

Abstract
Enzyme catalysis is central to almost all biochemical processes, speeding up rates of reactions to biological relevant timescales. Enzymes make use of a large ensemble of conformations in recognizing their substrates and stabilizing the transition states, due to the inherent dynamical nature of biomolecules. The exact role of these diverse enzyme conformations and the interplay between enzyme conformational dynamics and catalysis is, according to the literature, not well understood. Here, we use molecular dynamics simulations to study human cyclophilin A (CypA), in order to understand the role of enzyme motions in the catalytic mechanism and recognition. Cyclophilin A is a tractable model system to study using classical simulation methods, because catalysis does not involve bond formation or breakage. We show that the conformational dynamics of active site residues of substrate-bound CypA is inherent in the substrate-free enzyme. CypA interacts with its substrate via conformational selection as the configurations of the substrate changes during catalysis. We also show that, in addition to tight intermolecular hydrophobic interactions between CypA and the substrate, an intricate enzyme-substrate intermolecular hydrogen-bonding network is extremely sensitive to the configuration of the substrate. These enzyme-substrate intermolecular interactions are loosely formed when the substrate is in the reactant and product states and become well formed and reluctant to break when the substrate is in the transition state. Our results clearly suggest coupling among enzyme-substrate intermolecular interactions, the dynamics of the enzyme, and the chemical step. This study provides further insights into the mechanism of peptidyl-prolyl cis/trans isomerases and the general interplay between enzyme conformational dynamics and catalysis.
Introduction
The importance of enzymes in biology cannot be overstated. Enzymes are critical to a broad range of functions, including metabolism (1), gene regulation (2), cell survival (3), intracellular communication (4), and hormone regulation (5). They catalyze specific biochemical reactions, increasing reaction rates by many orders of magnitude to more biologically relevant timescales. Enzymes can act to form or break covalent bonds, perform acid-base chemistry, transfer functional groups, and switch configurations around bonds to yield isomers (6–8). Certain enzymes can perform these functions alone, while others need cofactors or prosthetic groups to assist in the catalytic function. Fully understanding the mechanism of action of enzymes could provide valuable insights into engineering proteins and designing new drugs.
In vitro experiments have provided valuable insights into the mechanisms of enzymes (9). However, detailed atomistic understanding of the mechanism along the catalytic pathway is not always possible with current experimental techniques. Therefore, computational simulations are routinely used to complement experiments (10), usually starting from well-characterized atomic x-ray crystal structures. Nonetheless, classical molecular dynamics (MD) presents several challenges in studying enzyme mechanisms with catalytic turnover times in the millisecond timescale. In addition to the submicrosecond timescale limitation, molecular dynamics cannot be used to study chemical reactions involving bond formation and breakage without the use of more demanding hybrid quantum-mechanical methods. As of this writing, it is not believed possible to directly simulate most enzymatic reactions without using some form of coarse-graining or advanced sampling techniques.
Peptidyl-prolyl cis-trans isomerases (PPIases) are a class of enzymes that take part in many cellular processes and catalyze their reactions without any bond formation or breakage. This characteristic makes them tractable and ideal to study using classical molecular dynamics. PPIases catalyze cis-trans isomerization of backbone peptidyl-prolyl ω-bonds of their various protein substrates. The reaction coordinate of the chemical step is defined by the backbone peptide ω-bond angle. The cis, trans, and transition-state configurations of the substrate along the reaction coordinate are well defined at ∼0°, ±180°, and 90°, respectively. Also, the cis-trans interconversion can be simulated directly using accelerated molecular dynamics, without any conformational bias, as was previously shown (11,12). PPIases consist of cyclophilins, FK-506-binding proteins, and parvulins (13). The tertiary structure of the catalytic domain of cyclophilins is structurally conserved among all of the familial isoforms (14). Human cyclophilin A (CypA), the smallest prototypic cyclophilin of ∼18 human isoforms, is the most studied and characterized isoform (15). Uncatalyzed prolyl cis-trans isomerization has an activation free-energy barrier of ∼20 kcal/mol (∼84 kJ/mol) (16) and a half-life on the second timescale (17,18). Human CypA speeds up the reaction rate from seconds to milliseconds (19). CypA catalyzes the peptide bond of a -X-Pro- motif (where X is any amino acid), and differences in catalytic turnover rates are mainly due to the identity of the amino acid in the X position (19,20).
Human CypA has a range of specific functions in vivo. The immunosuppressive drug cyclosporine A (CsA) binds to CypA, and the CypA-CsA complex inhibits calcineurin, suppressing the transcription of cytokine genes by inhibiting calcineurin’s native phosphatase function (13). CypA is the first human protein that has been found to be both enclosed within the HIV-1 virion and crucial for viral replication (21,22). An interaction between CypA and the HIV-1 capsid core protein, CAN, facilitates viral replication by accelerating destruction of the capsid (22,23). Hepatitis C virus also uses CypA to replicate by forming a critical contact with the HCV NS5B RNA polymerase (24). A role of CypA in signal transduction involves regulating the function of the prolactin receptor in mammary cells, impacting the interaction of the prolactin receptor with Janus-activated tyrosine kinase (25). Also, CypA can form a complex with Interleukin-2-tyrosine kinase inside of Jurkat T-cells, which is disrupted upon addition of CsA (26). Cyclophilins are also involved in protein folding (13) and oncogenesis (25,27).
The exact catalytic mechanism of CypA is not fully understood. Several hypotheses have been presented to explain the greater than five orders-of-magnitude speedup in prolyl isomerization by the enzyme (25,28–33). It has been suggested that conformational heterogeneity that occurs during enzyme catalysis provides the means by which an enzyme complements its substrate (34). Therefore, in order to fully understand the mechanism of CypA, there is a need to fully understand how enzymes make use of a large ensemble of conformations in recognition and catalysis at different points along the chemical step. We have therefore simulated the substrate-free enzyme and enzyme-substrate complexes of the cis, trans, and transition-state configurations of the substrate—three important segments along the catalytic pathway. We have also carried out accelerated MD simulations on the enzyme-substrate complex in order to freely sample cis-trans isomerization during catalysis and investigate the coupling between the conformational dynamics of CypA and the chemical step. Altogether, we carried out >2 μs of MD simulations in full atomistic detail, sampling conformational changes beyond the nanosecond timescale. Moreover, these computational approaches provide a way to study the short-lived ensemble of conformations of the enzyme-substrate transition-state complex. These studies provide further insight into the importance of enzyme flexibility in catalysis, as well as the coupling between the chemical step and the stabilizing polar and nonpolar intermolecular interactions.
Methods
All simulations were carried out using the AMBER 10 suite of programs (35) in explicit TIP3P water (36) using the PARM99SB (37) modified version of the force-field parameters from Cornell et al. (38). Additional modifications to the dihedral parameters for the peptide ω-bond angle were also employed (39). A 1.58 Å resolution x-ray crystal structure with PDB:1AWR was used for the simulations (23). An experimentally well-studied substrate analog, Ace-Ala-Ala-Pro-Phe-Nme (AAPF), was used in these studies (19,20,31,32,40–43). The Ace-AAPF-Nme substrate analog was introduced by keeping common backbone and side-chain atoms of the substrate analog (HAGPIA) in the PDB file and adding the missing atoms using the Xleap module in AMBER. The complex was then solvated with ∼5500 TIP3P water molecules and was neutralized with four chloride ions. The potential energy of the system was initially minimized for 1000 steps with a harmonic constraint of 100 kcal/mol/Å2 applied to the atoms of the protein, followed by two short (400-ps) MD simulations with harmonic constraints of 50 kcal/mol/Å2 and 25 kcal/mol/Å2, respectively, applied to all of the atoms of the protein. The system was then equilibrated for an additional 200 ps without any constraints using the isothermal-isobaric ensemble at 300 K and 1 bar. The Pmemd module in AMBER 10 was used to carry out all of the conventional MD simulations. An integration time step of 0.002 ps was used to integrate Newton’s equation of motion.
The SHAKE algorithm (44) was used to restrain all bonds involving a hydrogen atom during the simulations. Langevin dynamics was used to maintain the temperature at 300 K with a collision frequency of 1 ps−1. This temperature and a constant pressure of 1 bar were used throughout all simulations. The nonbonded cutoff distance was set to 9 Å during all simulations. The long-range electrostatic interactions were treated using particle-mesh Ewald summation (45–47). The ω-bond angle of the substrate in the transition-state complex was maintained at ∼90° using a flat bottom-well torsional restraint with a force constant of 1000 kcal/mol/rad2 between 89° and 91°. Restraints were not required to maintain the substrate in the trans and cis configurations in their enzyme-substrate complexes, because a high barrier separates the two low-energy states. The substrate in the crystal structure was in the trans configuration. Thus, the cis configuration of the substrate was equilibrated with the same torsional restraint in order to shift the substrate from the trans configuration to the cis, and subsequently simulated with no restraint.
The aggregate simulation time for all of the conventional MD simulations of substrate-free CypA and the enzyme-substrate complexes exceeded 1.5 μs. Four independent simulations were carried out for each enzyme-substrate complex. Each simulation was carried out for at least 110 ns. The first 10 nanoseconds were discarded as part of the equilibration phase. One long 350-ns simulation was carried out on substrate-free CypA, using a similar setup and equilibration procedure as was done for the substrate-bound complexes.
All accelerated MD simulations (48) were carried out using a modified version of the Pmemd module in AMBER 10, in order to accelerate the rate of cis-trans isomerization of the substrate while in complex with the enzyme. Eight independent accelerated MD simulations were carried out for a total of ∼1 μs of simulation time. The total torsional potential of the substrate was selectively boosted (11), using a boost energy, E, of 60 kcal/mol above the average total dihedral energy calculated after equilibration and a tuning parameter, α, of 10 kcal/mol. Each configuration was reweighted using the strength of the Boltzmann factor of the bias potential energy, eβΔV(r), calculated on-the-fly during the simulation to calculate the probability distributions (49).
Principal component analysis (PCA) was carried out using the Ptraj module in AMBER (35). The implementation of this method has been extensively discussed (50–54), and Ptraj was used to calculate and diagonalize the covariance matrix. The Ptraj module was also used to calculate the torsional angles, root-mean-square fluctuations, and hydrogen-bonding distances. For residues containing equivalent δ-carbons (such as Leu or Phe), the Cδ1 atom was selected when measuring the torsional angle. The binding free energies were estimated using the molecular mechanics/Poisson-Boltzmann surface-area approach (55,56). The relative changes in translational, rotational, and conformational entropies were assumed to be negligible in estimating the relative binding free energies.
Results and Discussion
Conformational selection in CypA recognition during catalysis
In general, conformational selection (57–60) and induced fit (61) can be used to describe the mechanism of enzyme-substrate recognition. Conformational selection implies that equilibria between weak- and tight-binding conformations of the substrate-free enzyme exist before substrate binding, whereas substrate binding is a prerequisite for the formation of a tightly bound enzyme-substrate complex in the induced-fit mechanism (62). However, these two mechanisms are limiting extremes for dynamical systems, and it is difficult to ascribe either one as the sole contributor to biomolecular recognition (59,63). Also, these two mechanisms characterize conformational changes in enzymes with little regard given to conformational changes in the substrate. It has been previously noted that the free-energy landscape of the enzyme and substrate are transformed upon complex formation (59). Thus, variation in the conformation of the substrate should also be considered when describing the conformational heterogeneity of the complex because the conformations of the enzyme can affect those of the substrate, and vice versa.
We have used principal component analysis (PCA) to characterize the mechanism of recognition of CypA upon binding the cis, trans, and transition-state configurations of the substrate. PCA allows us to project the conformational phase space sampled by the active site residues of the substrate-free and substrate-bound enzyme using a reduced set of degrees of freedom. It helps us to determine if the conformations of the active site residues of the substrate-free enzyme can effectively bind the substrate by revealing similarities between substrate-free and substrate-bound ensembles of the enzyme conformations (Fig. 1). Active site residues of CypA consist of Arg55, Phe60, Met61, Gln63, Ala101, Asn102, Ala103, Phe113, Leu122, and His126. These residues were selected because they are no more than 4 Å away from the peptide ω-bond angle and form the binding cavity for the substrate. Also, most of these residues are fully conserved across species and have been identified as participating in substrate turnover (64).
Figure 1.
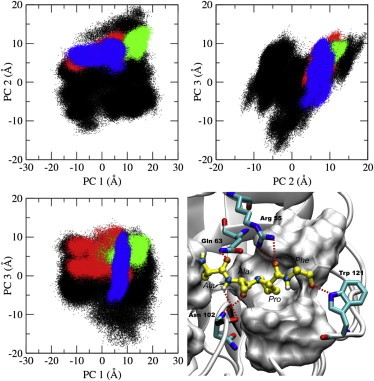
Principal component analysis (PCA) of substrate-free CypA and substrate-bound CypA complexes. The top three principal components’ dominant motions are shown, with each data point representing a conformation of the active site residues. Depicted are the substrate-free enzyme (black), the trans-CypA complex (red), the transition-state-CypA complex (green), the cis-CypA complex (blue), and the substrate-bound CypA complex with hydrophobic active-site residues (on white surface). Also depicted are the active site residues (Arg55, Gln63, and Asn102) and Trp121 that form hydrogen bonds with the substrate (sticks), and the substrate in the transition state configuration (yellow).
Fig. 1 shows that the active site residues of substrate-free CypA sample a large conformational space involving several rotameric states (Fig. 2 and see Fig. S1 in the Supporting Material). Upon binding the substrate, the active site residues of CypA lose a tremendous amount of conformational freedom that was present in the substrate-free enzyme. The phase space sampled by the active site residues of the enzyme-substrate complexes in the cis and trans configurations (ground states) of the substrate is slightly broader than that of the transition-state complex. The conformational-space of the transition-state ensemble is compact, with few differences in the enzyme-substrate intermolecular interactions from one conformation to the other. These enzyme-substrate intermolecular interactions involve several key hydrogen bonds, as shown in Fig. 1 D. These well-optimized hydrogen bonds localize the transition-state ensemble of the enzyme conformations to a single region of phase space, as we show later. It can be seen that the active site of the substrate-free enzyme also samples the majority of the transition-state conformations (Fig. 1).
Figure 2.
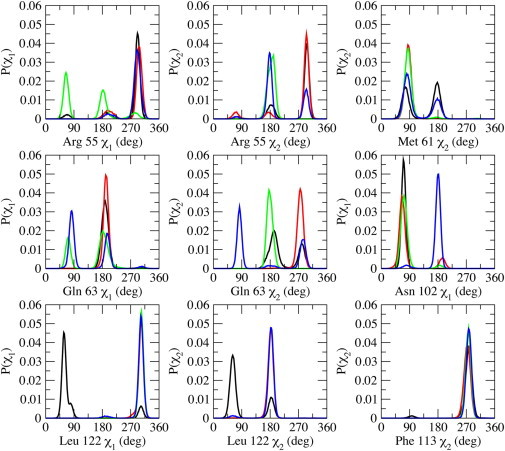
Probability distributions of side-chain torsional angles of active site residues, Arg55, Met61, Gln63, Asn102, Phe113, and Leu122 in substrate-free (black) and substrate-bound CypA: trans (red), transition state (green), and cis (blue). The χ1-angle corresponds to the atoms N-Cα-Cβ-Cγ, and the χ2-angle corresponds to the atoms Cα-Cβ-Cγ-Cδ.
The active site of the enzyme does not necessarily have to be induced to some exclusive conformations for catalysis to occur. The active site conformations of the ground-state complexes overlap quite well with each other, while the transition state shares a smaller subset of conformations with the ground states. The substrate-bound active site conformations of the enzyme are subsets of that of the substrate-free enzyme conformations based on the top three principal components (Fig. 1 and see Fig. S2) that represent ∼70% of the motions of the top 10 principal components of the active site residues (see Fig. S3). The connection between the substrate-free and substrate-bound active site conformations is indicative of an existing equilibrium between weak- and tight-binding conformations of the enzyme. Localization of the active-site residues of the bound ensembles is characteristic of a population shift toward a subset of the substrate-free enzyme conformations. The results suggest that CypA has evolved to complementarily shift its active-site conformation alongside the configuration of the substrate with only slight changes to the rotameric state of the active site residues as the reaction progresses. Whether CypA binds the substrate in the trans, transition-state, or cis configuration, the needed enzyme conformations already exist in the ensemble of the substrate-free enzyme. Thus, the binding mechanism of CypA is predominantly conformational selection, as was previously suggested by NMR studies (40,65).
Examining the rotameric states of the active site residues in the substrate-free and the different substrate configurations of the substrate-bound CypA suggests similar conclusions (Fig. 2). In general, each ensemble of the bound complexes has its own unique distribution of rotamers for the active site residues. Various intramolecular and intermolecular interactions in the enzyme affect the conformational preference of the active site residues, which are also dependent on the state of the substrate. The rotamers sampled by the active site residues of the substrate-free enzyme overlap with the rotamers sampled by the substrate-bound enzyme complexes, also demonstrating that the bound conformations of the enzyme are a subset of the free enzyme conformations. Active site residues that form hydrogen-bonding interactions with the substrate tend to sample more rotameric states than hydrophobic residues. Unlike the hydrophilic residues, the hydrophobic residues in the bound states of the enzyme predominantly sample a single rotameric state and are less sensitive to the configuration of the substrate.
The most flexible active site residues are Arg55, Gln63, and Asn102—three residues that form key hydrogen-bonding interactions with the substrate (Fig. 1 D). Arg55 has been shown to be important for substrate recognition and catalysis by forming a bifurcated hydrogen bond with the carbonyl oxygen of proline of the substrate (11,31,33). Gln63 and Asn102 also participate in hydrogen-bonding interactions with the substrate. However, only the hydrogen-bonding interactions between residues Arg55 and Asn102 and the substrate have been identified as being important in stabilizing the transition state (31). The backbone amine group of Asn102 forms a hydrogen bond with the carbonyl oxygen of Ala in the -Ala-Pro- motif only in the transition-state and cis enzyme-substrate complexes. The side-chain amide group of Gln63 forms a hydrogen bond with the carbonyl oxygen of the Ala residue preceding the -Ala-Pro- motif of the Ace-Ala-Ala-Pro-Phe-Nme substrate analog used in this study.
Phe113 sits at the base of the proline-binding pocket and has been suggested to play a key role in catalyzing cis-trans isomerization (40). Phe113 can rotate in the free enzyme, and rotation to the minor rotameric state was suggested to be coupled to the catalytic step (40). Our studies suggest that, once the substrate binds to the enzyme, the side chain of Pro in the substrate pushes directly against the phenyl group of Phe113. This keeps Phe113 localized to the same rotameric state regardless of the conformation of the substrate (Fig. 2 and see Fig. S1). The minor rotameric state of Phe113 seems to obstruct the proline-binding pocket. Interestingly, the less-active Ser99Thr mutation of CypA was shown to increase the population of the minor rotameric state of Phe113, which sits on top of Ser99 (40). Another hydrophobic residue, Leu122, in the active site forms intermolecular contact with the substrate in the proline-binding pocket and predominantly samples only one rotameric state in all of the substrate-bound enzyme complexes (Fig. 2). In the substrate-free enzyme, Leu122 participates in loose hydrophobic contacts with other active site residues in the proline-binding pocket, visiting more than one rotameric state.
Substrate binding alters the conformational dynamics of CypA beyond the active site
In addition to the conformational changes observed in the active site of CypA upon substrate binding, the dynamics and fluctuations of the enzyme beyond the active site are also altered, as shown in Fig. 3. Here, Fig. 3 shows the average percent-change in the root-mean-square fluctuations of the enzyme backbone atoms upon binding the substrate. Enzyme residues that become more localized upon binding have positive change; conversely, residues that become more flexible upon binding have negative change. The loop region containing residues 75–85 becomes overall less flexible upon binding the substrate in the cis and trans ground states (orange; Fig. 3). This region is part of a larger loop (residues 66–96) that has been reported to undergo fast conformational exchange in the substrate-free enzyme (40). It appears that Lys82 acts as a hinge for this loop, becoming more flexible upon binding the substrate in the transition-state configuration. This residue has also been found to undergo significant deviation upon binding in other studies (29,66). These results suggest that the motions of this loop have significant impact on complex formation. Several residues of CypA within the loop region 75–85 are well conserved across species, with the exception of residues Lys76, Glu81, and Glu84 (64). Another loop region (residues 147–155) exhibits smaller fluctuations upon substrate binding (violet; Fig. 3). This region, in combination with the α-helix from residues 136–146, acts as a hinge region to the two β-sheets contributing to the closed β-barrel fold. The stability of this loop region upon substrate binding suggests that the two β-sheets become more compact in the enzyme-substrate complex and less likely to separate as much as would be expected in the breathing motions of the substrate-free enzyme.
Figure 3.
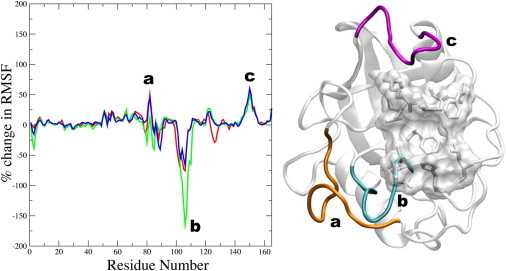
Average percent-change in root-mean-square fluctuations (left) of all the backbone atoms of CypA (right) upon substrate binding (relative to the substrate-free enzyme). A negative change signifies enhanced fluctuation upon substrate binding and a positive change signifies reduced fluctuation upon substrate binding. (Left) The trans complex (red), the transition-state complex (green), and the cis complex (blue). (Right) Enzyme regions with significant changes, depicted on the model structure, are (a) residues 75–85 (orange), (b) residues 101–110 (cyan), and (c) residues 147–155 (violet). The active site residues of CypA are also shown (transparent surface and stick representations).
Of particular interest is the loop region consisting of residues 101–110 (cyan; Fig. 3). This region becomes more flexible upon substrate binding and most flexible in the transition-state enzyme-substrate complex. Moreover, these residues are well conserved across different species (64) and have been shown to contribute to the dominant motions of CypA (67). The motion of this loop may be critical to catalytic turnover, and this motion may be required to introduce enough deviation to allow Asn102 to form hydrogen bonds with the substrate. This hypothesis may be tested experimentally by modifying the dynamics and flexibility of that loop to determine how the catalytic turnover rate is impacted. Very few deviations are apparent in the backbone of the active site residues upon binding the substrate in the different configurations, as opposed to the relatively large side-chain rearrangements that are observed. The results therefore suggest that the backbone of the active site in the substrate-free enzyme is preorganized to bind the substrate in its different configurations that requires rearrangements of several key side chains during catalysis. Also, many of these loop residues have been shown to contribute to the top three vibrational modes of CypA (64,66).
Enzyme-substrate intermolecular interactions are tightly coupled to the chemical step
It is well established that enzymes lower the free energy barrier of a reaction by stabilizing the transition state during catalysis (8). However, it is not always clear as to how exactly this barrier reduction is achieved. In the active site of CypA, the conformations that complement the transition state are very ordered, resulting in a low entropy, yet high affinity complex. This order is achieved due to key enzyme-substrate intermolecular interactions. The key intermolecular interactions of CypA include the hydrophobic contacts between the substrate and the active site cavity and the formation of several specific enzyme-substrate hydrogen bonds (Fig. 1 D). The smaller conformational space sampled by active site residues of the substrate-bound complexes (as compared to the substrate-free enzyme) is a result of the formation of these intermolecular interactions.
Initially, two main hydrogen bonds involving Arg55 and Asn102 of CypA were identified as being responsible for stabilizing the transition state relative to the ground state (11). These two hydrogen bonds flank the proline residue of the -Ala-Pro- motif of the substrate analog. In this work, we have identified at least four intermolecular hydrogen bonds between the enzyme and substrate that are deemed to be important in stabilizing the transition state (Figs. 1 D and 4). The bifurcated hydrogen bond between the guanidinium group of Arg55 and the carbonyl oxygen of proline in the -Ala-Pro- motif of the substrate continually forms and breaks in the ground (cis and trans) states. However, this hydrogen bond is always well formed in the transition-state complex and is almost never broken (Fig. 4 and see Fig. S4). While Arg55 can undergo fast conformational changes in the free enzyme (29), it becomes less mobile in the enzyme-substrate complexes, especially upon binding the transition state.
Figure 4.
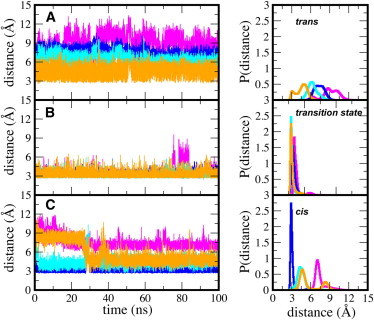
Enzyme-substrate intermolecular hydrogen bonds between CypA and the substrate in the (A) trans, (B) transition-state, and (C) cis configurations. Hydrogen bonds are measured from heavy atom to heavy atom. (Magenta curve) Contact between the Arg55 guanidinium carbon and the carbonyl oxygen of the substrate Pro. (Royal-blue curve) Contact between the backbone nitrogen of Asn102 and the carbonyl oxygen of Ala of the -Ala-Pro- motif. (Cyan curve) Contact between the backbone oxygen of Asn102 and nitrogen of Ala of the -Ala-Pro- motif. (Orange curve) Contact between the side-chain amino nitrogen of Gln63 and the carbonyl oxygen of Ala, preceding the -Ala-Pro- motif.
Similarly, Gln63 and Asn102 are involved in several hydrogen-bonding interactions with the substrate that are loosely formed in the cis and trans states, but well formed in the transition state. The side-chain amide proton of Gln63 forms a hydrogen bond with the carbonyl oxygen of the Ala residue preceding the -Ala-Pro- motif of the substrate (Fig. 1 D). This hydrogen-bonding interaction is difficult to form in the trans and cis states. However, in the transition state, it is difficult to break (Fig. 4 and see Fig. S4). The backbone amine group of Asn102 forms a tight hydrogen-bonding interaction with the carbonyl oxygen of alanine in the -Ala-Pro- motif of the substrate in the cis and transition states, but not in the trans state. We believe this tightly formed hydrogen-bond interaction between Asn102 and the cis configuration is partly responsible for the higher binding affinity of the cis configuration over the trans configuration to the enzyme, as shown below and previously observed computationally (11) and experimentally (29,65).
In the substrate-free enzyme, Arg55 can move freely. In the enzyme-substrate complexes of the cis and trans states, Arg55 can either orient downward, interacting with the substrate; or upward (away from the active site), interacting mainly with residue Asn149 in a loop (violet; Fig. 3). In one out of the four independent normal MD simulations carried out on the enzyme-substrate complex when the substrate is in the trans state, Arg55 is consistently in the downward position, interacting with the substrate. In the other three simulations, Arg55 spends most of the time upward, away from the active site and substrate. In all of the independent normal MD simulations of the enzyme-substrate complex when the substrate is in the cis state, Arg55 sampled both downward and upward conformations. The interaction between Arg55 and the substrate in the transition state complex was hardly ever broken in all four independent normal MD simulations. The results suggest that the behavior of Arg55 is sensitive to the state of the substrate and changes along the catalytic pathway. To fully understand the conformational preference of Arg55, and other key residues, and determine how the intermolecular interactions are coupled to the state of substrate during catalysis, we carried out accelerated MD simulations on the enzyme-substrate complex. Accelerated molecular dynamics allowed us to observe the back-and-forth cis-trans isomerization of the catalytic process while sampling the conformational space of the enzyme.
The hydrogen-bonding interaction between Arg55 and the substrate is coupled to the chemical step along the reaction coordinate, ω, as can be seen in Fig. 5. Also, it is clear from Fig. 5 that, in the trans state, Arg55 has to overcome an energetic barrier of ∼5 kcal/mol in order to switch between the upward and downward conformers, unlike the cis configuration. This barrier is small in the cis enzyme-substrate complex, and Arg55 in the transition-state complex consistently stays in the downward position due to optimized contact with the substrate. These results are consistent in all eight, independent, accelerated MD simulations that were carried out. Fig. 5 is an average of the eight independent runs. The barrier observed in the trans state also explains why the hydrogen-bonding interaction between Arg55 and the substrate in one of the four independent normal MD simulations of the trans complex stayed formed during the entire simulation. Our results suggest that the barrier separating the formed and unformed state of the Arg55 with the substrate in the enzyme-substrate complex of the trans state exists because Arg55 can equally form long-lasting interactions downward (with the substrate) and upward (with Asn149), corresponding to the two observed wells in Fig. 5. Because there is a barrier separating the formed and the unformed states of the intermolecular interaction between Arg55 and the trans substrate, these simulations can get trapped in the formed or unformed well for a long period of time. It is interesting to see that the configuration of the substrate can directly affect the enzyme dynamics. Fig. 5 also confirms that the hydrogen-bonding interaction between the guanidinium group of Arg55 and the carbonyl of proline in the -Ala-Pro- motif of the substrate can form and break in the trans and cis states and is well formed as the substrate goes through the transition state.
Figure 5.
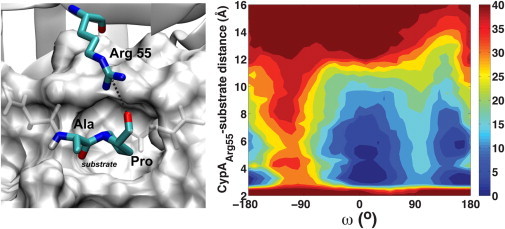
Coupling between the enzyme-substrate intermolecular hydrogen-bonding interaction with Arg55 and the chemical step. Contour plot (right) is in kcal/mol. Measured interaction (left) is depicted (dashed line). The -Ala-Pro- motif of the substrate is shown in the transition-state configuration, and the remaining residues of the substrate are also shown (white stick representation), as well as the active site residues (white surface representation).
The hydrogen-bonding interaction between Gln63 and the substrate is also coupled to the chemical step, as shown in Fig. 6. The hydrogen bond between Gln63 and the substrate readily forms and breaks when the substrate is in the trans or cis states, and stays consistently formed in the enzyme-substrate complex of the transition state (Fig. 6). Gln63 is completely conserved across the human cyclophilin isoforms (15) and across species (31,68), and our results suggest that it is primarily important for stabilizing the transition state of the substrate, along with Arg55 and Asn102, two other well-conserved amino acids of CypA (15,69,70). The suggested role of Gln63 in stabilizing the transition state can be tested by mutagenesis experiments or chemical modification of the side chain in order to abolish the hydrogen-bonding interaction with the substrate and measurements of the effect on the catalytic turnover rate.
Figure 6.

Coupling between the enzyme-substrate intermolecular hydrogen-bonding interaction with Gln63 and the chemical step. Contour plot (right) is in kcal/mol. Measured interaction (left) is shown (dashed line). The -Ala-Ala-Pro- motif of the substrate is shown with the -Ala-Pro- motif shown in the transition-state configuration, and the remaining residues of the substrate are also shown (white stick representation), as well as the active site residues (white surface representation).
CypA preferentially binds the substrate in the transition state
We have estimated the binding free energies of the enzyme-substrate complexes with different configurations of the substrate using the molecular mechanics/Poisson-Boltzmann surface area approach (55,56), in order to further understand how CypA speeds-up the rate of cis-trans isomerization (Fig. 7). The relative changes in conformational, translational, and rotational entropies are not included in these estimates and are assumed to be negligible. The average free energies of binding the substrate in the trans configuration are ∼−13.1 kcal/mol (∼−54.8 kJ/mol) in the simulation where Arg55 does not interact with the substrate and ∼−22.1 kcal/mol (∼−92.5 kJ/mol) in the simulation where Arg55 continuously interacts with the substrate, as shown in Fig. 7 A. The average free energies of binding the substrate in the cis and transition-state configurations are estimated to be ∼−24.1 kcal/mol (∼−100.8 kJ/mol) and ∼−31.4 kcal/mol (∼−131.4 kJ/mol), respectively. The enzyme binds the transition state better than the cis and trans states, as was previously shown (11,71). These results suggest that CypA is designed to preferentially bind and stabilize the transition state of the substrate. CypA lowers the free-energy barrier by ∼10 kcal/mol (∼42 kJ/mol), similar to estimates from Figs. 5 and 6 and in line with previous simulations and experiments (11,32,65), which is achieved by binding the transition-state configuration of the substrate better than the cis and trans states by this amount. Also, our results provide a quantitative estimate of the critical role of the interaction of Arg55 with the substrate in forming a stable enzyme-substrate complex.
Figure 7.
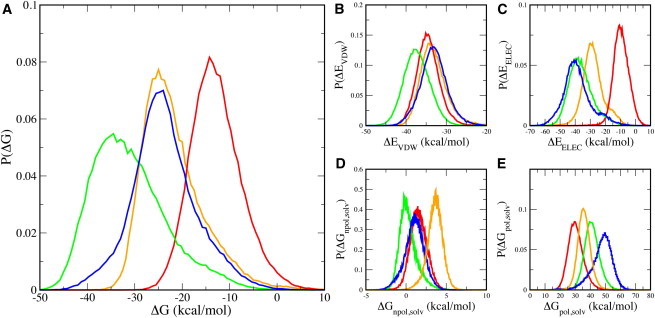
Probability distributions of free energies of binding the substrate in different configurations by CypA, when the substrate is in the trans (orange and red), transition-state (green), and cis (blue) configurations. Two sets of results are shown for the enzyme-substrate complex with the substrate in the trans configuration: one in which Arg55 maintains a hydrogen bond with the substrate (orange) and the other when Arg55 does not maintain a hydrogen bond with the substrate (red). The total free-energy change upon binding the different states of the substrate (A) and the individual components of the binding free energy change (B–E) are shown.
A breakdown of the energetic components (Fig. 7, B–E) reveals that the enzyme stabilizes the transition-state configuration using optimized van der Waals and electrostatic interactions. The trans substrate forms slightly better van der Waals contact with the enzyme than the cis substrate, but the trans substrate forms the worst electrostatic contacts with the enzyme out of the three configurations. Interestingly, when Arg55 forms its hydrogen bond with the substrate, the electrostatic contacts significantly improve (Fig. 7 C). The cis complex forms significantly better electrostatic contact with the enzyme than the trans complex, yet the cis complex forms the weakest van der Waals interactions. This implies that the cis complex formation is characterized by better hydrogen-bond formation, while the trans complex formation is characterized by a better grip on the proline ring. The difference in electrostatic contacts between the ground states is much greater than the difference in nonpolar contacts. This indicates that electrostatic contributions have the biggest impact on complex formation. The importance of electrostatic contacts in complex formation and catalytic turnover has been previously highlighted (72). Interestingly, and somewhat expected, the tighter the electrostatic interaction between the enzyme and the substrate (Fig. 7 C), the less favorable the change in the polar free energy of solvation (Fig. 7 E). However, the electrostatic and van der Waals interactions overcompensate for the unfavorable change in the polar solvation free energy. The change in nonpolar solvation free energy is relatively small (Fig. 7 D).
On the role of Trp121
Tryptophan 121 forms a hydrogen bond with the substrate, but does not help to carve out the hydrophobic proline-binding pocket. Therefore, it had not always been considered an active site residue, but mutating Trp121 has been shown to adversely affect the catalytic activity of CypA (42,73,74). The amine group of the indole ring can form a hydrogen bond with the backbone carbonyl oxygen of the residue (Phe) after proline of the -Ala-Pro- motif of the substrate (Fig. 8 C). The side chain of Trp121 predominantly populates a single rotameric state, mainly forming hydrophobic contact with the outside of the proline-binding pocket, and the hydrogen of the indole ring points toward the substrate (Fig. 8, A–C). Unlike the other residues that form hydrogen bonds with the substrate, the hydrogen-bonding interaction between Trp121 and the substrate is well formed in the trans enzyme-substrate complex and mostly formed in that of the transition state, but completely unformed in the complex of the cis state (Fig. 8, D and E). The variation in the hydrogen-bonding interaction of Trp121 is mainly dependent on the configuration of the substrate and not on the dynamics of the residue. A fluorescence study of substrate-free CypA and CypA-CsA complex resulted in a twofold increase in Trp121 fluorescence upon binding CsA, suggesting that Trp becomes more localized upon substrate binding (75). Therefore, our results suggest that Trp121 may be critical for recognizing the substrate in the trans configuration and help stabilize the transition state.
Figure 8.
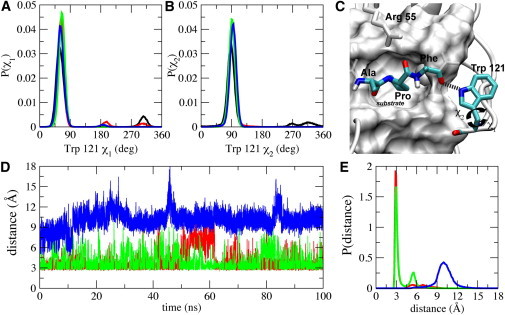
Backbone torsional-angle distributions of Trp121 and the intermolecular hydrogen bond between Trp121 and the substrate. Probability distributions of the side-chain torsional angle (A) χ1 and (B) χ2 of Trp121. (C) The intermolecular hydrogen bond (in dashed line) between the side-chain amine group of Trp121 and the carbonyl oxygen of Phe that is after the -Ala-Pro- motif. The substrate is shown in the transition-state configuration, and the rest of the substrate is also shown (white transparent stick representation), as well as the active site residues of CypA (white surface). (D) The intermolecular hydrogen bond between the indole NH of Trp121 and the carbonyl oxygen of Phe of the enzyme-substrate complexes when the substrate is in the trans (red), transition state (green), and cis (blue) configurations. (E) Probability distribution of the intermolecular hydrogen bond between the amine group of Trp121 and the substrate in the different configurations. (The color-code is the same as panel D.)
Trp121 is moderately conserved across human cyclophilin isoforms; however, it is well conserved across orthologous CypA species (15,68,69). Interestingly, human cyclophilin isoforms without tryptophan at this position are normally substituted with a histidine or a tyrosine, residues that can also form hydrogen bonds with their side chains (15). A Trp121Phe mutation, which abolishes this hydrogen-bonding interaction, causes CypA to bind cyclosporine with much lower affinity, resulting in CsA-resistant strains of Saccharomyces cerevisiae (73). The Trp121Ala (42) and Trp121Tyr (74) mutations retain 9% and 19% of wild-type catalytic activity, respectively; to the best of our knowledge, there is no experimental data for the catalytic activity of the human Trp121His mutant.
Conclusions
Molecular dynamics simulations of substrate-free CypA and the enzyme-substrate complexes when the substrate is in the trans, transition-state, and cis configurations are carried out to determine the relevant conformational changes and intermolecular interactions that impact catalysis. Our results suggest that CypA binds its substrates via conformational selection. The dynamics of active site residues of the substrate-bound CypA complexes are inherent in the substrate-free enzyme. Variations in the electrostatic and hydrophobic contacts are observed as the configuration of the substrate changes in the active site during catalysis. Presence of the substrate in the active site impacts the dynamics of certain key active site residues and loop regions of the enzyme. CypA stabilizes the transition state by forming optimized hydrophobic and electrostatic interactions with the substrate, making use of a preorganized network of hydrogen bonds. These hydrogen bonds form and break with ease in the ground states, but remain tightly formed in the transition state, localizing the enzyme conformations of the transition state to a relatively small conformational space. The enzyme therefore binds the substrate in the transition-state configuration more tightly than the substrate in the cis state, followed by the substrate in the trans state. The hydrogen-bonding interaction that is formed between Arg55 and the substrate is responsible for a large portion of the electrostatic interaction. This study provides a general atomistic insight into the interplay among enzyme conformational dynamics, recognition, and catalysis.
Acknowledgments
This work is supported in part by the National Science Foundation CAREER grant (No. MCB-0953061), the Georgia Cancer Coalition of the Georgia Research Alliance, and the Molecular Basis of Disease program at Georgia State University. L.C.M. is a Molecular Basis of Disease fellow. This work was also supported by Georgia State’s IBM System p7 supercomputer, acquired through a partnership of the Southeastern Universities Research Association and IBM supporting the SURAgrid initiative.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Srivastava D.K., Bernhard S.A. Metabolite transfer via enzyme-enzyme complexes. Science. 1986;234:1081–1086. doi: 10.1126/science.3775377. [DOI] [PubMed] [Google Scholar]
- 2.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 4.Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 5.Miller W.L. Early steps in androgen biosynthesis: from cholesterol to DHEA. Baillieres Clin. Endocrinol. Metab. 1998;12:67–81. doi: 10.1016/s0950-351x(98)80461-8. [DOI] [PubMed] [Google Scholar]
- 6.Babbitt P.C., Gerlt J.A. Understanding enzyme superfamilies. Chemistry as the fundamental determinant in the evolution of new catalytic activities. J. Biol. Chem. 1997;272:30591–30594. doi: 10.1074/jbc.272.49.30591. [DOI] [PubMed] [Google Scholar]
- 7.May S.W. Applications of oxidoreductases. Curr. Opin. Biotechnol. 1999;10:370–375. doi: 10.1016/S0958-1669(99)80067-6. [DOI] [PubMed] [Google Scholar]
- 8.Wolfenden R., Snider M.J. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 9.Kraut D.A., Carroll K.S., Herschlag D. Challenges in enzyme mechanism and energetics. Annu. Rev. Biochem. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 10.Warshel A. Computer simulations of enzyme catalysis: methods, progress, and insights. Annu. Rev. Biophys. Biomol. Struct. 2003;32:425–443. doi: 10.1146/annurev.biophys.32.110601.141807. [DOI] [PubMed] [Google Scholar]
- 11.Hamelberg D., McCammon J.A. Mechanistic insight into the role of transition-state stabilization in cyclophilin A. J. Am. Chem. Soc. 2009;131:147–152. doi: 10.1021/ja806146g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamelberg D., Shen T., McCammon J.A. Phosphorylation effects on cis/trans isomerization and the backbone conformation of serine-proline motifs: accelerated molecular dynamics analysis. J. Am. Chem. Soc. 2005;127:1969–1974. doi: 10.1021/ja0446707. [DOI] [PubMed] [Google Scholar]
- 13.Wang P., Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dornan J., Taylor P., Walkinshaw M.D. Structures of immunophilins and their ligand complexes. Curr. Top. Med. Chem. 2003;3:1392–1409. doi: 10.2174/1568026033451899. [DOI] [PubMed] [Google Scholar]
- 15.Davis T.L., Walker J.R., Dhe-Paganon S. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010;8:e1000439. doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer G. Chemical aspects of peptide bond isomerization. Chem. Soc. Rev. 2000;29:119–127. [Google Scholar]
- 17.Dugave C., Demange L. Cis-trans isomerization of organic molecules and biomolecules: implications and applications. Chem. Rev. 2003;103:2475–2532. doi: 10.1021/cr0104375. [DOI] [PubMed] [Google Scholar]
- 18.Grathwohl C., Wüthrich K. NMR studies of the rates of proline cis-trans isomerization in oligopeptides. Biopolymers. 1981;20:2623–2633. [Google Scholar]
- 19.Harrison R.K., Stein R.L. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry. 1990;29:3813–3816. doi: 10.1021/bi00468a001. [DOI] [PubMed] [Google Scholar]
- 20.Schmidpeter P.A.M., Jahreis G., Schmid F.X. Prolyl isomerases show low sequence specificity toward the residue following the proline. Biochemistry. 2011;50:4796–4803. doi: 10.1021/bi200442q. [DOI] [PubMed] [Google Scholar]
- 21.Sherry B., Zybarth G., Bukrinsky M. Role of cyclophilin A in the uptake of HIV-1 by macrophages and T lymphocytes. Proc. Natl. Acad. Sci. USA. 1998;95:1758–1763. doi: 10.1073/pnas.95.4.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braaten D., Luban J. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001;20:1300–1309. doi: 10.1093/emboj/20.6.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vajdos F.F., Yoo S., Hill C.P. Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci. 1997;6:2297–2307. doi: 10.1002/pro.5560061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterji U., Bobardt M., Gallay P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009;284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng J., Koblinski J.E., Clevenger C.V. Prolyl isomerase cyclophilin A regulation of Janus-activated kinase 2 and the progression of human breast cancer. Cancer Res. 2008;68:7769–7778. doi: 10.1158/0008-5472.CAN-08-0639. [DOI] [PubMed] [Google Scholar]
- 26.Brazin K.N., Mallis R.J., Andreotti A.H. Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin A. Proc. Natl. Acad. Sci. USA. 2002;99:1899–1904. doi: 10.1073/pnas.042529199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J., Kim S.S. An overview of cyclophilins in human cancers. J. Int. Med. Res. 2010;38:1561–1574. doi: 10.1177/147323001003800501. [DOI] [PubMed] [Google Scholar]
- 28.Bosco D.A., Eisenmesser E.Z., Kern D. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. USA. 2002;99:5247–5252. doi: 10.1073/pnas.082100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenmesser E.Z., Millet O., Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 30.Kamerlin S.C.L., Warshel A. At the dawn of the 21st century: is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fanghänel J., Fischer G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front. Biosci. 2004;9:3453–3478. doi: 10.2741/1494. [DOI] [PubMed] [Google Scholar]
- 32.Harrison R.K., Stein R.L. Mechanistic studies of peptidyl prolyl cis-trans isomerase: evidence for catalysis by distortion. Biochemistry. 1990;29:1684–1689. doi: 10.1021/bi00459a003. [DOI] [PubMed] [Google Scholar]
- 33.Li G., Cui Q. What is so special about Arg 55 in the catalysis of cyclophilin A? Insights from hybrid QM/MM simulations. J. Am. Chem. Soc. 2003;125:15028–15038. doi: 10.1021/ja0367851. [DOI] [PubMed] [Google Scholar]
- 34.Wolfenden R. Enzyme catalysis: conflicting requirements of substrate access and transition state affinity. Mol. Cell. Biochem. 1974;3:207–211. doi: 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
- 35.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco, CA: 2008. AMBER 10. [Google Scholar]
- 36.Jorgensen W., Chandrasekhar J., Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 37.Hornak V., Abel R., Simmerling C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornell W.D., Cieplak P., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 39.Doshi U., Hamelberg D. Reoptimization of the AMBER force field parameters for peptide bond (ω) torsions using accelerated molecular dynamics. J. Phys. Chem. B. 2009;113:16590–16595. doi: 10.1021/jp907388m. [DOI] [PubMed] [Google Scholar]
- 40.Fraser J.S., Clarkson M.W., Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trzesniak D., van Gunsteren W.F. Catalytic mechanism of cyclophilin as observed in molecular dynamics simulations: pathway prediction and reconciliation of x-ray crystallographic and NMR solution data. Protein Sci. 2006;15:2544–2551. doi: 10.1110/ps.062356406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zydowsky L.D., Etzkorn F.A., Walsh C.T. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992;1:1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kofron J.L., Kuzmic P., Rich D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry. 1991;30:6127–6134. doi: 10.1021/bi00239a007. [DOI] [PubMed] [Google Scholar]
- 44.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 45.Toukmaji A., Sagui C., Darden T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000;113:10913–10927. [Google Scholar]
- 46.Sagui C., Pedersen L.G., Darden T.A. Towards an accurate representation of electrostatics in classical force fields: efficient implementation of multipolar interactions in biomolecular simulations. J. Chem. Phys. 2004;120:73–87. doi: 10.1063/1.1630791. [DOI] [PubMed] [Google Scholar]
- 47.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 48.Hamelberg D., Mongan J., McCammon J.A. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 49.Shen T., Hamelberg D. A statistical analysis of the precision of reweighting-based simulations. J. Chem. Phys. 2008;129:034103–034109. doi: 10.1063/1.2944250. [DOI] [PubMed] [Google Scholar]
- 50.Balsera M.A., Wriggers W., Schulten K. Principal component analysis and long time protein dynamics. J. Phys. Chem. 1996;100:2567–2572. [Google Scholar]
- 51.Maisuradze G.G., Liwo A., Scheraga H.A. Principal component analysis for protein folding dynamics. J. Mol. Biol. 2009;385:312–329. doi: 10.1016/j.jmb.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Numata J., Wan M., Knapp E.W. Conformational entropy of biomolecules: beyond the quasi-harmonic approximation. Genome Informatics. 2007;18:192–205. [PubMed] [Google Scholar]
- 53.Hayward S., Kitao A., Gō N. Harmonic and anharmonic aspects in the dynamics of BPTI: a normal mode analysis and principal component analysis. Protein Sci. 1994;3:936–943. doi: 10.1002/pro.5560030608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skjaerven L., Martinez A., Reuter N. Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit. Proteins. 2011;79:232–243. doi: 10.1002/prot.22875. [DOI] [PubMed] [Google Scholar]
- 55.Kollman P.A., Massova I., Cheatham T.E., 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 56.Hou T., Wang J., Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai C.-J., Ma B., Nussinov R. Folding and binding cascades: shifts in energy landscapes. Proc. Natl. Acad. Sci. USA. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boehr D.D., Nussinov R., Wright P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Csermely P., Palotai R., Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010;35:539–546. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma B., Nussinov R. Enzyme dynamics point to stepwise conformational selection in catalysis. Curr. Opin. Chem. Biol. 2010;14:652–659. doi: 10.1016/j.cbpa.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koshland D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hammes G.G., Chang Y.-C., Oas T.G. Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA. 2009;106:13737–13741. doi: 10.1073/pnas.0907195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou H.-X. From induced fit to conformational selection: a continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys. J. 2010;98:L15–L17. doi: 10.1016/j.bpj.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agarwal P.K. Enzymes: an integrated view of structure, dynamics and function. Microb. Cell Fact. 2006;5:2. doi: 10.1186/1475-2859-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kern D., Kern G., Drakenberg T. Kinetic analysis of cyclophilin-catalyzed prolyl cis/trans isomerization by dynamic NMR spectroscopy. Biochemistry. 1995;34:13594–13602. doi: 10.1021/bi00041a039. [DOI] [PubMed] [Google Scholar]
- 66.Agarwal P.K. Role of protein dynamics in reaction rate enhancement by enzymes. J. Am. Chem. Soc. 2005;127:15248–15256. doi: 10.1021/ja055251s. [DOI] [PubMed] [Google Scholar]
- 67.Ramanathan A., Agarwal P.K. Evolutionarily conserved linkage between enzyme fold, flexibility, and catalysis. PLoS Biol. 2011;9:e1001193. doi: 10.1371/journal.pbio.1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agarwal P.K., Geist A., Gorin A. Protein dynamics and enzymatic catalysis: investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A. Biochemistry. 2004;43:10605–10618. doi: 10.1021/bi0495228. [DOI] [PubMed] [Google Scholar]
- 69.Yeh H.-Y., Klesius P.H. Channel catfish, Ictalurus punctatus, cyclophilin A and B cDNA characterization and expression analysis. Vet. Immunol. Immunopathol. 2008;121:370–377. doi: 10.1016/j.vetimm.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 70.Bergsma D.J., Eder C., Levy M.A. The cyclophilin multigene family of peptidyl-prolyl isomerases. Characterization of three separate human isoforms. J. Biol. Chem. 1991;266:23204–23214. [PubMed] [Google Scholar]
- 71.Doshi U., McGowan L.C., Hamelberg D. Resolving the complex role of enzyme conformational dynamics in catalytic function. Proc. Natl. Acad. Sci. USA. 2012;109:5699–5704. doi: 10.1073/pnas.1117060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Warshel A., Sharma P.K., Olsson M.H. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 73.Cardenas M.E., Lim E., Heitman J. Mutations that perturb cyclophilin A ligand binding pocket confer cyclosporin A resistance in Saccharomyces cerevisiae. J. Biol. Chem. 1995;270:20997–21002. doi: 10.1074/jbc.270.36.20997. [DOI] [PubMed] [Google Scholar]
- 74.Bosco D.A., Eisenmesser E.Z., Kern D. Dissecting the microscopic steps of the cyclophilin A enzymatic cycle on the biological HIV-1 capsid substrate by NMR. J. Mol. Biol. 2010;403:723–738. doi: 10.1016/j.jmb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 75.Gastmans M., Volckaert G., Engelborghs Y. Tryptophan microstate reshuffling upon the binding of cyclosporin A to human cyclophilin A. Proteins. 1999;35:464–474. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.