

Abstract
The ability of cells to sense and respond appropriately to changing environmental conditions is often mediated by signal transduction pathways that employ mitogen-activated protein kinases (MAPKs). In the yeast Saccharomyces cerevisiae, the high osmolarity glycerol (HOG) and the filamentous growth (FG) pathways are activated following hyperosmotic stress and nutrient deprivation, respectively. Whereas the HOG pathway requires the MAPK Hog1, the FG pathway employs the MAPK Kss1. We conducted a comprehensive screen of nearly 5,000 gene deletion strains for mutants that exhibit inappropriate cross-talk between the HOG and FG pathways. We identified two novel mutants, mnn10Δ and mnn11Δ, that allow activation of Kss1 under conditions that normally stimulate Hog1. MNN10 and MNN11 encode mannosyltransferases that are part of the N-glycosylation machinery within the Golgi apparatus; deletion of either gene results in N-glycosylated proteins that have shorter mannan chains. Deletion of the cell surface mucin Msb2 suppressed the mnn11Δ phenotype, while mutation of a single glycosylation site within Msb2 was sufficient to confer inappropriate activation of Kss1 by salt stress. These findings reveal new components of the N-glycosylation machinery needed to ensure MAPK signaling fidelity.
The ability of cells to sense and respond to changes in their environment is fundamental to cell survival. The mitogen-activated protein kinases (MAPKs) are commonly employed to transduce extracellular signals and to evoke specific intracellular responses (1). MAPKs are part of an evolutionarily-conserved three-tiered signaling cascade consisting of the MAPK, a MAPK kinase (MAPKK), and a MAPKK kinase (MAPKKK). In mammalian cells, MAPKs mediate responses to a variety of stimuli such as hormones, stresses, and cytokines, which promote cell proliferation, differentiation, and inflammation. Clearly, the signals that initiate these events must be transmitted in a tightly regulated manner. The complexities of these pathways, however, have hindered our understanding of MAPK regulation.
MAPK pathways are also present in the unicellular eukaryote Saccharomyces cerevisiae (hereafter, yeast). Due to its relative simplicity and the ease of genetic manipulation, yeast has emerged as an excellent model system for investigating the MAPK pathway and points of regulation. Indeed, studies in yeast have revealed key pathway components and improved our understanding of how these components are regulated (2).
Among the MAPK pathways in yeast, the most well-studied are the pheromone response pathway, the high osmolarity glycerol (HOG) pathway, and the filamentous growth (FG) pathway (detailed in Figure 1). Each of these pathways is activated by a different stimulus and employs a distinct MAPK. In the HOG pathway, high osmotic stress activates the three-component kinase cascade: Ste11 (or Ssk2/Ssk22), Pbs2, and the MAPK Hog1. Activated Hog1 promotes events leading to stress adaptation, including cell cycle arrest and increased glycerol production to restore osmotic balance (2–5). The filamentous growth pathway responds to nutrient deprivation by activating Ste11, Ste7, and the MAPK Kss1, which promotes increased cell-cell adhesion, increased cell-substratum adhesion, and an increased ability of cells to penetrate the substratum. These phenotypes may comprise a foraging response to allow non-motile cells to grow into their surroundings in a search for additional nutrients (2, 6, 7). In haploid yeast cells, this response is termed “invasive growth” (8), while in diploids it is termed “pseudohyphal growth” (9, 10). Finally, in the mating response pathway, a pheromone signal leads to the activation of Ste11, Ste7, and the MAPK Fus3, which initiates a series of changes that include cell cycle arrest, cytoskeletal rearrangements, and new gene transcription that lead to cell fusion and mating (2, 11–13).
Figure 1. Components of the HOG, FG and mating pheromone pathways.

Some components are omitted for brevity.
The HOG, FG, and mating pathways share several components (Figure 1) and yet they exhibit remarkable signal fidelity when stimulated individually. The prevalence of shared components raises the question of how pathway fidelity is regulated and maintained so that a signal transmitted though one pathway does not cross-activate another pathway. Prior work has revealed dynamic mechanisms that help these pathways function in a mutually exclusive manner. For instance, when exposed to a hyperosmotic environment, cells lacking Pbs2 or Hog1 exhibit sustained activation of Kss1 and undergo filamentous growth (14–17). This finding suggests that activated Hog1 normally phosphorylates proteins that serve to limit activation of the competing FG pathway and does so in a dynamically-regulated (stimulus-dependent) manner. Hog1 was later shown to repress Kss1 in part through phosphorylation of the MAPKKK adaptor Ste50 (18, 19).
In addition to dynamic feedback mechanisms, pathway fidelity can also be maintained by static mechanisms involving scaffold proteins. The Ste11 adaptor protein Ste50 behaves as a dynamic integrator of multiple signals (18–20). The kinase scaffold protein Ste5 was originally thought to serve as a signal insulator (2, 21), although current evidence indicates that it is also a dynamic regulator of Fus3 activity (22, 23). To identify new components that regulate cross-pathway signaling, we screened for mutants that display inappropriate MAPK responses in a high-salt environment. By this approach, we identified gene deletion mutants that exhibit hyperactivation of Kss1 in response to osmotic stress stimulation. These genes, MNN10 and MNN11, each encode a subunit of the Golgi mannosyltransferase complex needed for proper N-glycosylation of proteins (24). Deletion of a known glycoprotein (Msb2), or potential Msb2 binding partners at the plasma membrane (Sho1, Opy2), or downstream effectors of Msb2 (Ste20, Ste50, Ste11, Ste7) was sufficient to suppress hyperactivation of Kss1. Finally, mutation of a single putative N-glycosylation site within a single shared component, Msb2, led to inappropriate activation of Kss1, in the manner of mnn10Δ and mnn11Δ. We propose that N-glycosylation of Msb2 is needed for insulation of pathway signaling components and to promote MAPK signaling fidelity.
EXPERIMENTAL PROCEDURES
Strains and growth conditions
Standard procedures for the growth, maintenance and transformation of yeast and bacteria and for the manipulation of DNA were used throughout. Yeast Saccharomyces cerevisiae strains used in this study were BY4741 (MATa leu2 met15Δ his3-1 ura3Δ). Strains from the Yeast Knock-out (YKO) gene deletion library (25) were used for the initial genetic screen (Supporting Information, Table S1). Aside from Table S1, all of our analysis was done using mutant strains that had been reconstructed by PCR amplification of YKO genomic DNA and homologous recombination in BY4741. All double mutants were constructed by PCR amplification of LEU2, URA3, or HIS3 cassettes and homologous recombination in mnn11Δ. The mnn10Δ mnn11Δ double mutant is reported to be viable and to exhibit the same phenotype as the individual mutations (26). However, we have not been able to sporulate the heterozygous diploid in our strain background, and so we were not able to characterize the double mutant or to verify the earlier findings.
Cells were routinely grown at 30°C in synthetic complete dextrose (SCD) liquid medium (yeast nitrogen base w/o amino acids, ammonium sulfate, adenine, amino acids, dextrose) or in selective medium where needed for plasmid maintenance.
Plasmid construction
pRS313-MNN11, pRS316-SNF3, pRS315-SHO1, pRS313-RGT2, and pRS316-STE50 were constructed by PCR amplification from BY4741 genomic DNA, restriction digestion, and ligation into the indicated pRS-series vectors (ATCC). pRS316-MSB2 was constructed by PCR amplification of MSB2 from BY4741 genomic DNA and gap repair with pRS316 in yeast. Human influenza hemagglutinin (HA) epitope tags were introduced by site-directed mutagenesis to construct pRS316-SNF3-HA, pRS313-RGT2-HA, and pRS316-MSB2-HA. The FLAG epitope tag was introduced by site-directed mutagenesis to construct pRS315-SHO1-FLAG. pRS316-ste505A was constructed as described previously (18, 19). All N-glycosylation site mutations were constructed with the QuikChange site-directed mutagenesis kit (Stratagene), according to the manufacturer’s directions. Mutagenic oligonucleotides (plus complementary strand, not shown) were as follows: msb2N30A: 5′-CCT TCG ACT TTA TAT TCG GCG CTG GAA CGC AAC AAG CTC AGA GCC-3′; snf3N383A: 5′-CTA CGG TGT CAA TTT CTT CGC TAA GAC AGG AGT CAG TAA TAG-3′; rgt2N136A: 5′-GTT AAA ACC TAC ATT GCT CCG GCC CAT TCA TAT TTC ACC ACT AGC-3′; rgt2N385A: 5′-CTA TGG AGT TAA TTT TTT CGC CAA CAC AGG GGT GGA CAA C-3′; and sho1N59A: 5′-CCA TCT CAT CTG CAT CCA CCG CTG AAT CCT TCC CAC GTT TTA CTT GG –3′.
Genetic screen of the YKO gene deletion library
Cells from the YKO gene deletion library were inoculated into a 96-well plate containing 100 μL of SCD liquid medium in each well. After incubation overnight, 100 μL of SCD liquid medium was added to each culture, and the cells were grown for an additional 24 hr. Each mutant strain was spotted in duplicate onto agar plates made from SCD medium (2% agar) with either no salt, 0.5 M KCl, or 1.5 M KCl. 5 μL spots were made using a multichannel pipette.
After incubating the plates for 24 hr at 30°C, the colonies on each plate were scored for diminished growth in high salt. After incubating the plates for a minimum of two days, they were rinsed with deionized water and each colony was scored for penetration into the agar.
Cell-extract preparation and immunoblot analysis
Cells were grown overnight to saturation in SCD medium and re-inoculated into SCD and grown to A600nm ~1.0. Cells were then treated with 0.5 M KCl for 5 min (unless otherwise indicated), 1M sorbitol for 5 min, 3 μM α factor pheromone for 30 min, or left untreated at 30°C. Cells were harvested by addition of 100% trichloroacetic acid, centrifugation at 3000 x g for 2 min, washing with 500 μL of 10 mM NaN3 and storage at −80°C. Protein extracts were prepared by glass bead lysis in trichloroacetic acid as described previously (27). Protein concentration was determined by DC protein assay, according to the manufacturer’s instructions (Bio-Rad Laboratories). Protein extracts were resolved by 10% SDS-PAGE and immunoblotted with Phospho-p44/42 MAPK antibodies (9101L, Cell Signaling Technology) at 1:500, Phospho-p38 MAPK antibodies (4631L, Cell Signaling Technology) at 1:500, Fus3 antibodies (sc-6773, Santa Cruz Biotechnology) at 1:500, Hog1 antibodies (sc-6815, Santa Cruz Biotechnology), Kss1 antibodies (sc-28547, Santa Cruz Biotechnology) and glucose-6-phosphate dehydrogenase (G6PDH) antibodies (A9521, Sigma-Aldrich) at 1:10,000. Immunoreactive species were visualized by chemiluminescent detection (PerkinElmer Life Sciences LAS) of horseradish peroxidase-conjugated antibodies (170-5047 and 170-5046, Bio-Rad). Image densitometry was conducted consistently throughout using NIH ImageJ (28). Statistical analysis was done using GraphPad Prism 4 for at least three independent experiments.
Transcription reporter assay
Pheromone-dependent FUS1-lacZ transcription reporter assays were conducted as described previously (29). Cells transformed with pRS423-FUS1-lacZ (29) were grown to A600nm ~1.0, then dispensed at 90 μL per well into a 96-well plate and mixed with 10 μL of α-factor pheromone at the indicated concentrations for 90 min at 30°C. The reaction was started by adding 20 μL of FDG solution (130 mM PIPES pH 7.2, 0.25% Triton-X100, 0.5 mM fluorescein di-β-galactopyranoside [M0250, Marker Gene Technologies]) for 1 hr at 37°C. The reaction was stopped by adding 20 μL of 1 M sodium bicarbonate. Fluorescence was quantified using a fluorescence plate reader at 485 μm excitation, 580 μm emission (SpectraMax M5, Molecular Devices).
Salt-dependent FUS1-lacZ transcription reporter assays were conducted as described above, except cell cultures at A600nm ~1.0 were dispensed at 65 μL per well and mixed with 35 μL of KCl at the indicated concentrations for 90 min at 30°C. Each well was then mixed with 20 μL of FDG solution for 3 hr at 37°C. Salt-dependent CRE-lacZ transcription reporter assays were conducted in the same manner, except using the pRS423-CRE-lacZ reporter plasmid (30) and treating with FDG for 6 hr. Statistical analysis was done using GraphPad Prism 4 for at least three independent experiments.
RESULTS
Genome-wide screen reveals a new regulator of osmostress signaling
Under normal circumstances, the HOG pathway is strongly activated by osmotic stress. Under the same conditions, however, certain yeast mutants incorrectly cross-activate Kss1 and the filamentous growth pathway. Most notably, mutations in the MAPK Hog1, or its upstream activator Pbs2, grow invasively and penetrate their substratum when exposed to an osmotic stimulus. When grown on agar plates with salt, these cells cannot be easily rinsed off the plate (15).
To identify additional regulators of cross-pathway activation, we used the invasive growth assay to screen the complete YKO yeast gene deletion strain collection (25). The screen did not reveal any mutants that invaded the agar to the same degree as hog1Δ or pbs2Δ. Nevertheless, some mutants penetrated the agar in a weak but detectable manner. Upon retesting, all but one of the mutants grew invasively in the absence of salt and were not pursued (Supporting Information, Table S1). One remaining mutant, mnn11Δ, was invasive in the presence but not in the absence of salt and was characterized in detail.
Mnn11 impedes the activation of Kss1 by osmotic stress
We began by reconstructing the mnn11Δ strain and in this case observed no FG behavior, suggesting that the original YKO strain contained one or more enhancer mutations. Substratum penetration is often mediated by activation of the MAPK Kss1. To determine if the mnn11Δ mutation conferred an elevated MAPK response, we monitored the activity of Kss1 directly by immunoblotting with an antibody that recognizes the dually-phosphorylated, fully-activated form of the kinase (phospho-Kss1). Kss1 activity was indeed elevated in mnn11Δ as compared with wild-type cells, and activation was elevated further upon stimulation with 0.5 M KCl. Elevated MAPK activity was also evident in the reconstructed mnn11Δ strain, treated with either 0.5 M KCl or 1 M sorbitol (Figure 2A, Supporting Information Figure S1). Introduction of MNN11 on a single-copy plasmid restored normal Kss1 activity (pRS313-MNN11, Figure 2A).
Figure 2. Deletion of MNN11 results in hyperactivation of Kss1 and the filamentous growth pathway.

A) Activation of Kss1 and Hog1; Wild-type, mnn11Δ and pRS313-MNN11-transformed mnn11Δ cells were stimulated with 0.5 M KCl for the indicated times. Cell lysates were resolved by 10% SDS-PAGE. Phospho-Kss1 (p-Kss1) and phospho-Hog1 (p-Hog1) were detected by immunoblotting with phospho-p44/42 and phospho-p38 antibodies, which recognize the dually phosphorylated and activated forms of Kss1 and Hog1, respectively. G6PDH served as a loading control. (B) Activation of Kss1, Hog1 and Fus3; WT, mnn10Δ and mnn11Δ cells were stimulated with 0.5 M KCl for 5 min or with 3 μM α-factor pheromone for 30 min. Cell lysates were resolved by 10% SDS-PAGE. Specific antibodies were used to detect the activated form of Hog1, Kss1 and Fus3 (p-Fus3). Total Hog1 and Fus3 abundance were determined with Hog1 and Fus3 antibodies. G6PDH served as a loading control. All primary antibodies were recognized by chemiluminescent detection and quantified by scanning densitometry (ImageJ). The bottom panel shows averaged scanning densitometry data for three individual experiments. Error bars represent ± SEM. (C) Transcriptional activation (β-galactosidase activity) was measured spectrofluorometrically in wild-type (WT), mnn10Δ and mnn11Δ cells transformed with a plasmid containing either a pheromone-inducible reporter (FUS1-lacZ) or a salt-inducible reporter (CRE-lacZ). Transcription was induced by the addition of the indicated concentrations of KCl or α-factor pheromone. Data are the mean ± SE of four individual colonies measured in triplicate and presented as a percentage of wild-type maximum.
By immunoblotting with an antibody that recognizes Kss1, we determined that mnn11Δ cells express about 2-fold more Kss1 as compared to wild-type cells (Supporting Information, Figure S2). This difference in expression is likely due to the 2-fold transcriptional induction of KSS1 when the FG pathway is activated, for example using an activated allele of Msb2 (31). Higher Kss1 expression, however, cannot account for the 5-fold increase in Kss1 phosphorylation in the mnn11Δ strain. Taken together, these results show that Mnn11 regulates the filamentous growth pathway by limiting the activity of Kss1 under osmotic stress conditions.
Mnn11 selectively regulates Kss1
The findings above reveal that MNN11 is needed to prevent inappropriate activation of Kss1 by osmotic stress. To determine if Mnn11 regulates the functions of any other MAPKs, we examined the activity of Hog1, Fus3 and Kss1 following stimulation with either salt (KCl) or the pheromone α-factor. For these experiments we also tested an mnn10Δ mutant strain. MNN10, like MNN11, encodes an α-1,6-mannosyltransferase present in the lumen of the Golgi (24). Moreover, Mnn10 and Mnn11 are part of the same macromolecular complex, and both proteins are required for the extension of mannan chains on N-glycosylated proteins (24).
Once again we monitored activation of the MAPKs by immunoblotting with antibodies that recognize the phosphorylated, activated forms of each kinase. As shown in Figure 2B, the mnn10Δ and mnn11Δ mutants exhibited elevated phospho-Kss1 levels, in both the absence and presence of salt stimulus. In the stimulated cells, phospho-Kss1 was five- and seven-fold higher in mnn11Δ and mnn10Δ, respectively, as compared with wild-type cells. This increase in phospho-Kss1 was well above the two-fold higher expression of total Kss1, noted above (Supporting Information, Figure S2). In contrast, the mutants had no effect on the activation of the osmo-sensitive MAPK Hog1. Activation of the mating-specific MAPK Fus3 was likewise unaffected, although there was a modest elevation in Fus3 abundance. Thus, Mnn10 and Mnn11 serve to limit salt activation of Kss1 but not of other MAPKs.
We then examined whether Mnn10 or Mnn11 alter the response to α-factor pheromone (Figure 2B), which normally activates Fus3 and Kss1 but not Hog1. When stimulated with α-factor, Fus3 activation in the mutants was comparable to wild-type. Here, Kss1 activation increased about two-fold in mnn10Δ and mnn11Δ cells. This difference mirrors the two-fold higher expression of Kss1 (Supporting Information, Figure S2). Under no circumstance was Hog1 activated by pheromone. Hence, these data reveal that Mnn10 and Mnn11 limit the activation of Kss1 under salt stress conditions. These effects are highly selective, since the same mutants did not affect activation of Kss1 by pheromone. Moreover, these mutants did not affect activation of either Fus3 or Hog1 in cells treated with either salt or pheromone.
Our findings above indicate that Mnn10 and Mnn11 specifically limit activation of Kss1 by salt. One of the functions of MAPKs is to activate downstream transcription factors that induce the expression of a specific set of genes. To corroborate the MAPK activity data, we monitored gene induction downstream of Kss1. To this end, we used a reporter comprised of the β-galactosidase gene fused to the FUS1 promoter (FUS1-lacZ). FUS1 is strongly induced upon activation of either Fus3 or Kss1 (12). Fus3 is not activated by osmotic stress, however (Figure 2B), so under these conditions FUS1-lacZ induction is mediated solely by Kss1. As shown in Figure 2C, basal FUS1-lacZ expression was significantly higher in both mnn10Δ and mnn11Δ mutants, as compared to wild-type cells. This increase is consistent with the elevated Kss1 activity. However, in no case did we detect a further increase following treatment with 0.5 M KCl. The absence of any stimulus-dependent change in transcription-reporter activity contrasts with the marked increase in activation of Kss1 in the mutant strains. As shown in Figure 2A, Kss1 activation was quite transient, however, and may not have been sufficient to fully activate the downstream transcription factors. Just as transient activation of Kss1 is not sufficient to initiate the mating response in wild-type yeast (32), transient activation of Kss1 did not initiate mating responses in mnn10Δ or mnn11Δ cells. A second possibility is that, under these specific circumstances, Kss1 activates an alternative transcription program.
We then monitored FUS1-lacZ as a reporter for the pheromone response pathway mediated by Fus3 (Figure 2C). Once again the mnn10Δ and mnn11Δ strains exhibited elevated basal activity, but no further induction with α-factor pheromone, as compared with wild-type cells. Given that deletion of Mnn11/10 does not influence the activation status of Fus3 (Figure 2B), we would not expect these mutants to significantly alter the FUS1-lacZ response to pheromone. The pheromone response pathway is mediated primarily by the MAPK Fus3, and signals through Kss1 only when Fus3 is absent (2). When Fus3 is present, Kss1 activation is repressed (33). Finally, to examine transcriptional induction downstream of Hog1, we used a reporter comprised of the β-galactosidase gene fused to the CRE (cAMP-responsive element) promoter motif (CRE-lacZ) (30). As shown in Figure 2C, mnn10Δ and mnn11Δ mutants exhibited full induction of CRE-lacZ, although maximum induction occurred at slightly (<2-fold) lower concentrations of salt as compared to wild-type cells. Taken together, the transcription-reporter data support the conclusion that Mnn10 and Mnn11 restrict Kss1 activity under basal (unstimulated) conditions. Other unidentified mechanisms appear to limit transcriptional induction by Kss1 under salt-stimulated conditions. These mechanisms may account as well for the absence of a reproducible invasive growth phenotype noted for the mnn11Δ mutant.
Genetic analysis reveals potential targets of Mnn11
MNN11 was shown previously to be required for proper N-glycosylation of proteins. Given that the absence of MNN11 allows inappropriate activation of Kss1, we reasoned that deletion of a glycosylated target protein might suppress the observed mnn11Δ phenotype. To this end we deleted a series of candidate regulators in conjunction with the mnn11Δ mutation. We then monitored the activation state of Kss1, in the absence or presence of 0.5 M KCl and in the absence or presence of MNN11.
We began our analysis with components of the FG pathway that are shared with the salt-stress pathway, any of which could limit inappropriate activation of Kss1 by salt; these include Ste20 (MAPKKKK), Ste11 (MAPKKK), Ste50 (Ste11 adaptor), and Ste7 (MAPKK) (2). We also tested the pheromone receptor (Ste2) and the G protein β subunit (Ste4) since they too have the potential to activate Kss1. Of all these pathway components, only Ste2 is known to undergo N-glycosylation (34).
As shown in Figure 3, Kss1 was no longer activated by salt in the absence of Ste20, Ste50, Ste11, or Ste7. The mnn11Δste20Δ mutant did exhibit a slight elevation in basal Kss1 activation, presumably due to the presence of kinases related to Ste20 (Cla4 and Skm1). We were not able to test this hypothesis because the ste20Δcla4Δ double mutant is inviable (35). Nevertheless, mnn11Δste20Δ remained unresponsive to salt. In contrast, Kss1 was still hyperactivated by salt in the ste2Δmnn11Δ and ste4Δmnn11Δ double mutant strains, even though deletion of STE4 reduces basal signaling and expression of several pheromone pathway components, both upstream and including Kss1 (2). These data indicate that components in the FG pathway are needed for Kss1 hyperactivation (Ste20, Ste50, Ste11 and Ste7), whereas components unique to the pheromone response pathway (Ste2 and Ste4) are not. Taken together, these data suggest that Mnn11 targets a membrane protein other than Ste2, but one that is upstream of the protein kinases Ste20, Ste11, Ste7 and Kss1.
Figure 3. Mnn11 limits Kss1 activation through the filamentous growth pathway and not the pheromone response pathway.

Activation of Kss1 and Hog1; mnn11Δ and the indicated double gene deletion mutant cells were stimulated with 0.5 M KCl for 5 min. Cell lysates were resolved by 10% SDS-PAGE and immunoblotting. Specific antibodies were used to detect the dually phosphorylated, fully activated forms of Kss1 and Hog1 (p-Kss1 and p-Hog1). G6PDH served as a loading control. Data are representative of three individual experiments.
We next considered candidate targets of Mnn11 in the FG pathway. To this end we tested a group of cell surface proteins known to act upstream of Kss1 (36); these include Sho1, Msb2, Opy2 and Hkr1. Sho1 is a transmembrane protein that can form a hetero-oligomeric complex with the signaling mucin Msb2 (2, 31, 37). Both proteins are likely to be N-glycosylated and function at the head of both the FG and HOG pathways (2, 31, 36, 38, 39). Opy2 is an integral membrane protein that interacts with and recruits Ste50 to the plasma membrane; Opy2 is likewise necessary for both the FG and HOG responses (2, 20, 40). While it does not have any luminal N-glycosylation sites (N-X-S/T) (41, 42) and therefore cannot be targeted directly by Mnn11, Opy2 could associate with another glycosylated binding partner such as Msb2. Finally, Hkr1 is another signaling mucin that functions as an osmosensor in complex with Sho1 (36).
Once again we constructed double mutant strains lacking Mnn11 and each of the candidate target proteins. We then exposed these mutant strains to hyperosmotic stress and immunoblotted for activated Kss1. As shown in Figure 4, Kss1 was hyperactivated by salt in the hkr1Δmnn11Δ strain. Kss1 activation was also slightly elevated in cells lacking only Hkr1, consistent with its role as a negative regulator of the FG response (39). In contrast, Kss1 was no longer activated by salt in the msb2Δmnn11Δ, sho1Δmnn11Δ, or opy2Δmnn11Δ strains. These data suggest that Mnn11 targets Msb2, Sho1, or Opy2. However, while these proteins are components of the FG pathway, only Msb2 and Sho1 have consensus N-glycosylation sites. Thus, the effects of Mnn11 on Opy2 are likely to be indirect.
Figure 4. Mnn11 may target the glycoproteins Msb2 or Sho1, or a glycoprotein associated with Opy2.

Activation of Kss1 and Hog1; mnn11Δ and the indicated double mutant cells were stimulated with 0.5 M KCl for 5 min. Cell lysates were resolved by 10% SDS-PAGE and immunoblotting. Specific antibodies were used to detect the dually phosphorylated, fully activated forms of Kss1 and Hog1 (p-Kss1 and p-Hog1). G6PDH served as a loading control. The panels on the right show quantitation for those conditions that diminished Kss1 activity. The bottom panel is a composite image from a single exposure of a single gel. Error bars represent ± SEM.
We then considered proteins that act in conjunction with Opy2, which may in turn be regulated by Mnn11. It was proposed recently that Opy2 is phosphorylated and regulated by the casein kinases Yck1 and Yck2 (20). These kinases appear to play a role in pathway fidelity, since unphosphorylated Opy2 activates Kss1 in preference to Hog1, while phosphorylated Opy2 activates Hog1 in preference to Kss1 (20). Yck1 and Yck2, in turn, are thought to be regulated by Snf3 and Rgt2. These proteins are 12-pass transmembrane glucose sensors responsible for inducing the expression of hexose transporters (20, 43, 44). Thus, there is an established functional relationship between the two glucose sensors, Opy2, and pathway fidelity. Indeed, the deletion of both Snf3 and Rgt2 together, but not deletion of either alone, was able to fully suppress Kss1 hyperactivation in the mnn11Δ background (Figure 5). In fact, we observed a greater salt-dependent activation of Kss1 in the snf3Δrgt2Δ strain compared with the mnn11Δsnf3Δrgt2Δ strain. This observation lends further support to the idea that the glucose sensors Snf3 and Rgt2 might potentially be in the same pathway with Mnn11, given the diminution of Kss1 activity when Mnn11 is absent.
Figure 5. Both Snf3 and Rgt2 are required to limit the activation of Kss1 by osmotic stress.
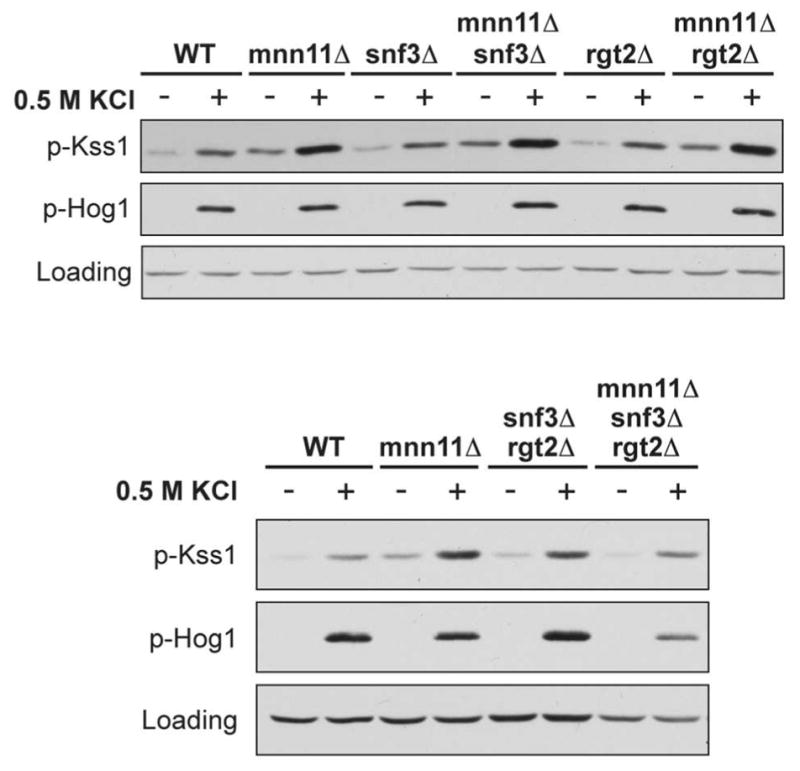
Activation of Kss1 and Hog1; mnn11Δ and the indicated double or triple mutant cells were stimulated with 0.5 M KCl for 5 min. Cell lysates were resolved by 10% SDS-PAGE and immunoblotting. Specific antibodies were used to detect the dually phosphorylated, fully activated forms of Kss1 and Hog1 (p-Kss1 and p-Hog1). G6PDH served as a loading control. Data presented are representative of three individual experiments.
Analysis of glycosylation site mutants
The data presented above reveal several proteins required to manifest the mnn11Δ phenotype. Among these are plasma membrane proteins that contain N-glycosylation sites. These include the glucose sensors Snf3 and Rgt2, as well as the putative osmosensors Msb2 and Sho1. Having shown that these proteins are needed to confer Kss1 hyperactivation in salt-treated mnn11Δ cells, we sought to identify specific sites of glycosylation that may be targeted by Mnn11. For each of the mutant strains we introduced the corresponding gene, expressed from a single copy plasmid, in which all potential N-glycosylation sites had been replaced with alanine: Asn-383 in Snf3 (snf3N383A), Asn-136 and Asn-385 in Rgt2 (rgt2N136A+N385A), Asn-59 in Sho1 (sho1N59A), and Asn-30 in Msb2 (msb2N30A). Notably, aside from Asn-30, Msb2 has six additional potential N-glycosylation sites, which have all been previously mutated (Msb2-6N/A) without an effect on basal FUS1-lacZ expression (40). We then exposed these cells to hyperosmotic stress and immunoblotted for activated Kss1. Of these mutants, only msb2N30A was able to restore Kss1 hyperactivation to the corresponding deletion strain (msb2Δ) when either expressed from a plasmid (Figure 6A) or integrated into the genome at the MSB2 locus (Figure 6B). Although we repeatedly and consistently observed Kss1 hyperactivation following construction of the msb2N30A mutant strains, the hyperactivation phenotype was less consistent as we propagated the strains, possibly as a consequence of genetic reversion or the acquisition of suppressor mutations. Given this instability, we did not attempt to construct any double mutant strains containing msb2N30A. In contrast, Kss1 responded normally in cells expressing snf3N383A and rgt2N136A+N385A (snf3Δrgt2Δ strain) or sho1N59A (sho1Δ) (Figure 6). Thus, Msb2, Sho1 and either Snf3 or Rgt2 can limit inappropriate activation of Kss1 by salt. Of these, only Msb2 requires an intact putative N-glycosylation site to function properly as a pathway insulator.
Figure 6. Msb2 Asn-30 is a putative N-glycosylation site required to limit the activation of Kss1 by osmotic stress.

(A) Activation of Kss1 and Hog1; mnn11Δ, snf3Δrgt2Δ, sho1Δ, and msb2Δ mutant strains, transformed with pRS316-SNF3-HA, pRS313-RGT2-HA, pRS315-SHO1-FLAG, pRS316-MSB2-HA, or the indicated glycosylation-site mutants, were stimulated with 0.5 M KCl for 5 min. Cell lysates were resolved by 10% SDS-PAGE and immunoblotting. Specific antibodies were used to detect the dually phosphorylated, fully activated forms of Kss1 and Hog1 (p-Kss1 and p-Hog1). G6PDH served as a loading control. For the relevant uncomplemented deletion strains, refer to Figures 4 and 5. (B) Activation of Kss1 and Hog1 in wild-type, mnn11Δ or integrated msb2N30A mutant, stimulated with 0.5 M KCl for 5 min (69).
DISCUSSION
MAPK pathways often share common signaling components yet exhibit remarkably little cross-pathway activation. In yeast, salt stress leads to activation of the MAPK Hog1 but not Fus3, while mating pheromone activates Fus3 but not Hog1. Transient activation of Kss1 has been observed in response to pheromone (32) and osmotic stress (18, 45). However, neither stimulus is sufficient to invoke the filamentous growth response; this restriction is due in part to Fus3 phosphorylation of a transcription factor in the invasive pathway, Tec1. Tec1 is then desumoylated, ubiquitinated, and subsequently degraded (46–50). Another mechanism entails Hog1 phosphorylation of Ste50, one of several components shared by all three MAPK pathways. However, the role of Ste50 appears to be limited to cross talk between the FG pathway and the Sho1 branch of the HOG pathway, since Ste50 phosphorylation-site mutants sustain Kss1 signaling in ssk1Δ cells (18) more so than in wild-type cells, where both branches of the HOG pathway are functioning (20) (see Supporting Information, Figure S3). Nevertheless, the realization that Kss1 is activated by pheromone or osmotic stress, as well as by nutrient deprivation, challenged a long-held notion that cross-activation would occur only upon loss of Fus3 or Hog1 function. On this basis we initiated a genome-scale search for additional mutants that disrupt pathway fidelity. Our approach was modeled after previous efforts to analyze pathway specificity using comprehensive genomic (31, 45, 51, 52), proteomic (53), and expression profiling strategies (12, 31, 54, 55). This screen led to the identification of mnn11Δ from the YKO collection, which could penetrate its substratum in response to hyperosmotic stress. Although we could not recapitulate the original invasive-growth phenotype in a reconstructed mnn11Δ strain, we observed an increase in Kss1 phosphorylation, indicative of activation. Based on these findings we postulate the existence of additional, as yet unidentified, genes that limit invasive growth in response to Kss1. For example, this could explain why we did not identify MNN10 in our original screen.
Hence, our effort revealed two new mutants that exhibit inappropriate activation of Kss1; when MNN10 or MNN11 is absent, Kss1 is hyperphosphorylated in response to salt stress. In comparison to wild type, the abundance of Kss1 is elevated by two-fold in the deletion strain (Figure S2). Phospho-Kss1 is likewise elevated but increases an additional 2.5-fold under salt-stimulated conditions (Figure 2B). Notably, this salt-dependent increase in phospho-Kss1 is evident only in the mutant strains. We conclude that Mnn11/10 limit the overall expression of Kss1 under basal conditions and also limit the activation of Kss1 under salt stress conditions. The same gene mutations do not alter the response of Kss1 to pheromone, and they do not alter the response of Fus3 or Hog1 to either pheromone or salt.
MNN10 and MNN11 encode α-1,6-mannosyltransferases associated with the Golgi apparatus. These enzymes are responsible for extending the mannan chains on all N-glycosylated proteins destined for the cell surface (24). Deletion of either MNN10 or MNN11 leads to a reduction in the length of mannan chains on N-glycosylated proteins (24, 56). Based on the known function of Mnn10 and Mnn11, we postulated that one or more N-glycosylated proteins serve to limit the activity of Kss1 under hyperosmotic conditions and that the substrate must be glycosylated in order to function properly. We subsequently established that deletion of a known glycoprotein (Msb2) is sufficient to suppress hyperactivation of Kss1 in the mnn11Δ mutant. Finally we asked whether loss of a particular N-glycosylation site would lead to hyperactivation of Kss1, even in the absence of Mnn11. N-glycosylation-site mutations in Sho1, Snf3, or Rgt2 had no effect on Kss1. In contrast, mutation of a single candidate N-glycosylation site (Asn-30) in Msb2 led to inappropriate activation of Kss1, in the manner of mnn11Δ.
Previous studies have indicated that protein glycosylation is important for proper MAPK signaling, particularly within the FG pathway (40, 57–59). One study reported high basal activation of Kss1 in cells bearing partial loss-of-function alleles of genes needed for O-linked and N-linked glycosylation (57). Another study reported FUS1-lacZ induction when a specific type of O-glycosylation was absent and when the cells were treated with tunicamycin, a pharmacological inhibitor of protein N-glycosylation. That analysis pointed to Msb2 as the most likely target for O-glycosylation, and that some combination of O- and N- glycosylation was needed to maintain signal fidelity (40). Our study advances earlier findings by identifying a specific mannosyltransferase activity as well a single N-glycosylation site that is evidently needed for Msb2 to function as a pathway insulator.
While it is well established that Msb2 functions at the head of the FG MAPK pathway (31, 36), a long-standing question is how Msb2 initiates a signaling cascade leading to Kss1. The MSB2 gene was originally identified as a high-copy suppressor of temperature sensitive alleles of cdc42 and cdc24 (60). Cdc42 is a Rho family GTPase, while Cdc24 is an exchange factor that activates Cdc42. Whereas Msb2 associates with Cdc42 (31), Sho1 binds Cdc24 (37). Thus, assembly of Msb2 with Sho1 could bring Cdc42 into close proximity with its activator (36). Once activated, Cdc42 stimulates a protein kinase cascade beginning with Ste20 (61, 62) and culminating with Kss1 (31, 36). It is also well established that diminished nutrient availability leads to filamentous growth in yeast (6–8, 10, 63). Indeed, our data implicates a new role for the glucose sensors Snf3 and Rgt2 in the filamentous growth pathway, as these proteins are needed to confer Kss1 hyperactivation in salt-treated cells. However, it is not established how the pathway can detect changes in nutrient levels, levels that must fall somewhere between what is needed to support vegetative growth and what triggers entry into stationary phase (6). One possibility is that a reduction in glucose availability results in diminished glycosylation of key signaling proteins (64). In this regard, Msb2 is an attractive target of regulation. Perturbations to the Msb2 extracellular domain were shown previously to trigger Kss1 activation (31, 37, 40). Our own data reveal a critical role for a candidate N-glycosylation site in Msb2, Asn-30, as well as for two mannosyltransferases, Mnn10 and Mnn11, in dictating the activity of Kss1.
While much has been learned, important questions remain. How does protein glycosylation in general, and Mnn10/11 in particular, regulate Msb2? Synthesis of N-glycans begins in the endoplasmic reticulum (ER), where a core oligosaccharide structure is assembled on nascent proteins during their translocation into the ER lumen (65). These N-glycosylated proteins are then delivered to the Golgi apparatus, where the oligosaccharide core structure is elaborated with mannose in one of two ways. Proteins located on endomembranes of the internal organelles have a small N-linked glycan structure with only a few mannoses added onto the core oligosaccharide. Proteins that are incorporated into the cell wall and/or plasma membrane, however, have a large mannan structure that consists of a backbone of about 50 mannose residues with short side branches (56). Mnn10 and Mnn11 are part of the M-pol II complex in the Golgi. M-pol II is dedicated to extending the mannan chain on all N-glycosylated proteins, and within this complex both Mnn10 and Mnn11 are thought to be responsible for cellular α-1,6-mannosyltransferase activity (24). When either of these enzymes is absent, the consequence is a shorter mannan chain on N-glycosylated proteins (24, 66).
How might nutrient limitation affect Msb2 glycosylation? Both Mnn10 and Mnn11 use mannose as a substrate. Mannose is derived from glucose-6-phosphate and glucose. As glucose become limiting, mannose synthesis will likely be diminished, and this could prevent Mnn10 and Mnn11 from properly modifying Msb2. Therefore, if under-glycosylated Msb2 represents the activated form of the protein, the absence of Mnn10 or Mnn11 could be mimicking the events that occur normally following nutrient deprivation. The consequence in either case is Kss1 activation. Given the similarity between Msb2 and the signaling mucin MUC1 in animals, and given that both proteins activate MAP kinases (reviewed in (67, 68)), the mechanisms reported in yeast could point to similar processes in humans.
Further studies are needed to determine whether Msb2 is modified by Mnn10 or Mnn11 directly and whether Msb2 has a large mannan structure characteristic of M-pol II substrates. Addressing this question rigorously would require the reconstitution of M-pol II components Mnn11, Mnn9, Anp1, Mnn10, and Hoc1 (24), as well as the ability to detect and quantify the properly modified and mis-modified forms of Msb2. Indeed, our efforts to monitor Msb2 modifications by immunoblotting were impeded by the poor detection of our epitope-tagged Msb2, the induction of Msb2 following Kss1 activation, and the existence of proteolytically-processed forms of the protein in vivo (31, 37).
In summary, we have identified novel mutations that exhibit hyperactivation of Kss1 in response to osmotic stress stimulation. Our findings indicate that Mnn10 and Mnn11 act specifically to limit activation of Kss1 by an inappropriate stimulus. We propose that N-glycosylation of Msb2 in particular is needed for insulation of pathway signaling components. Together, these findings reveal new proteins and processes needed for proper MAPK signaling in cells.
Supplementary Material
Acknowledgments
Funding Source Statement: Supported by NIH grant GM080739 (to HGD).
We wish to thank Sarah Clement, Justin English and Paul Cullen for their helpful advice and members of the Dohlman lab for their guidance and support.
ABBREVIATIONS AND TEXTUAL FOOTNOTES
- CRE
Cyclic adenosine monophosphate -responsive element
- FG
filamentous growth
- G6PDH
glucose-6-phosphate dehydrogenase
- HOG
high osmolarity glycerol
- MAPK
mitogen-activated protein kinase
- MAPKK
mitogen-activated protein kinase kinase
- MAPKKK
mitogen-activated protein kinase kinase kinase
- MAPKKKK
mitogen-activated protein kinase kinase kinase kinase
- SCD
synthetic complete dextrose
- YKO
yeast knockout
Footnotes
Supporting information available. One table and four figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Turjanski AG, Vaque JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- 2.Chen RE, Thorner J. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochimica et biophysica acta. 2007;1773:1311–1340. doi: 10.1016/j.bbamcr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- 4.Hohmann S. Osmotic stress signaling and osmoadaptation in yeasts. Microbiology and molecular biology reviews: MMBR. 2002;66:300–372. doi: 10.1128/MMBR.66.2.300-372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Rourke SM, Herskowitz I. Unique and redundant roles for HOG MAPK pathway components as revealed by whole-genome expression analysis. Molecular biology of the cell. 2004;15:532–542. doi: 10.1091/mbc.E03-07-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cullen PJ, Sprague GF., Jr Glucose depletion causes haploid invasive growth in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13619–13624. doi: 10.1073/pnas.240345197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuchin S, Vyas VK, Carlson M. Role of the yeast Snf1 protein kinase in invasive growth. Biochemical Society transactions. 2003;31:175–177. doi: 10.1042/bst0310175. [DOI] [PubMed] [Google Scholar]
- 8.Roberts RL, Fink GR. Elements of a single MAP kinase cascade in Saccharomyces cerevisiae mediate two developmental programs in the same cell type: mating and invasive growth. Genes & development. 1994;8:2974–2985. doi: 10.1101/gad.8.24.2974. [DOI] [PubMed] [Google Scholar]
- 9.Brown CM, Hough JS. Elongation of yeast cells in continuous culture. Nature. 1965;206:676–678. doi: 10.1038/206676a0. [DOI] [PubMed] [Google Scholar]
- 10.Gimeno CJ, Ljungdahl PO, Styles CA, Fink GR. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell. 1992;68:1077–1090. doi: 10.1016/0092-8674(92)90079-r. [DOI] [PubMed] [Google Scholar]
- 11.Chang F, Herskowitz I. Identification of a gene necessary for cell cycle arrest by a negative growth factor of yeast: FAR1 is an inhibitor of a G1 cyclin, CLN2. Cell. 1990;63:999–1011. doi: 10.1016/0092-8674(90)90503-7. [DOI] [PubMed] [Google Scholar]
- 12.Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR, Tyers M, Boone C, Friend SH. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287:873–880. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- 13.Yu L, Qi M, Sheff MA, Elion EA. Counteractive control of polarized morphogenesis during mating by mitogen-activated protein kinase Fus3 and G1 cyclin-dependent kinase. Molecular biology of the cell. 2008;19:1739–1752. doi: 10.1091/mbc.E07-08-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall JP, Cherkasova V, Elion E, Gustin MC, Winter E. The osmoregulatory pathway represses mating pathway activity in Saccharomyces cerevisiae: isolation of a FUS3 mutant that is insensitive to the repression mechanism. Molecular and cellular biology. 1996;16:6715–6723. doi: 10.1128/mcb.16.12.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Rourke SM, Herskowitz I. The Hog1 MAPK prevents cross talk between the HOG and pheromone response MAPK pathways in Saccharomyces cerevisiae. Genes & development. 1998;12:2874–2886. doi: 10.1101/gad.12.18.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westfall PJ, Thorner J. Analysis of mitogen-activated protein kinase signaling specificity in response to hyperosmotic stress: use of an analog-sensitive HOG1 allele. Eukaryotic cell. 2006;5:1215–1228. doi: 10.1128/EC.00037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patterson JC, Klimenko ES, Thorner J. Single-cell analysis reveals that insulation maintains signaling specificity between two yeast MAPK pathways with common components. Science signaling. 2010;3:ra75. doi: 10.1126/scisignal.2001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao N, Zeng Y, Elston TC, Dohlman HG. Control of MAPK specificity by feedback phosphorylation of shared adaptor protein Ste50. The Journal of biological chemistry. 2008;283:33798–33802. doi: 10.1074/jbc.C800179200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagiec MJ, Dohlman HG. Checkpoints in a yeast differentiation pathway coordinate signaling during hyperosmotic stress. PLoS genetics. 2012;8:e1002437. doi: 10.1371/journal.pgen.1002437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto K, Tatebayashi K, Tanaka K, Saito H. Dynamic control of yeast MAP kinase network by induced association and dissociation between the Ste50 scaffold and the Opy2 membrane anchor. Molecular cell. 2010;40:87–98. doi: 10.1016/j.molcel.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 21.Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2004;25:1465–1476. doi: 10.1016/j.peptides.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 22.Inouye C, Dhillon N, Thorner J. Ste5 RING-H2 domain: role in Ste4-promoted oligomerization for yeast pheromone signaling. Science. 1997;278:103–106. doi: 10.1126/science.278.5335.103. [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- 24.Jungmann J, Rayner JC, Munro S. The Saccharomyces cerevisiae protein Mnn10p/Bed1p is a subunit of a Golgi mannosyltransferase complex. The Journal of biological chemistry. 1999;274:6579–6585. doi: 10.1074/jbc.274.10.6579. [DOI] [PubMed] [Google Scholar]
- 25.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 26.Bartkeviciute D, Sasnauskas K. Disruption of the MNN10 gene enhances protein secretion in Kluyveromyces lactis and Saccharomyces cerevisiae. FEMS yeast research. 2004;4:833–840. doi: 10.1016/j.femsyr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Lee MJ, Dohlman HG. Coactivation of G protein signaling by cell-surface receptors and an intracellular exchange factor. Current biology: CB. 2008;18:211–215. doi: 10.1016/j.cub.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 29.Hoffman GA, Garrison TR, Dohlman HG. Analysis of RGS proteins in Saccharomyces cerevisiae. Methods in enzymology. 2002;344:617–631. doi: 10.1016/s0076-6879(02)44744-1. [DOI] [PubMed] [Google Scholar]
- 30.Tatebayashi K, Yamamoto K, Tanaka K, Tomida T, Maruoka T, Kasukawa E, Saito H. Adaptor functions of Cdc42, Ste50, and Sho1 in the yeast osmoregulatory HOG MAPK pathway. The EMBO journal. 2006;25:3033–3044. doi: 10.1038/sj.emboj.7601192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cullen PJ, Sabbagh W, Jr, Graham E, Irick MM, van Olden EK, Neal C, Delrow J, Bardwell L, Sprague GF., Jr A signaling mucin at the head of the Cdc42- and MAPK-dependent filamentous growth pathway in yeast. Genes & development. 2004;18:1695–1708. doi: 10.1101/gad.1178604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabbagh W, Jr, Flatauer LJ, Bardwell AJ, Bardwell L. Specificity of MAP kinase signaling in yeast differentiation involves transient versus sustained MAPK activation. Molecular cell. 2001;8:683–691. doi: 10.1016/s1097-2765(01)00322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao N, Yildirim N, Nagiec MJ, Parnell SC, Errede B, Dohlman HG, Elston TC. Combined computational and experimental analysis reveals MAP kinase-mediated feedback phosphorylation as a mechanism for signaling specificity. Molecular biology of the cell. 2012 doi: 10.1091/mbc.E12-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mentesana PE, Konopka JB. Mutational analysis of the role of N-glycosylation in alpha-factor receptor function. Biochemistry. 2001;40:9685–9694. doi: 10.1021/bi0108507. [DOI] [PubMed] [Google Scholar]
- 35.Cvrckova F, De Virgilio C, Manser E, Pringle JR, Nasmyth K. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes & development. 1995;9:1817–1830. doi: 10.1101/gad.9.15.1817. [DOI] [PubMed] [Google Scholar]
- 36.Tatebayashi K, Tanaka K, Yang HY, Yamamoto K, Matsushita Y, Tomida T, Imai M, Saito H. Transmembrane mucins Hkr1 and Msb2 are putative osmosensors in the SHO1 branch of yeast HOG pathway. The EMBO journal. 2007;26:3521–3533. doi: 10.1038/sj.emboj.7601796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vadaie N, Dionne H, Akajagbor DS, Nickerson SR, Krysan DJ, Cullen PJ. Cleavage of the signaling mucin Msb2 by the aspartyl protease Yps1 is required for MAPK activation in yeast. The Journal of cell biology. 2008;181:1073–1081. doi: 10.1083/jcb.200704079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hao N, Behar M, Parnell SC, Torres MP, Borchers CH, Elston TC, Dohlman HG. A systems-biology analysis of feedback inhibition in the Sho1 osmotic-stress-response pathway. Current biology: CB. 2007;17:659–667. doi: 10.1016/j.cub.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 39.Pitoniak A, Birkaya B, Dionne HM, Vadaie N, Cullen PJ. The signaling mucins Msb2 and Hkr1 differentially regulate the filamentation mitogen-activated protein kinase pathway and contribute to a multimodal response. Molecular biology of the cell. 2009;20:3101–3114. doi: 10.1091/mbc.E08-07-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang HY, Tatebayashi K, Yamamoto K, Saito H. Glycosylation defects activate filamentous growth Kss1 MAPK and inhibit osmoregulatory Hog1 MAPK. The EMBO journal. 2009;28:1380–1391. doi: 10.1038/emboj.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bause E, Hettkamp H. Primary structural requirements for N-glycosylation of peptides in rat liver. FEBS letters. 1979;108:341–344. doi: 10.1016/0014-5793(79)80559-1. [DOI] [PubMed] [Google Scholar]
- 42.Marshall RD. The nature and metabolism of the carbohydrate-peptide linkages of glycoproteins. Biochemical Society symposium. 1974:17–26. [PubMed] [Google Scholar]
- 43.Pasula S, Chakraborty S, Choi JH, Kim JH. Role of casein kinase 1 in the glucose sensor-mediated signaling pathway in yeast. BMC cell biology. 2010;11:17. doi: 10.1186/1471-2121-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moriya H, Johnston M. Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1572–1577. doi: 10.1073/pnas.0305901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shock TR, Thompson J, Yates JR, 3rd, Madhani HD. Hog1 mitogen-activated protein kinase (MAPK) interrupts signal transduction between the Kss1 MAPK and the Tec1 transcription factor to maintain pathway specificity. Eukaryotic cell. 2009;8:606–616. doi: 10.1128/EC.00005-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bao MZ, Schwartz MA, Cantin GT, Yates JR, 3rd, Madhani HD. Pheromone-dependent destruction of the Tec1 transcription factor is required for MAP kinase signaling specificity in yeast. Cell. 2004;119:991–1000. doi: 10.1016/j.cell.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 47.Bruckner S, Kohler T, Braus GH, Heise B, Bolte M, Mosch HU. Differential regulation of Tec1 by Fus3 and Kss1 confers signaling specificity in yeast development. Current genetics. 2004;46:331–342. doi: 10.1007/s00294-004-0545-1. [DOI] [PubMed] [Google Scholar]
- 48.Chou S, Huang L, Liu H. Fus3-regulated Tec1 degradation through SCFCdc4 determines MAPK signaling specificity during mating in yeast. Cell. 2004;119:981–990. doi: 10.1016/j.cell.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Dohlman HG. Pheromone-regulated sumoylation of transcription factors that mediate the invasive to mating developmental switch in yeast. The Journal of biological chemistry. 2006;281:1964–1969. doi: 10.1074/jbc.M508985200. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Abu Irqeba A, Ayalew M, Suntay K. Sumoylation of transcription factor Tec1 regulates signaling of mitogen-activated protein kinase pathways in yeast. PloS one. 2009;4:e7456. doi: 10.1371/journal.pone.0007456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chavel CA, Dionne HM, Birkaya B, Joshi J, Cullen PJ. Multiple signals converge on a differentiation MAPK pathway. PLoS genetics. 2010;6:e1000883. doi: 10.1371/journal.pgen.1000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin R, Dobry CJ, McCown PJ, Kumar A. Large-scale analysis of yeast filamentous growth by systematic gene disruption and overexpression. Molecular biology of the cell. 2008;19:284–296. doi: 10.1091/mbc.E07-05-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu T, Shively CA, Jin R, Eckwahl MJ, Dobry CJ, Song Q, Kumar A. A profile of differentially abundant proteins at the yeast cell periphery during pseudohyphal growth. The Journal of biological chemistry. 2010;285:15476–15488. doi: 10.1074/jbc.M110.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madhani HD, Galitski T, Lander ES, Fink GR. Effectors of a developmental mitogen-activated protein kinase cascade revealed by expression signatures of signaling mutants. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12530–12535. doi: 10.1073/pnas.96.22.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Breitkreutz A, Boucher L, Breitkreutz BJ, Sultan M, Jurisica I, Tyers M. Phenotypic and transcriptional plasticity directed by a yeast mitogen-activated protein kinase network. Genetics. 2003;165:997–1015. doi: 10.1093/genetics/165.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munro S. What can yeast tell us about N-linked glycosylation in the Golgi apparatus? FEBS letters. 2001;498:223–227. doi: 10.1016/s0014-5793(01)02488-7. [DOI] [PubMed] [Google Scholar]
- 57.Cullen PJ, Schultz J, Horecka J, Stevenson BJ, Jigami Y, Sprague GF., Jr Defects in protein glycosylation cause SHO1-dependent activation of a STE12 signaling pathway in yeast. Genetics. 2000;155:1005–1018. doi: 10.1093/genetics/155.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cullen PJ, Xu-Friedman R, Delrow J, Sprague GF. Genome-wide analysis of the response to protein glycosylation deficiency in yeast. FEMS yeast research. 2006;6:1264–1273. doi: 10.1111/j.1567-1364.2006.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee BN, Elion EA. The MAPKKK Ste11 regulates vegetative growth through a kinase cascade of shared signaling components. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12679–12684. doi: 10.1073/pnas.96.22.12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bender A, Pringle JR. A Ser/Thr-rich multicopy suppressor of a cdc24 bud emergence defect. Yeast. 1992;8:315–323. doi: 10.1002/yea.320080409. [DOI] [PubMed] [Google Scholar]
- 61.Peter M, Neiman AM, Park HO, van Lohuizen M, Herskowitz I. Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. The EMBO journal. 1996;15:7046–7059. [PMC free article] [PubMed] [Google Scholar]
- 62.Leberer E, Wu C, Leeuw T, Fourest-Lieuvin A, Segall JE, Thomas DY. Functional characterization of the Cdc42p binding domain of yeast Ste20p protein kinase. The EMBO journal. 1997;16:83–97. doi: 10.1093/emboj/16.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu H, Styles CA, Fink GR. Elements of the yeast pheromone response pathway required for filamentous growth of diploids. Science. 1993;262:1741–1744. doi: 10.1126/science.8259520. [DOI] [PubMed] [Google Scholar]
- 64.Dennis JW, Nabi IR, Demetriou M. Metabolism, cell surface organization, and disease. Cell. 2009;139:1229–1241. doi: 10.1016/j.cell.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 66.Hernandez LM, Ballou L, Alvarado E, Tsai PK, Ballou CE. Structure of the phosphorylated N-linked oligosaccharides from the mnn9 and mnn10 mutants of Saccharomyces cerevisiae. The Journal of biological chemistry. 1989;264:13648–13659. [PubMed] [Google Scholar]
- 67.Cullen PJ. Post-translational regulation of signaling mucins. Current opinion in structural biology. 2011;21:590–596. doi: 10.1016/j.sbi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cullen PJ. Signaling mucins: the new kids on the MAPK block. Critical reviews in eukaryotic gene expression. 2007;17:241–257. doi: 10.1615/critreveukargeneexpr.v17.i3.50. [DOI] [PubMed] [Google Scholar]
- 69.Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nature biotechnology. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.