

Abstract
In previous work transduction of Escherichia coli with phage λ particles carrying packaged plasmids was shown to provide complete off-to-on expression of plasmid-borne genes (Cronan, J. E. 2003. J. Bacteriol. 185, 6522–6529). The plasmids used contained the phage λcos site (and hence are cosmids) and were very efficiently packaged into λ phage particles in vivo upon induction of λ lysogens having an inactivated cos site. However, a shortcoming was that the stocks contained a variable fraction of infectious λ phage which arose by recombinational repair of the inactive cos site. I report that the construction of E. coli strains that eliminate the background of infectious phage and show that the system can be utilized to express proteins by the phage T7 RNA polymerase/pET vector system.
Keywords: Phage lambda, cosmid, DNA packaging, Protein expression
1. Introduction
The determinants for processive packaging of phage λ DNA reside in the 223 bp minimal cos region (Miwa and Matsubara, 1982) that contains the site of cleavage generating the λ cohesive ends. Plasmids of sufficient length that contain the λcos region (called cosmids) can be packaged into phage particles either in vitro or in vivo. Although in vitro packaging is often used for construction of genome libraries, the titers obtained are very low. In vivo packaging of cosmids by induction of λ lysogens is well studied and is known to result in particles that contain either cosmid DNA or phage DNA (Miwa and Matsubara, 1983; Umene et al., 1978; Umene et al., 1979). However, there was little utilization of this approach due to the moderate yields of packaged cosmids and the very large excess of infectious phage particles (Jacobs et al., 1986; Miwa and Matsubara, 1983; Umene et al., 1978; Umene et al., 1979). Previously I reported λ lysogens having an inactivated cos site that packaged cosmid DNAs much more efficiently than λ DNA to give much higher titers of cosmid transducing particles (Cronan, 2003). I now report improvements to the packaging strain that result in packaged cosmid preparations with no detectable infectious phage. Such preparations can be used to provide T7 RNA polymerase or, conversely, to introduce a gene driven by a T7 promoter into a strain that expresses T7 RNA polymerase.
In the prior work I constructed two lysogenic strains carrying λ phages in which the cos site had been inactivated by insertion of an antibiotic cassette between the filled-in cohesive ends thereby disrupting the strict spacing of the cos elements required for packaging (Cronan, 2003). The prophages also encoded a temperature-sensitive cI repressor to allow ready induction of phage replication and a nonsense mutation in the S gene required for cell lysis which facilitates preparation of high titer stocks (Arber et al., 1983). Upon thermal induction of phage replication the phage excises from the bacterial chromosome and commences rolling circle DNA replication to produce the concatemeric substrates required for DNA packaging (Casjens, 2011; Rao and Feiss, 2008). In the presence of λ replication most common cloning vector plasmids shift from their normal theta replication into the rolling circle mode to give concatamers containing many head-to-tail copies of the plasmid (Miwa and Matsubara, 1983). If the concatamers contain a cos region and are sufficiently large, they become assembled into phage particles (Casjens, 2011; Feiss et al., 1982). Although disruption of the cos region of the lysogen resulted in greatly increased cosmid packaging efficiencies, the preparations contained significant and variable levels of infectious phage (Cronan, 2003). The infectious phage resulted from repair of the phage cos region by homologous recombination with either the cos region of the cosmid or the cos of one of the cryptic defective prophages resident in the E. coli K-12 chromosome. This recombination was catalyzed by the extremely efficient λ Red-Gam system which is required for rolling circle replication (Miwa and Matsubara, 1983) and thus cannot be eliminated. Since the recombinase could not be eliminated, the only means to prevent recombination was removal of the cos regions that provide the substrates for recombinational repair. Although elimination of the phage cos site appeared to be a straightforward means to prevent recombination with cosmids, this seemed of little avail due to the defective cryptic prophages resident in the E. coli K-12 genome (Casjens, 2003), two of which contain cos sites (plus extensive flanking regions) that are essentially identical to that of λ. E. coli K-12 strains in which all the defective prophages have been deleted were reported in 2002 (Kolisnychenko et al., 2002) but those strains have proprietary restrictions that prevent modification of the chromosome. However, Wang and coworkers (Wang et al., 2010) recently reported an E. coli K-12 strain, BW25113 Δ9, which lacks all nine cryptic prophages and have provided this strain for unrestricted use. Given this strain the only substrate for recombinational repair of cos would the cosmid to be packaged and this could be eliminated by deletion of the minimal cos sequence of the prophage. This was done by isolation of derivatives of strain BW25113 Δ9 that carried a single prophage followed by replacement of the cos sequence with a chloramphenicol resistance cassette by homologous recombination catalyzed by the λ Red recombineering proteins.
2. Material and Methods
2.1 Construction of cos prophages
The bacterial strains, plasmids and PCR primers used are given in Table 1. Three different cosmid packaging host strains were constructed from strain BW25113 Δ9. For construction of the prophage carrying a chloramphenicol resistance cassette with flanking FRT sites, the primers (Table 1) were λds-L Rz in which the first 36 nt correspond to nt 48391 to 48426 of the λ genome (Genbank J02459.1) and λds-R Nu1 in which the first 35 nt correspond to the complement of nt 148 to 182 of the λ genome with the remaining primer nt being those of the primers used for the pKD3 (Genbank AY048742.1) chloramphenicol resistance cassette by Datsenko and Wanner (Datsenko and Wanner, 2000). For construction of the prophage lacking FRT sites, the primers were Rz no FRT and Nu1 no FRT which contain the same λ sequences as above but resistance cassette sequences located between the FRT sites and the resistance gene. The chloramphenicol resistance cassette was removed from the FRT-containing prophage by transformation with pCP20 (Cherepanov and Wackernagel, 1995; Datsenko and Wanner, 2000). The transformants were grown overnight at 33°C in liquid medium lacking antibiotics and then plated at 30 °C for isolation of colonies sensitive to chloramphenicol and ampicillin.
Table 1.
E. coli strains, plasmids and primers
| E. coli strains | Relevant characteristics | Reference or source |
|---|---|---|
| BW25113 | No FRT scars, all nine K-12 prophages | (Datsenko and Wanner, 2000) |
| BW25113 Δ9 | Δrac ΔCP4-57 ΔCPS-53 ΔDLP12 ΔQin Δe14 ΔCP4-6 ΔCPZ-55ΔCP4-44 ΔKm |
(Wang et al., 2010) |
| MC1061 | Δ(araA-leu)7697 Δ(codB-lacI) | (Casadaban and Cohen, 1980) |
| DY329 | W3110 ΔlacU169 nadA::Tn10 gal490 λcI857 Δ(cro-bioA) | (Yu et al., 2000) |
| MG1655 | Wild type | CGSC |
| MM294 | Wild type | CGSC |
| LE392 | tryT (supF58) | Promega |
| Tuner | BL21 (DE3) ΔlacZY | Novagen |
| CY2113 | BW25113 Δ9 nadA::Tn10 gal490 λcI857 Δ(cro-bioA) | This work |
| CY2115 | BW25113 Δ9 λcI857 Sam7 Δcos::FRT-cml-FRT. | This work |
| CY2116 | λ-resistant derivative of CY2115 | This work |
| CY2119 | BW25113 Δ9 λcI857 Sam7 Δcos::cml. | This work |
| pCY922 | Cosmid derivative of pSTL28 | This work |
|
| ||
|
Plasmids
| ||
| pTara | paraBAD T7 gene 1, p15a origin chloramphenicol resistant | (Wycuff and Matthews, 2000) |
| pCY566 | High copy number cos resource plasmid carrying the minimal cos region flanked by multiple cloning sites. | (Cronan, 2003) |
| pCY915B | Cosmid derivative of pTara | This work |
| pET28b | T7 promoter expression plasmid | Novagen |
| pMC1871 | Promoter-less lacZY plasmid | (Casadaban et al., 1983) |
| pCY913 | pET28b expressing pMC1871 lacZY | This work |
| pCY914 | Cosmid derivative of pCY913 | This work |
| pSTL28 | pET28b carrying Bacillus cereus bioC gene | (Lin, 2012) |
2.2. Preparation and assay of phage particles
The media and procedures for cosmid packaging and titering were as described previously (Arber et al., 1983; Cronan, 2003) except that glucose was omitted from the medium and the harvested cells were washed with λdil before lysis. In a few cases the lysates were concentrated by centrifugal ultrafiltration using 100 kDa MWCO units (Vivaspin 6, GE Healthcare) and resuspended in λ dilution buffer lacking gelatin. The titers of packaged cosmids were generally measured on strain MM294 grown with maltose to increase the number of λ receptors (Cronan, 2003) although titers were independent of the host strain. Generally 0.1 ml of an early stationary phase culture was incubated with 0.1 ml a serial dilution of the stock of packaged cosmids for 30 min at room temperature before spreading on LB plates containing the appropriate antibiotic. The plates were incubated overnight at 37°C. Infectious phage titers were obtained on strain LE392 using standard procedures (Arber et al., 1983). The plates were incubated overnight at 42°C.
2.3. Cosmid Constructions
Several cosmids were constructed by insertion of the minimal cos fragment of plasmid pCY566 (Cronan, 2003) into plasmid restriction sites by use of the polylinker sequences that flank the cos region. For example cosmid pCY915B was constructed by ligation of the pCY566 NsiI-PstI cos fragment into the NsiI site of pTara, a p15a origin chloramphenicol resistant plasmid that expresses T7 RNA polymerase under arabinose control (Wycuff and Matthews, 2000). A plasmid encoding E. coli lacZ under control of a T7 promoter was constructed by ligation of the promoterless lacZ BamHI fragment of pMC1871 (Casadaban et al., 1983) to BamHI digested pET28b (Novagen) followed by screening of the kanamycin-resistant transformants on medium containing IPTG and X-gal. This plasmid (pCY913) was converted to a cosmid by insertion of the XhoI-HindIII fragment of pCY566 into the same sites of the pCY913 plasmid to give pCY914. The bioC cosmid pCY922 was constructed by inserting the BamHI-BglII fragment of pCY566 into the BglII site located upstream of the T7 promoter of pET28b which contains a synthetic gene encoding Bacillus cereus BioC (Lin, 2012).
2.4. Assays and media
β-Galactosidase assays were done essentially as described by Miller (Miller, 1992). Transductions with phage P1vir were done by standard procedures except that the plates were incubated at 30 °C. The minimal medium used to score the nadA and bioA markers was glucose M9 medium (Miller, 1992) supplemented with 0.2% Vitamin Assay Casamino Acids (Difco). When required nicotinic acid was added to 5 μg/ml and biotin to 4 nM. Luria-Bertani broth and 2XYT broth and Luria-Bertani agar media and (Miller, 1992) were used in the other manipulations.
3. Results
3.1 Construction of cos lysogens of strain BW25113 Δ9
The first attempts to replace the prophage cos region of a λ prophage resident in strain BW25113 Δ9 were compromised by the presence of tandem prophages inserted into the host attB site. Although documented long ago (Arber, 1960; Folkmanis and Freifelder, 1972; Freifelder et al., 1975; Freifelder and Kirschner, 1971; Freifelder and Levine, 1975) it is not generally appreciated that 40–60% of lysogens formed upon λ infection of E. coli are polylysogens. Since polylysogens provide multiple sites for recombination with the recombineering antibiotic cassette, recombination with a prophage of polylysogen would be favored over recombination with a single lysogen. Indeed, selection for replacement of cos with a chloramphenicol resistance cassette in lysogens of strain BW25113 Δ9 resulted in strains that still produced infectious λ phage indicating the presence of another prophage having a functional cos site. Another source of background in strain BW25113 Δ9 was the five copies of the “scar” sequences resulting from elimination of the defective prophages (Wang et al., 2010). The Datsenko-Wanner antibiotic resistance cassettes are flanked by FRT sites and upon excision of the cassette by site specific recombination a scar of 82- to 85-bp remains (Datsenko and Wanner, 2000). Since only limited homology is required for gene disruption using λ Red recombination, a new PCR fragment can recombine either at the newly targeted gene or at the scar of an earlier gene disruption (Datsenko and Wanner, 2000). Thus, the incoming PCR fragment designed to replace cos could recombine into the prophage(s) or into one of the BW25113 Δ9 scars. Hence, a method was needed to select for recombinants in which the chloramphenicol resistant cassette recombined with a monolysogen and thereby eliminate the undesired background products of recombination with scars, defective prophages and polylysogens. This was done by construction of strain CY2113, a derivative of strain BW25113 Δ9 that contained a single defective λ prophage with E. coli chromosomal makers located on either side of attB. This strain allowed selection of the desired constructs following transduction with phage P1vir lysates grown on pools of candidates. Strain CY2113 was constructed by P1 transduction of strain BW25113 Δ9 with a lysate of strain DY329 (Yu et al., 2000). Tetracycline-resistant transductants were selected and screened for temperature sensitive growth plus the ability to grow on minimal medium only upon supplementation with both nicotinic acid and biotin.
Strain DY329 carries a defective λcI857 prophage deleted for all genes from cro through attR (Patterson et al., 1993; Yu et al., 2000) plus part of the host bioA gene. The strain also carries a Tn10 insertion in nadA, a gene located on the side of the prophage opposite bioA. The bioA deletion and nadA::Tn10 insertion result in requirements for biotin and nicotinic acid, respectively. The strain DY329 prophage is readily transduced by phage P1 due to its small size, inability to excise from the bacterial chromosome and lack of most of the λ host-killing functions (Patterson et al., 1993).
In two parallel experiments λcI857 Sam7 lysogens of strains BW25113 and BW25113 Δ9 carrying the recombineering plasmid pKD46 (Datsenko and Wanner, 2000) were constructed. Strain BW25113 contains the standard nine E. coli K-12 defective prophages, but lacks scar sequences (Datsenko and Wanner, 2000) whereas strain BW25113 Δ9 lacks defective prophages but contains five scars (Wang et al., 2010). Both strains were used because the relative efficiencies of recombination with the scars versus defective prophages were unknown. The lysogens were grown and the pKD46-encoded Red recombination system was induced with arabinose followed by electroporation with a PCR product consisting of the pKD3 chloramphenicol cassette flanked by sequences bracketing the minimal cos site (Datsenko and Wanner, 2000). Phage P1vir lysates were grown on pools of the recombinants and used to transduce strain CY2113 with selection for chloramphenicol resistance plus growth in the absence of nicotinic acid and biotin (Fig. 1B). This selection eliminated recombinants formed by insertion into scar or defective prophage sequences. Moreover, selection for repair of both the nadA::Tn10 and bioA lesions ensured that the transductants contained only a single λcI857 Sam7 Δcos::cml prophage. This is because in order to carry both the nadA and bioA genes plus the 49.25 kbp Δcos::cml λ prophage, a transduced fragment of >74 kbp is required. Phage P1 carries a maximum of 110–115 kbp of DNA (Sternberg, 1990) and thus the second prophage of a dilysogen would result in omission of either nadA or bioA from the transduced chromosomal fragment.
Fig. 1.

Selection and analysis of single Δcos::cml lysogens.
Panel A. Some of the possible products of lysogenization and subsequent cos disruption in strain BW25113 Δ9 and its parent BW25113. The genome of strain BW25113 Δ9 contains five copies of the FRT scar sequence (two are shown as black rectangles) whereas the BW25113 genome contains nine different defective prophages, two of which contain cos regions essentially identical to that of phage λ (grey rectangles). Lysogenization of both strains produces both single lysogens (top lines) and polylysogens. Recombineering was done using the λred system and a PCR product (inverted triangle) containing a chloramphenicol resistance gene (cml) bracketed by sequences flanking cos. Chloramphenicol resistant colonies can be formed not only be by the intended insertion into a λ prophage, but into a scar site (strain BW25113 Δ9) or into the cos region of a defective prophage (strain BW25113). Multiple insertions are also possible. Phage P1 was grown on pools of chloramphenicol resistant colonies and used to transduce strain CY2113 in order to obtain lysogens that contained a single λ Δcos::cml prophage.
Panel B. Within the colony pools of panel A are cells that carry a single λ prophage where chloramphenicol resistance cassette replaced the minimal cos region. Most of the phage P1 particles that carry cat-containing bacterial genome segments will be too large (due to polylysogeny or carry segments of the genome which cannot repair the nadA and bioA mutations of strain CY2113. In the successful transductions the incoming prophage (top line) lies within an E. coli genome segment transduced by a phage P1 particle. Host RecA-catalyzed recombination with the chromosome of strain CY2113 carrying the defective prophage of strain DY329 (middle line) results in chloramphenicol resistance (CML) and restores function of the nadA and bioA genes (bottom line). The defective prophage contains λ genes from cI to int (Patterson et al., 1993). The cI repressor encoded by the defective prophage prevents the transduced Δcos::cml prophage from undergoing lytic development. A deletion of about 38.2 kbp removed the right side of the λ prophage from cro through attR (Patterson et al., 1993) and hence the prophage lacks cos and the lysis (S gene) region. The deletion also removed the last 35 codons of bioA. Some genes of the λ prophages are given on the solid lines whereas host genes are given on the broken lines. PL and PR indicate the early left and right promoters of λ while attL and attR indicate the left and right λ attachment sites.
Panel C. Colony PCR analysis of the cos regions of strains CY2115 (lane 1), CY2119 (lane 2) and CY2120 (lane 3). The molecular weight standard (Std) was the 1 kb Plus Ladder (Invitrogen). The sizes of the relevant bands are given in bp. The calculated sizes of the PCR products were 1888 bp, 1746 bp and 956 bp. Sequencing of the cloned PCR products confirmed the calculated values. The dark ovals on the gel are the tracking dye.
As expected all tested transductants were sensitive to temperature and tetracycline. One of these was retained as strain CY2115. Moreover, temperature-induced cultures of strain CY2115 lysed only upon chloroform treatment, the phenotype of λSam7 lysogens. Upon curing of the strain CY2115 prophage by heat pulse induction (Enquist and Weisberg, 1977) all cured colonies were sensitive to chloramphenicol. The ideal strain for packaging of cosmids would be one resistant to phage λ in order to avoid adsorption of packaged cosmids to bacterial debris during storage. Thus, several isolates were cross-streaked with λvir to select λ resistant strains. One of these strains is CY2116. The chloramphenicol cassette of strain CY2116 was subsequently removed by Flp mediated recombination (Cherepanov and Wackernagel, 1995) to give strain CY2120.
In a second approach primers hybridizing between the FRT sites and the chloramphenicol resistance gene of pKD3 were used such that the PCR product would lack FRT sites (and hence the means for cassette removal) and when electroporated into a λcI857 Sam7 lysogen of strain BW25113 Δ9, the only sequence homologous to the PCR product was the cos region of the prophage. Recombination with scars was thereby precluded but at cost of the ability to remove the antibiotic cassette. The prophages of several of these recombinants were transduced into strain CY2113 tested as above and one was made λ resistant to give strain CY2119.
Finally the cos regions of strains CY2115, CY2119 and CY2120 were amplified by colony PCR using primers (Amp-For and Amp-Rev) that primed from λ sequences flanking cos (Fig. 1B). The PCR products were inserted into vector pCR4 Topo (Invitrogen) and sequenced using the standard T7 and T3 promoter primers. Each strain gave a single PCR product of the size expected and sequencing of the PCR products demonstrated the expected junctions for each of the deletion events.
3.2 Cosmid packaging by strain CY2120
In the prior work (Cronan, 2003) the cosmids expressed the proteins of interest from E. coli promoters. To expand the system, phage T7 promoters were tested. Much of the work was performed with cosmid pCY915B, a derivative of the p15a origin T7 RNA polymerase expression plasmid, pTara (Wycuff and Matthews, 2000). pCY915B was constructed by insertion of the minimal cos region of plasmid pCY566 immediately downstream of the pTara araC gene. This chloramphenicol resistant cosmid expressed T7 polymerase from the arabinose inducible araBAD promoter. Strain CY2120 carrying pCY915B was grown at 30°C to late log phase then shifted to high temperatures to induce λ rolling circle replication. Following several hours of induction the cells were collected by centrifugation, washed, concentrated 100-fold and lysed with chloroform. Following DNAse treatment and centrifugation the lysates were assayed for their ability to transduce strain MM294 to chloramphenicol resistance and titered for infectious phage on strain LE392 which encodes a tRNA suppressor that restores synthesis of functional S protein. A cosmid transduction unit (ctu) is an antibiotic resistant colony formed upon transduction of a great excess of host bacteria whereas infectious phage were assayed as plaque forming units (pfu). Packaging of cosmid pCY915B routinely gave lysates containing >2 × 1011 ctu/ml with <10 pfu/ml (Table 2). Hence, all phage particles contained cosmid DNA and the lysates were free of infectious phage particles. As observed previously (Cronan, 2003) efficient packaging was obtained with cosmids having other replication origins (ColE1, RSF1030) or antibiotic resistance determinants (kanamycin, ampicillin) (Table 2). The cosmids tested varied between 8.2 and 9.4 kbp in length and given the size requirement for λ DNA packaging (~37 to 52 kb) they would be packaged into λ particles as linear cosmid pentamers or hexamers. Note that the ctu titers obtained in the present work were similar to those obtained in the prior work using cos disruption prophages (Cronan, 2003) however the titers of infectious particles were decreased by as much as 1010 -fold.
Table 2.
Purity of the packaged cosmid preparations
| Lysate | Cosmid transductants (ctu/ml) | Infectious phage (pfu/ml) |
|---|---|---|
| pCY915B prep 1 | 7.9 × 1011 | <10 |
| pCY915B prep 2 | 1.1 × 1012 | <10 |
| pCY915B prep 3 | 2.5 × 1011 | <1 |
| pCY915B prep 4 | 3.2 × 1011 | <1 |
| pCY923 | 2.4 × 1011 | <10 |
| pCY914 | 5.0 × 1011 | <10 |
| pCY894cos | 2.7 × 1011 | <10 |
The preparations of packaged cosmid pCY915B were obtained over the course of three months.
The other cosmids were packaged for reasons unrelated to this report.
3.3. Optimization of cosmid transduction of the T7 RNA polymerase gene
Expression of T7 RNA polymerase in E. coli strains carrying a T7 promoter is toxic and quickly blocks host transcription, probably by starving the host RNA polymerase of nucleotides (Studier and Moffatt, 1986). Phage λ infection is known to cause a transient depolarization of the host cell (Daugelavichius et al., 1987), the repair of which may require host RNA synthesis. Hence, a tightly controlled system, that of pTara (Wycuff and Matthews, 2000), was used to allow the transduced cells to recover membrane potential before induction of T7 RNA polymerase expression. To readily assay expression from the T7 promoter, the E. coli lacZ gene was inserted into the expression vector plasmid pET28b and the resulting plasmid, pCY913, was transformed into the ΔlacZYA E. coli K-12 strain MC1061. This strain was grown to mid log phase and transduced with cosmid particles containing pCY915B (5 ctu/viable cell was used to ensure that virtually all of the cells received a λ particle). The culture was concentrated to increase the rate of λ particle adsorption. After adsorption the transduced cultures were diluted to about 4 × 108 cells/ml and allowed to double. A strain MC1061 derivative stably transformed with both cosmid pCY915B and plasmid pCY913 was grown in parallel, but not transduced. Both cultures were then diluted to 4–5 ×108 cells/ml in LB medium containing chloramphenicol, arabinose (to induce T7 RNA polymerase expression) and IPTG (to relieve LacI repression of LacZ expression) and allowed to grow for several h prior to β-galactosidase assays (Fig. 2). Transduction of the T7 RNA polymerase cosmid was found to give levels of lacZ expression equal or greater than those given by the resident T7 RNA polymerase plasmid (Fig. 2A). Other experiments examined the effects of the presence of maltose, IPTG and chloramphenicol during induction (Table 3). Wycuff and Mathews (Wycuff and Matthews, 2000) reported that expression of proteins encoded by a pET vector with pTara as the T7 RNA polymerase source required only arabinose induction despite the fact that the T7 promoter was under LacI control and LacI was overexpressed. This argued that high levels of T7 RNA polymerase efficiently displace LacI. Indeed addition of IPTG to arabinose-induced cultures had essentially no effect (Table 3). The presence of maltose had, at most, a modest effect. The rationale of chloramphenicol addition was that this would prevent overgrowth of the cultures by those cells that failed to adsorb a λ particle. Note that although the level of gene expression was insensitive to the ctu/cell ratio (Fig. 2A), it is important to accurately determine the cell concentration. This is best done by assaying colony formation of dilutions of a log phase culture of known optical density measured using a properly calibrated spectrophotometer (Koch, 1970).
Fig. 2.

Expression levels of cosmid encoded lacZ genes introduced by transduction or resident in the host strain. In panel A strain MC1061 carrying plasmid pCY913 was transduced with particles carrying cosmid pCY915B at either 5 or 10 ctu/cell (left two sets of columns) and compared with the same strain carrying both the pET-lacZ plasmid and resident pCY915B (which have compatible origins and selectable markers). A strain carrying pET-lacZ and the empty vector from which pTara was derived was also tested. Expression was assayed 3.5 h after induction. Ara denotes the presence or absence of 0.2% arabinose. In panel B lacZ cosmid pCY914 was either transduced (5 ctu/cell) into the ΔlacZY host strain Tuner (which encodes T7 RNA polymerase on the chromosome under control of LacI) as described in Table 3 or was resident in Tuner. β-Galactosidase expression in the presence or absence of 1 mM IPTG was assayed 3.5 h after induction.
Table 3.
β-Galactosidase production upon transduction with packaged pCY915B; Influence of IPTG and maltose on arabinose induction.
| Ctu/cell | Ara | IPTG | Cml | Maltose | Miller Units |
|---|---|---|---|---|---|
| 0 | − | − | − | − | ND |
| 0 | + | + | − | − | ND |
| 5 | + | − | − | − | 51,300 |
| 5 | + | − | + | − | 48,430 |
| 5 | + | + | − | − | 46,760 |
| 5 | + | + | + | − | 45,090 |
| 5 | + | − | − | + | 45,001 |
| 5 | + | − | + | + | 43,420 |
| 5 | + | + | − | + | 33,400 |
| 5 | + | + | + | + | 28,390 |
| 5 | + | − | − | − | 40,800 |
| 5 | + | − | + | − | 42,084 |
Strain MC1061carrying pCY913 was transduced with λ particles containing pCY915B. The culture was grown to mid log phase in 2xYT medium containing maltose, the cells were harvested by centrifugation, washed with 2xYT medium and supended in one-tenth volume of 2xYT containing 2 mM MgCl2. The λ particles were then added and the culture shaken at 37°C for 30 min then diluted 10-fold into 2xYT and maltose was added or omitted as shown. In all but the last two rows the designated addition of arabinose, IPTG and/or chloramphenicol was done after the transduced culture OD600 had increased by 70%. In the bottom two rows the absorbance of the transduced culture had increased 2.1-fold and thus the culture was diluted with an equal volume of 2xYT and treated as designated. The cultures were incubated for 3 h following induction and then assayed for β-galactosidase (Miller, 1992). ND, not detected.
The cosmid transduction system provides a new tool to express those proteins that are extremely toxic to E. coli. As discussed below this may have advantages over prior approaches. A variation of the method would be to use cosmid transduction to introduce a cosmid carrying a gene under control of a T7 promoter into a strain that expresses T7 RNA polymerase. We have tested this reverse strategy by converting pCY913, the pET-LacZ plasmid used above, to a cosmid, pCY914. The cosmid was packaged into λ particles which were used to transduce a strain called Tuner, a commercial ΔlacZY derivative of the classical pET vector expression strain BL21(DE3). The transduced culture was compared to a culture of strain Tuner stably transformed with the cosmid. The two cultures produced comparable levels of β-galactosidase upon induction when induced with IPTG (Fig. 2B).
3.4 Expression of a toxic protein
Plasmid pCY922 which expresseds the toxic BioC protein from a T7 promoter was transformed into the wild type E. coli K-12 strain MG1655. BioC methylates the free carboxyl of malonyl-thioesters to form a methyl ester and thereby blocks fatty acid synthesis by starvation for the malonyl thioesters required for fatty acid elongation resulting in inhibition of membrane lipid synthesis and growth (Lin, 2012). This strain was transduced with λ particles carrying pCY915B as above and total cell extracts were then assayed for BioC overproduction by SDS-PAGE (Fig. 3). BioC was effectively expressed.
Fig. 3.
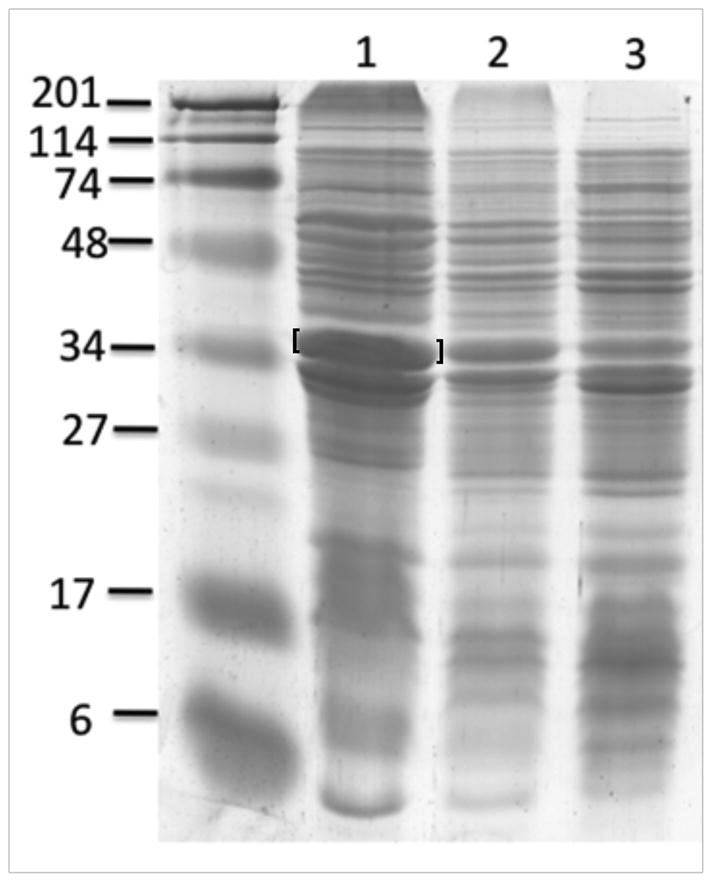
Production of BioC, a toxic protein. Strain MG1655 containing pCY922 was grown to mid log phase in maltose LB medium containing 2 mM MgCl2 and split into thirds. In lane 1 the culture was transduced with packaged particles of pCY915B (5 ctu/cell). After 45 min at 37°C arabinose was added to 0.2% and 4 h later the cells were recovered by centrifugation. The culture of lane 2 was similarly treated but did not receive transducing particles whereas the culture of lane 3 was transduced as in lane 1, but no arabinose was added. The left hand lane contains protein standards. The gel was 12% SDS-PAGE run in the Tricine buffer system. The bracketed band at ca. 34 kDa is BioC. The band immediately below the BioC band may be a protease-processed form of BioC since strain MG1655 contains the OmpT protease which the E. coli B (BL21) strain generally used for protein production lacks.
4. Discussion
The seemingly straightforward construction of strain BW25113 Δ9 lysogenized with a λ prophage lacking the minimal cos site required more manipulations than anticipated. This was due to the presence in the available strains of sequences other than the intended target of a single λ prophage inserted into attB. The abundance of polylysogens was another complicating factor.
The key tool in solving these problems was the defective prophage-containing chromosomal segment of strain DY329 in which selectable and non-revertible host markers are present on both sides of a defective λ prophage. Transduction of this segment into strain BW25113 Δ9 gave strain CY2113 that allowed efficient selection of valid Δcos::cml prophages from the background of polylysogens and other undesired insertions. Moreover, the λcI repressor encoded by the CY2113 prophage prevented lytic growth of λ prophages subsequently introduced by P1 transduction during selection for Δcos::cml prophages. The CY2113 prophage lacks the λ late genes and thus the S7 and Δcos mutations of the incoming prophage cannot be repaired by recombination.
The original disrupted cos prophages (Cronan, 2003) were constructed to remedy a situation in our work on lipoic acid synthesis in E. coli (Zhao et al., 2003) and provides an illustration of one use of in vivo cosmid packaging. Genetic analyses indicated that this covalently attached cofactor is assembled on its cognate proteins. To test this in vitro we wished to accumulate the proposed octanoyl-protein intermediate in the absence of the sulfur insertion enzyme, LipA. The straightforward approach was a tightly controlled promoter, but in testing six such promoters in their least active states, all were found to produce sufficient LipA for normal growth of a ΔlipA strain. The obvious alternative approach was to introduce the lipA gene after accumulation of the intermediate. However, essentially all of the cells must receive the gene because any that did not would give background in the chemical analyses to follow. The efficiency required ruled out chemical transformation (only about 10% of cells take up DNA) and electroporation (half of the cells are killed) and hence we developed the cosmid procedure. In the lipoic acid synthesis work we coped with the infectious phages that contaminated the cosmid particle preparations by constructing a λ lysogen of the recipient strain (Zhao et al., 2003). This step can now be eliminated by use of the new packaging strain.
Studier and coworkers(Studier and Moffatt, 1986; Studier et al., 1990) developed a powerful and widely used system to direct high level and selective expression of plasmid-borne genes using T7 RNA polymerase. However, genes whose products are toxic to growth of E. coli host strains remain problematical (Studier and Moffatt, 1986; Studier et al., 1990). Although expression and utilization of T7 RNA polymerase can be tightly regulated, residual expression of toxic proteins can lead to cell death or loss (or rearrangement) of the encoding plasmid such that the protein is not produced. In extreme cases expression plasmids cannot be introduced into host cells encoding T7 RNA polymerase; the transformants perish during colony formation (Studier and Moffatt, 1986; Studier et al., 1990). As a general remedy for toxicity a λ phage called CE6 that encodes T7 RNA polymerase was constructed (Studier and Moffatt, 1986) and is commercially available (thereby illustrating the incidence of toxic proteins). λCE6 infection allows plasmids encoding toxic genes driven by a T7 promoter to be carried in any λ-sensitive E. coli strain lacking T7 RNA polymerase. λCE6 infection results in a transient pulse of T7 RNA polymerase synthesis before transcription from host and phage promoters is shut off by massive transcription of the gene of interest by T7 polymerase (Studier and Moffatt, 1986). Although λCE6 infection can give protein expression levels similar to those obtained in host strains expressing plasmid\or chromosomally-encoded T7 RNA polymerase, phage stocks must be prepared, purified and titered. Another drawback is that an appreciable fraction of the host cells remain uninfected by λCE6 (Studier and Moffatt, 1986). These cells continue growth (unlike the infected cells) and upon prolonged incubation take over cultures. This dictates a brief period of protein expression in order to retain the selective expression of target proteins given by the T7 system (Studier and Moffatt, 1986). However, appreciable overexpression of some proteins requires long expression times (sometimes overnight). λCE6 utilization is also hampered by a present day lack of experience with phage techniques due to replacement of λ vectors by high-capacity plasmid vectors (e.g., bacterial artificial chromosomes). In contrast, plasmid cloning is the dominant technique of molecular genetics and packaging of T7 RNA polymerase-encoding plasmids into λ phage particles provides a replacement for λCE6. Moreover, the cells that fail to become transduced are sensitive to chloramphenicol and thus addition of the antibiotic will prevent their growth allowing long intervals of protein expression, a marked advantage over λCE6.
In the previous paper (Cronan, 2003) I argued that cosmid transduction would be a very rapid process, but the reader should note that this is not the case. The prior argument was based on the work of Schwartz (Schwartz, 1976). However, more recent data (Moldovan et al., 2007; Shao and Wang, 2008) indicate that the rates of λ adsorption reported by Schwartz are in error and that the actual rates are 10 to 100-fold lower. Since no original data were given in the Schwartz report, the source of error is unknown (Dr. Schwartz is deceased). It is possible that Schwartz used a phage of superior adsorbing ability (the λ isolate was not stipulated). If so, that phage has been lost. The present day laboratory wild-type λ, called λ Papa, is a recombinant that adsorbs more poorly than the original λ isolated from E. coli K-12. The λ Papa isolate lacks the auxiliary tail fiber due to a frameshift in the stf gene (Hendrix and Duda, 1992). However, although restoration of the auxiliary tail fiber increases the adsorption rate of the laboratory λ by 7 to 8-fold (Shao and Wang, 2008), the rates are still far below those reported by Schwartz.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant AI15650 from the National Institute of Allergy and Infectious Diseases (NIAID). I thank Vandana Chakravartty and Steven Lin for their interest and assistance.
Abbreviations are
- Ctu
cosmid transducing unit
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arber W. Polylysogeny for bacteriophage lambda. Virology. 1960;11:250–272. [Google Scholar]
- Arber W, et al. Experimental Methods for Use with Lambda. In: Hendrix R, et al., editors. Lambda II. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1983. [Google Scholar]
- Casadaban MJ, et al. Beta-galactosidase gene fusions for analyzing gene expression in Escherichia coli and yeast. Methods Enzymol. 1983;100:293–308. doi: 10.1016/0076-6879(83)00063-4. [DOI] [PubMed] [Google Scholar]
- Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- Casjens SR. The DNA-packaging nanomotor of tailed bacteriophages. Nat Rev Microbiol. 2011;9:647–657. doi: 10.1038/nrmicro2632. [DOI] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Cronan JE. Cosmid-based system for transient expression and absolute off-to-on transcriptional control of Escherichia coli genes. J Bacteriol. 2003;185:6522–6529. doi: 10.1128/JB.185.22.6522-6529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugelavichius R, et al. Formation of ion channels in the Escherichia coli cytoplasmic membrane after exposure to bacteriophages T4 and lambda. Biokhimiia. 1987;52:1059–1067. [PubMed] [Google Scholar]
- Enquist LW, Weisberg RA. A genetic analysis of the att-int-xis region of coliphage lambda. J Mol Biol. 1977;111:97–120. doi: 10.1016/s0022-2836(77)80117-4. [DOI] [PubMed] [Google Scholar]
- Feiss M, et al. Cosmid DNA packaging in vivo. Gene. 1982;17:123–130. doi: 10.1016/0378-1119(82)90064-6. [DOI] [PubMed] [Google Scholar]
- Folkmanis A, Freifelder D. Studies on lysogeny in Escherichia coli with bacteriophage lambda. Physical observation of the insertion process. J Mol Biol. 1972;65:63–73. doi: 10.1016/0022-2836(72)90492-5. [DOI] [PubMed] [Google Scholar]
- Freifelder D, et al. Requirements for insertion of bacteriophage lambda DNA into the DNA of Escherichia coli. J Mol Biol. 1975;91:401–408. doi: 10.1016/0022-2836(75)90268-5. [DOI] [PubMed] [Google Scholar]
- Freifelder D, Kirschner I. The formation of homoimmune double lysogens of phage lambda and the segregation of single lysogens from them. Virology. 1971;44:633–637. doi: 10.1016/0042-6822(71)90378-3. [DOI] [PubMed] [Google Scholar]
- Freifelder D, Levine EE. The formation of polylysogens during infection of Escherichia coli with bacteriophage lambda. Virology. 1975;63:428–437. doi: 10.1016/0042-6822(75)90315-3. [DOI] [PubMed] [Google Scholar]
- Hendrix RW, Duda RL. Bacteriophage lambda PaPa: not the mother of all lambda phages. Science. 1992;258:1145–1148. doi: 10.1126/science.1439823. [DOI] [PubMed] [Google Scholar]
- Jacobs WR, et al. In vivo repackaging of recombinant cosmid molecules for analyses of Salmonella typhimurium, Streptococcus mutans, and mycobacterial genomic libraries. Infect Immun. 1986;52:101–109. doi: 10.1128/iai.52.1.101-109.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AL. Turbidity measurements of bacterial cultures in some available commercial instruments. Anal Biochem. 1970;38:252–359. doi: 10.1016/0003-2697(70)90174-0. [DOI] [PubMed] [Google Scholar]
- Kolisnychenko V, et al. Engineering a reduced Escherichia coli genome. Genome Res. 2002;12:640–647. doi: 10.1101/gr.217202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. Thesis. Microbiology. University of Illinois; Urbana-Champaign: 2012. Biotin synthesis in Escherichia coli. [Google Scholar]
- Miller JH. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Miwa T, Matsubara K. Identification of sequences necessary for packaging DNA into lambda phage heads. Gene. 1982;20:267–279. doi: 10.1016/0378-1119(82)90045-2. [DOI] [PubMed] [Google Scholar]
- Miwa T, Matsubara K. Formation of oligomeric structures from plasmid DNA carrying cos lambda that is packaged into bacteriophage lambda heads. J Bacteriol. 1983;153:100–108. doi: 10.1128/jb.153.1.100-108.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan R, et al. On kinetics of phage adsorption. Biophys J. 2007;93:303–15. doi: 10.1529/biophysj.106.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson TA, et al. Improved bacterial hosts for regulated expression of genes from lambda pL plasmid vectors. Gene. 1993;132:83–87. doi: 10.1016/0378-1119(93)90517-7. [DOI] [PubMed] [Google Scholar]
- Rao VB, Feiss M. The bacteriophage DNA packaging motor. Annu Rev Genet. 2008;42:647–681. doi: 10.1146/annurev.genet.42.110807.091545. [DOI] [PubMed] [Google Scholar]
- Schwartz M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J Mol Biol. 1976;103:521–536. doi: 10.1016/0022-2836(76)90215-1. [DOI] [PubMed] [Google Scholar]
- Shao Y, Wang IN. Bacteriophage adsorption rate and optimal lysis time. Genetics. 2008;180:471–482. doi: 10.1534/genetics.108.090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg N. Bacteriophage P1 cloning system for the isolation, amplification, and recovery of DNA fragments as large as 100 kilobase pairs. Proc Natl Acad Sci U S A. 1990;87:103–107. doi: 10.1073/pnas.87.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Studier FW, et al. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Umene K, et al. Packaging of ColE1 DNA having a lambda phage cohesive end site. Mol Gen Genet. 1978;159:39–45. doi: 10.1007/BF00401746. [DOI] [PubMed] [Google Scholar]
- Umene K, et al. Lambda bacteriophage-mediated transduction of ColE1 deoxyribonucleic acid having a lambda bacteriophage-cohesive end site: selection of packageable-length deoxyribonucleic acid. J Bacteriol. 1979;139:738–477. doi: 10.1128/jb.139.3.738-747.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Cryptic prophages help bacteria cope with adverse environments. Nat Commun. 2010;1:147. doi: 10.1038/ncomms1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wycuff DR, Matthews KS. Generation of an AraC-araBAD promoter-regulated T7 expression system. Anal Biochem. 2000;277:67–73. doi: 10.1006/abio.1999.4385. [DOI] [PubMed] [Google Scholar]
- Yu D, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, et al. Assembly of the covalent linkage between lipoic acid and its cognate enzymes. Chem Biol. 2003;10:1293–1302. doi: 10.1016/j.chembiol.2003.11.016. [DOI] [PubMed] [Google Scholar]