop-out. Bivariate correlations were performed between the BOLD signal change during the NoGo>Go contrast and the fear-potentiated startle variables. All statistical analyses were conducted with SPM and SPSS software packages.
3. Results
3.1 Demographic and clinical characteristics of sample
The demographic and clinical data of the sample are shown in Table 1. All participants were African American women, matched for age and trauma exposure. The PTSD+ women had significantly higher current symptoms of PTSD than the PTSD− women. However, the groups did not differ on measures of state or trait anxiety immediately prior to the MRI scan.
Table 1.
Demographic and clinical data in the PTSD− and PTSD+ groups
| PTSD− (n=21) | PTSD+ (n=20) | ||
|---|---|---|---|
|
| |||
| Demographics | M (SE) | M (SE) | ANOVA |
| Age | 39.8 (2.8) | 36.6 (3.3) | F=0.56, p=0.49 |
| Sex | 100% female | 100% female | N/A |
|
Trauma exposure
|
|||
| Childhood maltreatment (CTQ) | 38.1 (4.0) | 42.3 (3.4) | F=0.60, p=0.56 |
| Lifetime trauma exposure (TEI) | 2.4 (0.4) | 2.8 (0.4) | F=0.50, p=0.52 |
|
Clinical assessments
|
|||
| PTSD symptoms (PSS) | 7.4 (2.2) | 24.0 (1.8) | F=39.72, p<0.0001 |
| Trait Anxiety (STAI-T) | 39.4 (2.6) | 42.0 (1.9) | F=0.64, p=0.39 |
| State Anxiety (STAI-S) | 36.4 (2.7) | 39.9 (2.0) | F=1.03, p=0.60 |
Abbreviations: PTSD=Posttraumatic Stress Disorder; CTQ=Childhood Trauma Questionnaire; TEI=Traumatic Events Inventory; PSS=PTSD Symptom Scale; STAI=State-Trait Anxiety Inventory; STAI-T=Trait; STAI-S=State
3.2 Go/NoGo functional MRI
The behavioral data on the response pad to the Go and NoGo trials were highly accurate in both groups, i.e., the error rate was very low (Go trials=94.4% correct, no group difference, F<1.0; NoGo trials=89.4% correct, no group difference, F<1.0). Therefore we collapsed the incorrect and correct trials for each type into a single contrast NoGo>Go. The whole brain analyses of BOLD signal change during this contrast, with an uncorrected p-value threshold set at <.005 revealed significantly greater activation in the vmPFC in the PTSD− group compared to the PTSD+ group (Z=3.09, p=.001, 15 voxels, Talairach coordinates: x=4, y=42, z=−5), see Figure 1A and Table 2. We repeated the analysis with a small volume correction for the vmPFC ROI (anatomically-based seed coordinates based on Etkin and Wager (2007) and found the same effects of PTSD (Z=3.07, p(FDR corr)=.009). We then extracted the BOLD signal value from these coordinates, and compared it between diagnostic groups using a one-way ANOVA. This analysis also showed significantly less BOLD signal change in the vmPFC in the PTSD+ group relative to the PTSD− group (F(1,40)=7.94, p=.008), see Figure 1B. We repeated this analysis in an ANCOVA with age, level of childhood maltreatment and lifetime trauma, and time since trauma added as covariates, and the effect of PTSD remained significant (F(1,33)=7.04, p=.01). Finally, in order to test whether the correct behavioral response during the task affected the results, we performed a separate ANCOVA covarying for % correct responses on the NoGo trial, and the effect of PTSD on the BOLD signal was still significant (F(1,38)=8.47, p=.006), with no interaction with the rate of correct response.
Figure 1.

A) Brain activation in the NoGo>Go contrast, in the PTSD− minus PTSD+ condition. B) Mean extracted BOLD signal change between the PTSD− and PTSD+ groups. Statistical p-value shown for small volume ROI in the vmPFC. PTSD=Posttraumatic Stress Disorder. BOLD=Blood Oxygen Dependent Level.
Table 2.
Results of the whole brain and region of interest analyses for the NoGo>Go trials contrast
| PTSD−>PTSD+ | cluster size | Z | p | Talairach coordinates | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Whole brain analyses (p<0.005 uncorr) | x | y | z | |||
| vmPFC/rostral ACC (BA32) | 15 | 3.09 | 0.001 (uncorr) | 4 | 42 | −5 |
| right PFC (BA32) | 7 | 2.95 | 0.002 (uncorr) | 20 | 40 | 2 |
| right ACC, PFC (BA32) | 5 | 2.63 | 0.004 (uncorr) | 12 | 39 | 2 |
| Region of interest (p<0.05 FDRcorr) | ||||||
|
| ||||||
| vmPFC/rostral ACC (BA32) | 5 | 3.07 | 0.009 (FDRcorr) | 0 | 42 | −9 |
|
| ||||||
| PTSD−<PTSD+ | ||||||
|
| ||||||
| No activation | ||||||
Abbreviations: PTSD=Posttraumatic Stress Disorder; PFC=Prefrontal Cortex; vmPFC=Ventromedial PFC; ACC=Anterior Cingulate Cortex
3.3 Fear-potentiated startle
Both groups showed significant potentiation of the startle response to the CS+ relative to the NA trials, PTSD− (F(1,15)=10.00, p=.006), PTSD+ (F(1,12)=7.26, p=.02). We then assessed differential fear conditioning by comparing the difference score of the startle magnitude (CS minus NA) for CS+ and CS−. Figure 2A shows the results of differential fear conditioning between diagnostic groups. A repeated-measures ANOVA of fear-potentiated startle during the late acquisition phase with trial type (CS+, CS−) as a within-groups variable showed that the PTSD− group demonstrated significant discrimination between the CS+ and CS− (F(1,15)=5.58, p=.03), while the PTSD+ group did not (F(1,12)=2.14, p=.17). This suggests that the PTSD− group was able to appropriately inhibit startle to the CS− safety signal, whereas the PTSD+ group was not.
Figure 2.

Fear-potentiated startle in the PTSD− and PTSD+ groups during A) late acquisition and B) extinction. Abbreviations: CS=Conditioned Stimulus; CS+=reinforced CS, CS−=nonreinforced CS. PTSD=Posttraumatic Stress Disorder.
We then examined fear extinction to the CS+ across the three phases of extinction (early, mid, late) within each diagnostic group. Figure 2B shows the results of extinction in both groups. As was the case during acquisition, the PTSD− group showed significant reduction in fear-potentiated startle across the phase variable (F(2,24)=7.55, p=.003), whereas the PTSD+ group did not successfully extinguish the fear response (F(2,20)=2.55, p=.10).
3.4 Association between Go/NoGo circuitry and inhibition of fear
Bivariate correlations between the extracted BOLD signal during the NoGo>Go contrast and fear conditioning variables revealed a significant negative correlation between vmPFC activation and fear expression during inhibitory trials for the entire sample. Specifically, greater fear-potentiated startle to the CS− during late fear acquisition (r=−.44, p=.02) and the CS+ during late extinction (r=−.70, p=.0002) were associated with less vmPFC activation. Fear potentiation to the CS+ during acquisition or the early- and mid- phases of extinction, which are associated with high fear expression rather than impaired inhibition, were not significantly correlated with the BOLD signal in the vmPFC.
In order to examine whether fear inhibition independently contributed to vmPFC activation, we performed a hierarchical regression model by adding age in the first step, PTSD status in the second step, and fear-potentiated startle to the inhibition trials (i.e., safety signal and late extinction) in the final step. The overall model was significant (F(4,23)=7.25, p=.001), and accounted for 60.4% of the variance in vmPFC activation. Impaired fear inhibition predicted vmPFC activation beyond age and PTSD status (Fchange(2,19)=9.57, p=.001), and alone accounted for 39.9% of the variance. Interestingly, when fear inhibition was added to the model, PTSD diagnosis no longer significantly predicted decreased vmPFC activation.
We also included degree of fear-potentiated startle to the safety signal as a regressor in a SPM analysis of the NoGo>Go contrast condition in a whole brain analysis with a threshold of p<.001 (uncorrected). BOLD activation in the vmPFC was negatively correlated with startle to the CS− (Z=3.73, p<.001,19 voxels, Talairach coordinates: x=4, y=31, z=−9), as shown in Figure 3. We repeated the analysis within the vmPFC ROI and replicated the negative correlation with startle to the safety signal (Z=3.46, p(FDRcorr)=.01).
Figure 3.
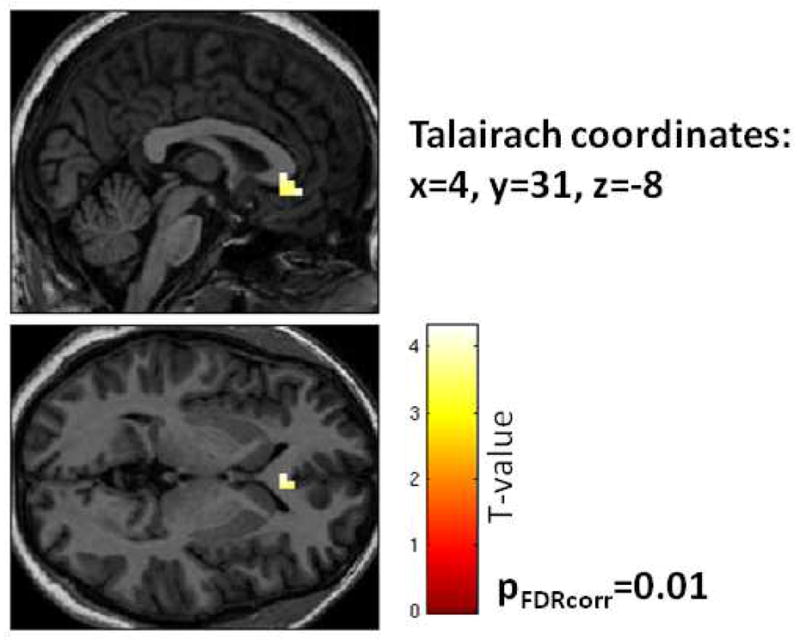
Brain activation in the NoGo>Go contrast, with fear-potentiated startle to the safety signal (CS−) used as a regressor in a negative correlation. Statistical p-value shown for small volume ROI in the vmPFC.
4. Discussion
This study used the Go/NoGo task in a functional MRI scan to assess response inhibition in a sample of highly traumatized African American women with and without PTSD. As in previous studies that have used this paradigm with PTSD populations, the PTSD group showed decreased activation in regions of the prefrontal cortex during the behavioral inhibition condition compared to controls (Falconer et al., 2008; Carrion et al., 2008). This is the first study, to the best of our knowledge that has used this paradigm during fMRI with a traumatized female African American sample.
The main finding in this study was the correlation of activity in the prefrontal cortex during the response inhibition task to inhibition of fear-potentiated startle to a conditioned safety cue outside the scanner. Given that the Go/NoGo task does not have an emotional component, it would seem that the neural circuitry for both kinds of inhibitory behaviors may be overlapping. Specifically, the brain region that was activated during the Go/NoGo task and that was correlated to fear inhibition is located primarily within the vmPFC (Brodmann area 32), and may include parts of the rACC (Brodmann area 24). As seen in Figures 1A and 3, the voxels with the highest BOLD signal change are immediately anterior to the corpus callosum and slightly ventral to the genu. This area has been shown to be activated during extinction recall, which also involves inhibition of conditioned fear (Milad et al., 2007), and during emotional conflict tasks (Etkin et al., 2006). The association between conditioned fear inhibition and vmPFC/rACC activation allow for the use of more cost-efficient, non-invasive methods of assessing the neural underpinnings of fear regulation, which is emerging as a putative biomarker for PTSD (Jovanovic et al., 2010a).
The vmPFC/rACC region may offer a target for novel PTSD treatment approaches. An area slightly more ventral to the rACC and below the corpus callosum, the subgenual cingulate (Brodmann area 25), has been used as a target in deep brain stimulation to relieve treatment-resistant depression (Holtzheimer and Mayberg, 2010), with long-term positive outcomes (Kennedy et al., 2011). There is an emerging body of literature assessing treatment-related structural and functional changes in the neural underpinnings of mental disorders. An early study for example, using single photon emission computed tomography (SPECT) imaging pre- and post-treatment with selective serotonin reuptake inhibitors (SSRIs), found significant changes in ACC and hippocampus after 12 weeks of treatment (Carey et al., 2004). In a recent study of Iraq and Afghanistan combat soldiers with PTSD who underwent exposure therapy, increased neural activation in the rACC in response to treatment was associated with greater clinical improvement, even in the absence of large changes in PTSD symptoms (Roy et al., 2010). These studies suggest neuroplasticity in the PFC with the potential for treatment-related modifications in activity (e.g., successfully attenuating amygdala-driven fear responses). Such findings offer great promise for improving available treatments for PTSD. The current study suggests that atypical patterns of fear inhibition during fear conditioning pre- and post-treatment may reflect these neuroanatomical changes. Therefore, these methods may have clinical application in providing a non-invasive technique for evaluating changes in the brain resulting from treatment or early intervention.
Several study limitations should be noted. First, the design of the study task resulted in very low error rates on the response pad and thus prohibited separate analyses of incorrect and correct responses to the NoGo trials; these two responses may differentially engage inhibitory circuits. Future research may use a paradigm that allows for the examination of neural activity during unsuccessful inhibition (Leibenluft et al., 2007). Next, the study did not include a non-traumatized comparison group. Although normative data would be interesting, the primary aim of the study was to examine neural correlates of psychopathology post trauma exposure, rather than the effects of trauma per se. The ANCOVA covarying for level of trauma exposure in both groups suggested that the effects on brain activation were not related to the trauma, but rather PTSD. Finally, although participants in this study represent an understudied population in the PTSD literature, the circumscribed demographic profile of this population may limit generalizability of these findings to other traumatized populations.
In conclusion, the effects of exposure therapy, which is one of the most successful psychotherapeutic approaches to PTSD, may be related to increasing fear inhibition. Given that exposure therapy is, in part, based on extinction learning, which activates the vmPFC, this premise would provide a likely neural mechanism of action. The regression analysis suggested that fear inhibition may mediate the relationship between PTSD and vmPFC activation, since PTSD status was no longer a significant predictor of activity once fear inhibition was added to the regression. Facilitating fear extinction, which the current study and our previous work has shown to be impaired in PTSD patients (Glover et al., 2011), may produce therapeutic modifications to underlying neural connectivity by increasing inhibition of fear.
Acknowledgments
Role of funding source:
This research was supported by funding from NIMH (MH071537 to K.J.R.; MH092576 to T.J.), National Centers for Research Resources (M01 RR00039), the Burroughs-Wellcome Foundation, NARSAD, and Howard Hughes Medical Institute (K.J.R.).
We would like to thank Allen Graham and the staff of the Grady Trauma Project for help with subject recruitment, and Robert Smith, III, at the Bioimaging Technology Center for his assistance with imaging.
Footnotes
Conflict of Interest:
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- APA. Diagnostic and statistical manual of mental disorders (DSM-IV) Washington, D.C: American Psychiatric Association; 1994. [Google Scholar]
- Bernstein DP, Fink L. Childhood Trauma Questionnaire A retrospective self-report manual. San Antonio, TX: The Psychological Corporation; 1998. [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 Polymorphisms and Childhood Abuse With Risk of Posttraumatic Stress Disorder Symptoms in Adults. Journal of the Americal Medical Association. 2008;299(11):1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W, Gillespie CF, Berg T, Evces M, Newport DJ, Stowe ZN, Heim CM, Nemeroff CB, Schwartz A, Cubells JF, Ressler KJ. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Archives General Psychiatry. 2008;65(2):190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant RA, Felmingham K, Whitford TJ, Kemp A, Hughes G, Peduto A, Williams LM. Rostral anterior cingulate volume predicts treatment response to cognitive-behavioural therapy for posttraumatic stress disorder. Journal of Psychiatry & Neuroscience. 2008;33(2):142–146. [PMC free article] [PubMed] [Google Scholar]
- Carey P, Warwick J, Niehaus D, van der Linden G, van Heerden B, Harvey B, Seedat S, Stein D. Single photon emission computed tomography (SPECT) of anxiety disorders before and after treatment with citalopram. BioMed Central Psychiatry. 2004;4(1):30. doi: 10.1186/1471-244X-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion VG, Garrett A, Menon V, Weems CF, Reiss AL. Posttraumatic stress symptoms and brain function during a response-inhibition task: an fMRI study in youth. Depression and Anxiety. 2008;25(6):514–526. doi: 10.1002/da.20346. [DOI] [PubMed] [Google Scholar]
- Corbo V, Clément M-H, Armony JL, Pruessner JC, Brunet A. Size Versus Shape Differences: Contrasting Voxel-Based and Volumetric Analyses of the Anterior Cingulate Cortex in Individuals with Acute Posttraumatic Stress Disorder. Biological Psychiatry. 2005;58(2):119–124. doi: 10.1016/j.biopsych.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Eagle D, Bari A, Robbins T. The neuropsychopharmacology of action inhibition: cross-species translation of the stop-signal and go/no-go tasks. Psychopharmacology. 2008;199(3):439–456. doi: 10.1007/s00213-008-1127-6. [DOI] [PubMed] [Google Scholar]
- Etkin A, Egner T, Peraza DM, Kandel ER, Hirsch J. Resolving emotional conflict: a role for the rostral anterior cingulate cortex in modulating activity in the amygdala. Neuron. 2006;51(6):871–882. doi: 10.1016/j.neuron.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Etkin A, Wager T. Functional Neuroimaging of Anxiety: A Meta-Analysis of Emotional Processing in PTSD, Social Anxiety Disorder, and Specific Phobia. American Journal of Psychiatry. 2007;164(10):1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer E, Bryant R, Felmingham KL, Kemp AH, Gordon E, Peduto A, Olivieri G, Williams LM. The neural networks of inhibitory control in posttraumatic stress disorder. Journal of Psychiatry & Neuroscience. 2008;33(5):413–422. [PMC free article] [PubMed] [Google Scholar]
- Foa EB, Tolin DF. Comparison of the PTSD Symptom Scale-Interview Version with the Clinician Administered PTSD Scale. Journal of Traumatic Stress. 2000;13(2):181–191. doi: 10.1023/A:1007781909213. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Holmes AP, Worsley K, Poline J. Statistical parametric maps in functional imaging: a general linear approach. Human Brain Mapping. 1995;2:189–210. [Google Scholar]
- Glover EM, Phifer JE, Crain DF, Norrholm SD, Davis M, Bradley B, Ressler KJ, Jovanovic T. Tools for translational neuroscience: PTSD is associated with heightened fear responses using acoustic startle but not skin conductance measures. Depression and Anxiety. 2011;28(12):1058–1066. doi: 10.1002/da.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley CA, Fischl B, Phelps EA. Brain Structure Correlates of Individual Differences in the Acquisition and Inhibition of Conditioned Fear. Cerebral Cortex. 2011;21(9):1954–62. doi: 10.1093/cercor/bhq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberlein KA, Hu X. Simultaneous acquisition of gradient-echo and asymmetric spin-echo for single-shot Z-shim: Z-SAGA. Magnetic Resonance Medicine. 2004;51:212–216. doi: 10.1002/mrm.10680. [DOI] [PubMed] [Google Scholar]
- Hester R, Fassbender C, Garavan H. Individual Differences in Error Processing: A Review and Reanalysis of Three Event-related fMRI Studies Using the GO/NOGO Task. Cerebral Cortex. 2004;14(9):986–994. doi: 10.1093/cercor/bhh059. [DOI] [PubMed] [Google Scholar]
- Holtzheimer PE, III, Mayberg HS. Deep Brain Stimulation for Treatment-Resistant Depression. American Journal of Psychiatry. 2010;167(12):1437–1444. doi: 10.1176/appi.ajp.2010.10010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Kazama A, Bachevalier J, Davis M. Impaired Safety Signal Learning May be a Biomarker of PTSD. Neuropharmacology. 2012;62(2):695–704. doi: 10.1016/j.neuropharm.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD. Neural mechanisms of impaired fear inhibition in posttraumatic stress disorder. Frontiers in Behavioral Neuroscience. 2011;5 doi: 10.3389/fnbeh.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, Ressler KJ. Impaired fear inhibition is a biomarker of PTSD but not depression. Depression and Anxiety. 2010a;27(3):244–251. doi: 10.1002/da.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Blanding NQ, Phifer JE, Weiss T, Davis M, Duncan E, Bradley B, Ressler K. Fear potentiation is associated with hypothalamic-pituitary-adrenal axis function in PTSD. Psychoneuroendocrinology. 2010b;35:846–857. doi: 10.1016/j.psyneuen.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SH, Giacobbe P, Rizvi SJ, Placenza FM, Nishikawa Y, Mayberg HS, Lozano AM. Deep Brain Stimulation for Treatment-Resistant Depression: Follow-Up After 3 to 6 Years. American Journal of Psychiatry. 2011;168(5):502–10. doi: 10.1176/appi.ajp.2010.10081187. [DOI] [PubMed] [Google Scholar]
- Leibenluft E, Rich BA, Vinton DT, Nelson EE, Fromm SJ, Berghorst LH, Joshi P, Robb A, Schachar RJ, Dickstein DP, McClure EB, Pine DS. Neural circuitry engaged during unsuccessful motor inhibition in pediatric bipolar disorder. American Journal of Psychiatry. 2007;164(1):52–60. doi: 10.1176/ajp.2007.164.1.A52. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Martis B. Neuroimaging Studies of Emotional Responses in PTSD. Annals of the New York Academy of Sciences. 2006;1071(1):87–109. doi: 10.1196/annals.1364.009. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quinn BT, Pitman RK, Orr SP, Fischl B, Rauch SL. Thickness of ventromedial prefrontal cortex in humans is correlated with extinction memory. Proceedings of the National Academy of Sciences. 2005;102(30):10706–10711. doi: 10.1073/pnas.0502441102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of Fear Extinction in Humans Activates the Ventromedial Prefrontal Cortex and Hippocampus in Concert. Biological Psychiatry. 2007;62(5):446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Norrholm SD, Jovanovic T, Olin IW, Sands LA, Karapanou I, Bradley B, Ressler KJ. Fear extinction in traumatized civilians with posttraumatic stress disorder: relation to symptom severity. Biological Psychiatry. 2011;69(6):556–563. doi: 10.1016/j.biopsych.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov IP. Conditioned Reflexes. Oxford University Press; 1927. [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43(6):897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry Models of Posttraumatic Stress Disorder and Extinction: Human Neuroimaging Research-Past, Present, and Future. Biological Psychiatry. 2006;60(4):376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB, Orr SP, Pitman RK. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biological Psychiatry. 2000;47(9):769–776. doi: 10.1016/s0006-3223(00)00828-3. [DOI] [PubMed] [Google Scholar]
- Rougemont-Bücking A, Linnman C, Zeffiro TA, Zeidan MA, Lebron-Milad K, Rodriguez-Romaguera J, Rauch SL, Pitman RK, Milad MR. Altered Processing of Contextual Information during Fear Extinction in PTSD: An fMRI Study. CNS Neuroscience & Therapeutics. 2010;17(4):227–36. doi: 10.1111/j.1755-5949.2010.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy MJ, Francis J, Friedlander J, Banks-Williams L, Lande RG, Taylor P, Blair J, McLellan J, Law W, Tarpley V, Patt I, Yu H, Mallinger A, Difede J, Rizzo A, Rothbaum B. Improvement in cerebral function with treatment of posttraumatic stress disordera. Annals of the New York Academy of Sciences. 2010;1208(1):142–149. doi: 10.1111/j.1749-6632.2010.05689.x. [DOI] [PubMed] [Google Scholar]
- Schwartz AC, Bradley RL, Sexton M, Sherry A, Ressler KJ. Posttraumatic stress disorder among African Americans in an inner city mental health clinic. Psychiatric Services. 2005;56(2):212–215. doi: 10.1176/appi.ps.56.2.212. [DOI] [PubMed] [Google Scholar]
- Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, Peters PM, Metzger LJ, Dougherty DD, Cannistraro PA, Alpert NM, Fischman AJ, Pitman RK. Regional Cerebral Blood Flow in the Amygdala and Medial Prefrontal Cortex During Traumatic Imagery in Male and Female Vietnam Veterans With PTSD. Archives General Psychiatry. 2004;61(2):168–176. doi: 10.1001/archpsyc.61.2.168. [DOI] [PubMed] [Google Scholar]
- Speilberger CD, Vagg PR. Psychometric Properties of the STAI: A Reply to Ramanaiah, Franzen, and Schill. Journal of Personality Assessment. 1984;48(1):95–97. doi: 10.1207/s15327752jpa4801_16. [DOI] [PubMed] [Google Scholar]