

Abstract
Disruption of the integrity of the plasma membrane by amyloidogenic proteins is linked to the pathogenesis of a number of common age-related diseases. Although accumulating evidence suggests that adverse environmental stressors such as unbalanced levels of metal ions may trigger amyloid-mediated membrane damage, many features of the molecular mechanisms underlying these events are unknown. Using human islet amyloid polypeptide (hIAPP, aka amylin), an amyloidogenic peptide associated with β-cell death in type 2 diabetes, we demonstrate that the presence of Ca2+ ions inhibits membrane damage occurring immediately after the interaction of freshly dissolved hIAPP with the membrane, but significantly enhances fiber-dependent membrane disruption. In particular, dye leakage, quartz crystal microbalance, atomic force microscopy, and NMR experiments show that Ca2+ ions promote a shallow membrane insertion of hIAPP, which leads to the removal of lipids from the bilayer through a detergent-like mechanism triggered by fiber growth. Because both types of membrane-damage mechanisms are common to amyloid toxicity by most amyloidogenic proteins, it is likely that unregulated ion homeostasis, amyloid aggregation, and membrane disruption are all parts of a self-perpetuating cycle that fuels amyloid cytotoxicity.
Introduction
Islet amyloid polypeptide (IAPP) is a member of a class of peptides that have the ability to form insoluble, β-sheet-rich aggregates known as amyloid (1). Amyloid deposits frequently accumulate in the tissues of patients with common age-related degenerative diseases, and have been linked to impaired cellular functioning. In particular, IAPP-rich amyloid deposits are found in the extracellular space of the islets of Langerhans in patients with type 2 diabetes mellitus (T2DM) but not in the islets of nondiabetic age-matched patients (2). IAPP aggregates are known to be toxic to β cells (3–6), supporting the idea that they play a key role in the depletion of pancreatic islets in T2DM (7,8). This linkage among IAPP aggregation, β-cell toxicity, and T2DM is supported by the correlation between the toxicity of IAPP peptides derived from different species and the prevalence of a diabetes-like pathology (9). For example, whereas exogenous human IAPP (hIAPP) is strongly cytotoxic when applied to cells, the corresponding rat sequence (rIAPP) is not (10,11). Although rats do not normally develop T2DM, mice transgenic for hIAPP develop a diabetes-like pathology if fed a high-fat diet (9). These findings suggest that β-cell failure caused by hIAPP is likely to a play a role in the development of T2DM in humans (12).
It has been suggested that hIAPP exerts its toxicity by damaging the integrity of phospholipid membranes (13–15). The mechanism of membrane disruption is still debated, and confusion exists regarding both the mechanism of membrane disruption and the species of hIAPP involved (16,17). Several studies have proposed that membrane disruption is initiated by the binding of prefibrillar oligomers to the membrane surface, either through the formation of transmembrane oligomeric pores (13,18–20) or by a nonspecific ion permeation process (18). Other studies have proposed that membrane disruption is caused by membrane fragmentation through the growth of amyloid fibrils on the membrane surface (21–23). Recently, it was proposed that both mechanisms are not mutually exclusive and both are likely to be involved in the process of membrane disruption by IAPP (24). A number of reports have further shown that IAPP–membrane interactions are significantly affected by several factors, such as membrane electrostatic properties, metal ions, pH, and IAPP concentration (25,26).
Although the notion that hIAPP is toxic under certain conditions has largely been accepted, the proximal molecular events that link the formation of toxic IAPP aggregates, membrane damage, and the induction of apoptotic events into β cells are still under intense debate. It has been demonstrated that amyloid peptides can destabilize Ca2+ homeostasis in cells by forming unregulated cation-selective channels across the plasma membrane (the channel hypothesis) (27,28). In line with the cation channel hypothesis, it has been reported that IAPP induces substantial intracellular [Ca2+]i elevations in GT1-7 cell lines (29). Furthermore, an increase of [Ca2+]i levels in diabetic subjects has also been reported (30,31). Notably, dysregulation of [Ca2+]i in the cells of several tissues occurs spontaneously during aging (32,33). Impaired intracellular Ca2+ signaling is known to affect a number of membrane-related functions, including membrane domain organization, vesicular trafficking, and membrane fusion (34,35), as well as the binding of cytosolic proteins to membranes that contain negatively charged phospholipids. These data suggest a possible correlation among Ca2+ dyshomeostasis, abnormal hIAPP–membrane interaction, and, eventually, impaired hIAPP trafficking. Previous studies have shown that Ca2+ ions may differently affect the binding properties of hIAPP and rIAPP to model lipid membranes (36). However, despite considerable interest in this field, a rigorous description of these phenomena is lacking.
Here, we evaluate the effect of Ca2+ on the interaction of hIAPP with membrane using a combination of physicochemical techniques. In particular, our results show that the presence of Ca2+ ions strongly modulates the type of membrane disruption that is induced by hIAPP. Using dye-leakage experiments, quartz crystal microbalance with dissipation monitoring (QCM-D), and NMR, we observed that a high concentration of Ca2+ decreases the ability of prefibrillar hIAPP species to insert into the bilayer and disrupt the membrane. On the contrary, the presence of Ca2+ enhances the membrane disruption from the elongation of hIAPP fibers on the surface of membranes by a detergent-like mechanism. Membrane fragmentation was confirmed by the detection of small lipid aggregates that are characteristic of a detergent-like mechanism. Taken as a whole, the results suggest that Ca2+ concentration could be one of the cofactors that are able to modulate the mechanism of membrane disruption, favoring one mechanism at the expense of the other.
Although the model membranes used in this study are a simplification of the complexity of a biological membrane, we believe that our results will help elucidate the largely debated mechanism that underlies the toxicity of hIAPP, which is essential for enabling the design of specific drugs to prevent and/or alleviate T2DM.
Materials and Methods
Materials
Full-length hIAPP and rIAPP with an amidated C-terminus (>95% purity) were purchased from Anaspec (San Jose, CA) and used for all experiments except those involving NMR spectroscopy. For the NMR experiments, we used uniformly 15N-labeled hIAPP in the free acid form (rPeptide, Bogart, GA). 1,2-Palmitoyl-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-palmitoyl-oleoyl-sn-glycero-3-phospho-L-serine (POPS), 1-myristoyl-2-hydroxy-sn-glycero-3-phospho-(1'-rac-glycerol) (LysoPg), and N-dodecylphosphocholine (DPC) were purchased from Avanti Polar Lipids with a purity of 99% and used without further purification. 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), 6-carboxyfluorescein, thioflavin T (ThT), MnCl2, CaCl2, MgCl2, and HEPES were purchased from Sigma-Aldrich (St. Louis, MO) with a purity of at least 99% and used for buffer preparation. Peptides were dissolved in HFIP (1.5 mg/ml) before use to break up any preformed aggregates present in the solution. Aliquots of the peptide stock solution were lyophilized overnight. Next, the lyophilized powder was dissolved in the buffer or, alternatively, added to lipid dispersions up to the desired lipid/peptide molar ratio as reported elsewhere (37). Preformed hIAPP fibers were used for some measurements and prepared by incubating 0.1 mg of hIAPP in 500 μL of buffer overnight. The same HEPES buffer was used in all experiments. All buffer solutions were filtered by using a 0.2 μm filter before use.
Preparation of Model Membranes
Model membranes were prepared as described elsewhere (38). Briefly, lipid solutions in CHCl3 were dried under a stream of dry nitrogen gas and evaporated under high vacuum to dryness in a round-bottomed flask. To obtain multilamellar vesicles (MLVs), the resulting lipid film was hydrated with an appropriate amount of HEPES buffer (10 mM buffer, 100 mM NaCl, pH 7.4) and dispersed by vigorous stirring in a water bath set at 4°C above the gel–liquid crystalline phase transition temperature of the lipid. The final concentration of lipids was 200 μM in each case. To obtain large unilamellar vesicles (LUVs), the MLVs were extruded through polycarbonate filters (pore size = 100 nm or 1000 nm; Nuclepore, Pleasanton, CA) mounted in a mini-extruder (Avestin, Ottawa, ON, Canada) fitted with two 0.5 ml Hamilton gastight syringes (Hamilton, Reno, NV). Samples were typically subjected to 23 passes through two filters in tandem as recommended elsewhere (39). An odd number of passages were performed to avoid contamination of the sample by vesicles that might not have passed through the filter.
Membrane Leakage Experiments
Membrane leakage experiments were carried out by using 6-carboxyfluorescein-filled LUVs of POPC/POPS (molar ratio 7:3, final concentration 200 μM). Dye-filled LUVs of POPC/POPS were prepared by hydrating the dry lipid film with the buffer solution containing 6-carboxyfluorescein (10 mM Hepes, 70 mM 6-carboxyfluorescein, pH 7.4) according to the procedure described above. To remove nonencapsulated 6-carboxyfluorescein, after extrusion, the solution containing LUVs was placed on a Sephadex G50 gel exclusion column (Sigma-Aldrich) and eluted using the final buffer (10 mM Hepes, 100 mM NaCl, pH 7.4). The first colored band containing the separated dye-containing vesicles was collected. The use of 10 mM Hepes, 70 mM 6-carboxyfluorescein, pH 7.4 inside the LUVs, and of Hepes 10 mM, 100 mM NaCl, pH 7.4 in the outside buffer ensured that the osmotic pressure would not destabilize the LUVs over time (Fig. S1 in the Supporting Material). The final concentration of lipids was checked by using the Stewart assay (40). Samples were prepared initially by diluting the dye-filled vesicles solution with buffer solution (10 mM HEPES buffer solution, 100 mM NaCl, pH 7.4) to a final concentration of 200 μM. Finally, 1 μL of the peptide stock solution was added to 100 μL of LUVs solution (final peptide concentration 2 μM, lipid concentration 200 μM, and a CaCl2 or MgCl2 concentration of 50, 100, 200, or 400 μM where present). Preformed fibers, where present, were added at a concentration of 1μM. Membrane damage was quantified by detecting the increase in fluorescence emission intensity of 6-carboxyfluorescein due to its dilution (dequenching) in buffer as a consequence of the membrane leakage. Experiments were carried out in Corning 96-well, nonbinding surface plates. Time traces were recorded with a BioTek Synergy 2 plate reader using a 494 excitation filter and a 520 emission filter at room temperature, and samples were shaken for 10 s before each read. The fraction leaked was calculated as follows:
where I is the emission intensity of the sample, I0 is the emission intensity obtained in the absence of peptide (baseline control), and I100 is the emission intensity obtained after adding Triton X-100 (a detergent), which acted as a positive control to give 100% leakage. All measurements were done in triplicate.
NMR Paramagnetic Quenching
The degree of solvent exposure when hIAPP was bound to membranes in the presence of Ca2+ was estimated by paramagnetic quenching with MnCl2. The degree of quenching was determined by comparing 2D 15N-1H band-selective optimized flip-angle short transient (SOFAST)-heteronuclear multiple-quantum correlation (HMQC) spectra of hIAPP bound to the detergent LysoPG in the presence of increasing concentrations of MnCl2. LysoPG was selected because of its relatively close structural similarity to the phospholipids used in this work (structure shown in Fig. S2); the affinity of calcium for the PG headgroup is roughly similar to that of PS (41). The isotopically labeled peptide used in this experiment was obtained from a recombinant source (rPeptide) and is not amidated at the C-terminus, unlike the physiologically expressed peptide used in the remainder of the experiments; however, the effect of amidation on membrane binding is relatively minor (42). All NMR spectra were collected on a 900 MHz Bruker Avance NMR spectrometer equipped with a triple-resonance z-gradient cryogenic probe optimized for 1H detection. The SOFAST technique was used for fast data acquisition to reduce the probability of artifacts arising from aggregation (43). Two-dimensional (2D) SOFAST-HMQC spectra of IAPP in solution (20 mM Tris buffer, 25 or 50 mM NaCl, pH 7.4, 80 μM hIAPP) were acquired using 128 t1 experiments, four scans, and a 100 ms recycle delay. In the absence of detergent, data were recorded at 4°C to avoid the fast aggregation and line broadening that occurs at higher temperatures in the absence of detergent. Experiments with lysoPC and DPC were recorded as above but with 64 scans and at 20°C, using a 156 μM concentration of hIAPP (78 μM for experiments with Mg2+) and 25 mM lysoPG or 100 mM DPC. Experiments with calcium were recorded using 12.5 mM CaCl2, 20 mM Tris buffer, and 25 mM NaCl at pH 7.4. Experiments without calcium were recorded under the same condition except that 50 mM NaCl was used.
Lipid Sedimentation Assay
The amount of membrane fragmentation that occurred during amyloid fiber formation was determined by a sedimentation assay using centrifugation to sediment intact vesicles, followed by analysis of the supernatant to measure concentrations of lipids removed from the membrane. Solutions of LUVs of POPC/POPS (molar ratio 7:3, final concentration 1 mg/ml) were prepared as described above. Larger LUVs with a diameter of 1000 nm were used for these experiments because they can be sedimented easily at low speeds by centrifugation. For each sample, hIAPP (final concentration 15 μM) was incubated with 500 μL of the LUV solution in the presence or absence of 100 μM Ca2+ for 24 hr to form amyloid fibers. To remove vesicles that were not broken by fibers, the samples were centrifuged for 40 min at 14,000 rpm, and the supernatant, which contains lipids embedded with fibers, was collected to measure the amount of lipid in solution.
The concentration of lipids in the supernatant was measured using the Stewart assay, a colorimetric method based on the ability of phospholipids to form a complex with ammonium ferrothiocyanate (40). A calibration curve was first created for each LUV lipid composition using a series of chloroform solutions with a known amount of lipids (volume 1 ml) treated with 1 ml of a solution containing ferric chloride and ammonium thiocyanate. Each solution was then shaken for 20 s and the organic phase was collected. The optical density of these standard solutions was read with an UV-VIS spectrophotometer at 461 nm and plotted versus the lipid concentration to obtain a linear calibration curve. The supernatant from centrifugation of lipid samples was treated similarly after first diluting to 500 μL of chloroform before the extraction step. Solutions without hIAPP were analyzed as a control. All measurements were made in triplicate.
QCM-D Measurements
QCM-D experiments were performed on a Q-Sense D300 quartz microbalance (Q-Sense AB, Gothenburg, Sweden). The 5 MHz QCM sensor crystals with gold-on-chromium electrodes (area size 0.189 cm2) covered by silicon dioxide were purchased from Q-Sense AB. The measurement chamber (80 μL of volume) operated at 25 ± 0.1°C in 10 mM HEPES buffer, 100 mM NaCl, pH 7.4. The instrument allowed simultaneous measurements of frequency, F, and energy dissipation, D, for the fundamental resonance frequency (n = 1, i.e., F = ∼5 MHz) and the first three overtones (n = 3, 5, and 7, corresponding to f = ∼15, ∼25, and ∼35 MHz, respectively). The resolution in F and D is ± 0.1 Hz and 1×10−7, respectively. The data were collected in Qsoft 301 and analyzed in Qtools version 1.1.0.2 (both provided by Q-Sense AB). POPC/POPS (molar ratio 7:3) supported lipid bilayers (SLBs) were used. All other experimental conditions were unchanged with respect to the membrane leakage experiments.
Atomic Force Microscopy Measurements
Film morphology and its roughness at the nanometer scale were investigated by atomic force microscopy (AFM) on a Multimode Nanoscope IIIa (Digital Instruments, Santa Barbara, CA) with both 10 and 125 μm scanners. Images were acquired in tapping mode with a fluid cell filled with phosphate buffer solution. Sharpened silicon nitride probes (Veeco) with a radius of 30 nm and a nominal spring constant, k = 0.06 Nm−1, were employed. Tapping mode in air imaging was performed by using phosphorus N-doped silicon probes. The roughness estimation was made on scan areas of 10 × 10 μm2 by using Nanoscope RIII software in terms of the standard deviation of heights (Rq). POPC/POPS lipid mixtures (molar ratio 7:3) were used. All other experimental conditions were unchanged with respect to the membrane leakage experiments.
Results and Discussion
Ca2+ Ions Inhibit Dye Release Induced by Prefibrillar hIAPP but Enhance Membrane Disruption Induced by Elongation of Fibers
Although the mechanism of membrane disruption induced by hIAPP has not been fully elucidated, a two-step mechanism for disruption of the integrity of the membrane was recently proposed (24). The first step is correlated with the interaction of prefibrillar species of hIAPP with the membrane, and the second step is correlated with the elongation of the fiber on the membrane surface.
To assess the influence of Ca2+ on both steps, we measured the influence of Ca2+ on the ability of hIAPP to cause dye release from liposomes with and without preformed fibers to seed fiber elongation (Fig. 1). First, the damage induced by prefibrillar species of hIAPP was evaluated by measuring dye release directly after the addition of 2 μM of freshly dissolved hIAPP to 200 μM of 7:3 POPC/POPS liposomes. In all samples prepared this way, freshly dissolved hIAPP caused a rapid release of the dye immediately after the addition of hIAPP, which reached its maximum after 10 min (Fig. S3A). The addition of Ca2+ significantly lowered the amount of maximum dye released after the addition of freshly dissolved hIAPP (Fig. 1 A, blue bars). Calcium ions are known to interact strongly with the negatively charged headgroup of POPS lipid (44–46). This interaction leads to a decrease in the total negative charge of the membrane surface, which may diminish the electrostatic interaction of hIAPP with the membrane (25,47). Because Mg2+ is known to have both a smaller affinity for the POPS headgroup than Ca2+ and a different binding mode to lipids (48–50), we repeated the same experiments using Mg2+ instead of Ca2+ to determine the extent to which the difference in membrane disruption is mediated by the interaction of Ca+2 with the membrane. The amount of dye release was essentially unaffected by the presence of Mg2+ within the experimental error of the measurement (Fig. 1 A, red bars; p > .05), indicating that specific binding by Ca2+ is most likely responsible for this difference. It is known that the presence of Ca2+ does not affect the structure of the peptide as observed by circular dichroism experiments (36), but its presence does slightly reduce the rate of fiber formation as measured by ThT (Fig. S4).
Figure 1.
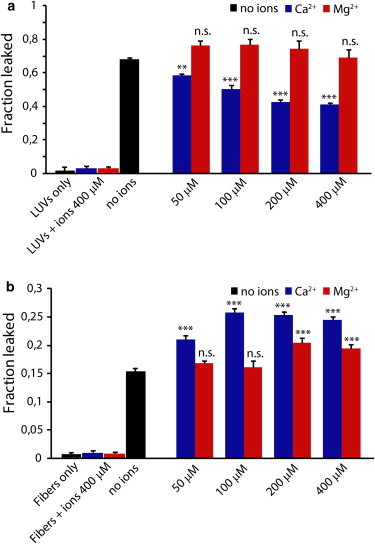
Dye release from model membranes induced by hIAPP in the presence and absence of calcium ions. (A and B) Values of dye release from 200 μM LUVs of POPC/POPS (molar ratio 7:3) in the absence (A) or presence (B) of 1 μM preformed hIAPP fibers, induced by 2 μM of freshly dissolved hIAPP after 30 min with the indicated ions at the indicated concentrations. All experiments were made in 10 mM HEPES buffer, 100 mM NaCl, pH 7.4 at 25°C. Results are the average of three experiments. The error bars represent the standard error of measurement.
The immediate membrane disruption caused by prefibrillar hIAPP is only one aspect of hIAPP’s effect on membranes. A second and frequently larger phase of membrane disruption occurs when fibers of IAPP and other amyloid proteins (51) begin to grow on the membrane (21,22,24,52). To evaluate the role of Ca2+ on this stage of membrane disruption, samples were first preincubated with preformed fibers of hIAPP and then the dye release was measured immediately following the addition of 2 μM freshly dissolved hIAPP to initiate fiber elongation (Fig. 1 B) (22). As expected, dye leakage was not observed for samples incubated with preformed fibers only (Fig. 1 B and Fig. S3B), suggesting that the fibers alone are not able to disrupt the membrane integrity. However, seeded fiber elongation of hIAPP caused a rapid increase of dye release within the first 10 min after the addition of monomers (Fig. S3B), similar to that observed with freshly dissolved hIAPP (Fig. S3A). However, Ca2+’s effect on membrane disruption by fiber elongation was the opposite of that observed with prefibrillar hIAPP. Although Ca2+ decreased the membrane disruption by prefibrillar hIAPP (Fig. 1 A, blue bars), it enhanced membrane disruption during fiber elongation (Fig. 1 B, blue bars). Similar experiments performed in the presence of Mg2+ instead of Ca2+ (Fig. 1 B, red bars) showed a significantly diminished effect that increased at higher concentrations of Mg2+, consistent with Mg2+ ions’ lower affinity for the membrane.
The total amount of dye released from the seeded reaction in both types of samples is lower than that released from the unseeded samples, most likely because much of the monomer is immediately sequestered into fibers that are not bound to the membrane and is therefore unavailable for fiber elongation on the membrane surface (53). This results in a lower effective concentration of monomer for fiber elongation and therefore a smaller extent of membrane disruption, as previously observed for hIAPP and Aβ1-40 (54,55).
Ca2+ Ions Hinder the Insertion of Prefibrillar hIAPP Species into the Hydrophobic Region of SLB and Promote the Fragmentation of the Bilayer during hIAPP Fiber Growth
To gain more insight into the impact of Ca2+ on the changes in the membrane physical properties caused by hIAPP binding, we monitored the exposure of SLBs composed of POPC/POPS (molar ratio 7:3) to hIAPP solutions by conducting QCM-D experiments in both the absence and presence of Ca2+ (Fig. 2). QCM-D experiments use a single SLB formed on top of a resonating SiO2 sensor to record the frequency shift (ΔF) and dissipation response (ΔD) of harmonics of the resonating frequency as a function of time after the addition of hIAPP (Fig. 2). The Sauerbrey relation states that the frequency shift (ΔF) is proportional to the mass adsorption of the ligand on the sensor surface (56). Although the Sauerbrey relation is technically valid only for thin, rigid films coupled without friction to the sensor surface, the loss of friction that occurs with mildly viscoelastic materials such as SLBs is minor. The frequency shift (ΔF) is therefore to a first approximation proportional to the change in mass absorbed, including any changes in hydration of the bilayer. The material properties of the bilayer are measured by the dissipation response (ΔD). A softer, more fluid bilayer with less internal cohesiveness between lipid molecules is more easily deformed and therefore will possess a higher ΔD than a more cohesive one (53). Each harmonic of the resonating frequency penetrates to a different depth away from the sensor surface. Higher harmonics probe relatively close to the surface of the chip, whereas lower harmonics extend beyond the SLB into the bulk solution (57). A comparison of response curves between harmonics can help localize the changes in the bilayer, although the resolution is not high enough to give atomic-level insight.
Figure 2.
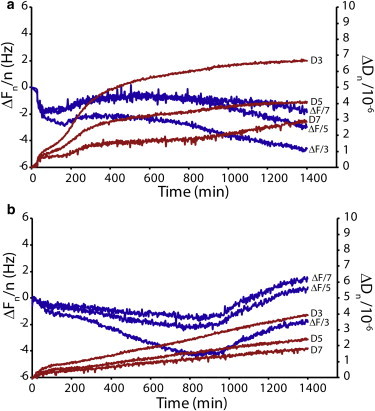
Interaction of hIAPP and SLBs studied by QCM-D. (A and B) QCM-D traces for POPC/POPS (7:3) SLBs exposed to 2 μM hIAPP (A) and 2 μM hIAPP and 100 μM Ca2+ ions (B). A comparison of the dissipation curves (ΔDn) shows that hIAPP interacts less with the hydrophobic core of the SLB in the presence of calcium ions. In the presence of Ca2+, a decrease in the mass adsorbed to the quartz crystal at ∼950 min (corresponding to the increase in ΔFn) indicates the removal of lipid from the bilayer after fiber formation. All experiments were performed in 10 mM HEPES buffer, 100 mM NaCl, pH 7.4 at 25°C.
We prepared SLBs by fusing SUVs on the supporting SiO2 substrates to form a continuous single SLB (58). SLBs formed in this manner are stable until hIAPP is added (see Fig. S5). QCM-D shows that the mechanism of interaction of IAPP with the membrane is complex but can be roughly divided into three steps:
1) For samples both with (Fig. 2 B) and without (Fig. 2 A) Ca2+, the ΔF signal decreases during the first 100 min after the addition of hIAPP. This corresponds to an increase in mass on the bilayer, and likely reflects the slow accumulation of prefibrillar IAPP on the membrane surface. During this phase, ΔD increases for both samples, showing that the membrane becomes substantially more viscous and therefore less cohesive after interaction with the protein. The ΔF decrease and ΔD increase are more evident in the absence of Ca2+, indicating that the adsorption of prefibrillar hIAPP to POPC/POPS membranes is comparatively more efficient in the absence of Ca2+. This is not surprising considering that Ca2+ is able to efficiently shield the negative charge on the membrane surface, which is known to enhance the binding of hIAPP (59), and this result matches that obtained with the dye-leakage assay (Fig. 1).
2) From 100 to ∼400 min, ΔF is almost flat, indicating that the surface is almost saturated in both samples. A minor increase in ΔF in the sample without Ca2+ is most likely correlated with the release of water from the surface of the SLB due to insertion of the peptide into the SLB, whereas the minor decrease in ΔF in the sample with Ca2+ most likely reflects the continuing absorption of the peptide to the SLB without the release of water. Interestingly, despite the absence of a significant shift in ΔF, the ΔD curve sharply increased for the sample without Ca2+, suggesting that a topological reorganization of the hIAPP within the SLB occurs to create a less cohesive, softer membrane. This increase in viscosity suggests that the hydrophobic core of the bilayer is disrupted, which may occur as a result of a time-dependent change toward deeper insertion of the peptide into the SLB due to aggregation (15,60,61). It must be stressed that because the additional mass uptake due to the adsorption of hIAPP is very small with respect to the mass of the SLB, the large increase of the dissipation factor is essentially due to the change of the viscosity of the SLB, rather than to a direct effect caused by the increase in mass from hIAPP adsorption. Although both samples show some degree of change in ΔD, the change is much larger in the absence of Ca2+ (Fig. 2 A), indicating that the degree of reorganization is more significant in the absence of Ca2+.
3) After 800 min, the sample without Ca2+ shows a decrease in ΔF with time, and a slight increase with time in the ΔD response (Fig. 2 A). This is on the same timescale as fiber formation and likely indicates an increase in the adsorbed mass on the membrane surface as a result of aggregation. On the other hand, in the presence of Ca2+, ΔF sharply increases after a lag time of ∼950 min (Fig. 2 B), indicating a significant loss of mass from the SLB at this time point in the presence of Ca2+.
The results obtained by dye leakage assays and QCM-D suggest that Ca2+ acts as a switch to hamper the interaction of the prefibrillar hIAPP species with the hydrophobic region of the membrane, and to promote the fragmentation of the membrane induced by hIAPP fibers. It should be noted that the loss of mass observed by the QCM-D and the increase in the ThT signal used to evaluate fiber formation (Fig. S4) occurs on a significantly different timescale. We attribute this difference to the different P/L ratio and the absence of shaking in the QCM-D experiment. Moreover, ThT measurements do not distinguish between fibers formed in solution and fibers formed on the surface of the bilayer, which can occur on a different timescale (54,55). Although the sudden decrease in mass in the Ca2+ is consistent with the onset of fiber formation and the consequent removal of the fibers from the surface, QCM cannot definitively establish the kinetics of fiber formation, because the ΔF signal is the sum of contributions from the SLB itself, hIAPP bound to the membrane, and a boundary layer of water coupled to the SLB.
As a control, we also performed QCM-D experiments by exposing the SLB films to rIAPP solution. rIAPP is not toxic to β cells and does not form fibers or induce dye release from lipid vesicles (8,36,62–64). The QCM-D results obtained with rIAPP (Fig. S6) were essentially stable in both the presence and absence of Ca2+ within the detection limit of the technique (i.e., ∼1 Hz). The very small weight fluctuations in the traces are due to Lundström-like adsorption-desorption of rIAPP on SLB (65).
Ca2+ Ions Hinder the Insertion of Monomeric hIAPP into lysoPG Micelles
The differences in dye-leakage rates and in the ΔD observed with QCM-D measurements, in the initial phase of membrane interaction, suggest that the prefibrillar states of hIAPP interact differently with membrane in the presence of calcium. To assess the effect of calcium on the positioning of prefibrillar states of hIAPP within the membrane, we employed paramagnetic quenching NMR experiments on 15N-labeled hIAPP bound to lysoPG (structure shown in Fig. S2) micelles in the presence and absence of calcium ions. Paramagnetic quenching experiments reveal the exposure of the peptide to solvent: if a residue is exposed on the surface of the micelle, the corresponding resonance will be broadened, but if it is buried within the micelle, the quenching agent will have little or no effect. The small size of detergent micelles ensures a low number of peptides bound per micelle, and therefore greatly stabilizes the monomeric form of hIAPP relative to oligomeric states.
An overlay of the 2D 15N-1H SOFAST-HMQC spectra of hIAPP in the absence and presence of 25 mM lysoPG is presented in Fig. S7. The spectrum of hIAPP bound to lysoPG is similar to that of IAPP bound to DPC micelles (Fig. S8), but differs significantly from that of IAPP in solution, indicating that the peptide is bound to lysoPG micelles both with and without Ca2+. In the absence of calcium ions, the addition of the paramagnetic quencher MnCl2 had a moderate line-broadening effect on the peaks observed in the NMR spectra, with many resonances clearly visible even in the presence of a high concentration (600 μM) of Mn2+ (projections of 2D spectra on the 1H chemical shift axis are shown in Fig. 3 A, and the full 2D spectra are shown in Fig. S9). The effect of Mn2+ is not uniform, with some resonances showing a greater degree of broadening than others (Fig. S9). The nonuniform, moderate effect of MnCl2 is in agreement with partial burial of the N-terminus of IAPP in the detergent micelle, as proposed in previous NMR structural studies of IAPP in anionic micelles (42,66).
Figure 3.
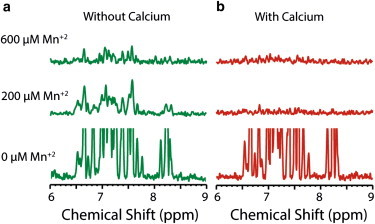
Paramagnetic quenching of IAPP bound to LysoPG in the absence and presence of calcium. (A and B) 1H chemical shift projections of 2D 15N-1H SOFAST-HMQC spectra of 150 μM IAPP bound to 25 mM LysoPG in the absence (A) and presence (B) of 12.5 mM calcium with the indicated MnCl2 concentration (full 2D NMR spectra are shown in Fig. S9 and Fig. S10). Manganese has a moderate effect on IAPP in the absence of calcium; the more-severe effect in the presence of calcium is an indication of greater solvent exposure of hIAPP when bound to the micelle. All experiments were performed in 20 mM Tris buffer, 25 mM NaCl, pH 7.4.
With the addition of calcium in a 1:2 molar ratio of Ca2+/LysoPG, the quenching effect of Mn2+ becomes much greater and the NMR peaks are totally quenched even at a low concentration (200 μM) of Mn2+ (Fig. 3 B and Fig. S10). Without the quencher, Ca2+ has little effect on the spectra of micelle-bound hIAPP (Fig. S11), which suggests that hIAPP remains bound to lysoPG in the presence of Ca2+. Paramagnetic quenching in the presence of Mg2+ is significantly diminished compared with the strong quenching observed with Ca2+ (Fig. S12), suggesting that the effect is specific to Ca2+. The stronger quenching effect of Mn2+ in the presence of Ca2+ is therefore likely due to the poorer penetration of the monomeric peptide into the micelle, although a contribution from the decrease in the micelle binding affinity due to Ca2+ cannot be rigorously excluded.
Ca2+ Promotes Disruption of Membrane by a Detergent-Like Mechanism
The QCM-D data show a loss of mass in the SLBs during fiber elongation when Ca2+ is bound to the membrane, suggesting that Ca2+ may enhance detergent-like membrane disruption by hIAPP. The detergent-like mechanism is characterized by fragmentation of the bilayer into a small, lipidic structure (vesicles or micelles). To test this hypothesis, we performed a sedimentation assay to separate intact bilayers from fragmented membranes. Freshly dissolved hIAPP (with 15 μM) was first incubated with POPC/POPS LUVs (molar ratio 7:3) for 24 hr to allow the formation of hIAPP fibers, and the samples were then centrifuged to eliminate intact vesicles. The concentration of lipid in the supernatant solution was then quantified with the Stewart assay (Fig. 4). A significantly higher concentration of lipid was detected in the supernatant in samples incubated in the presence of 200 μM Ca2+ (p < .001) compared with those without Ca2+, confirming the increase of membrane fragmentation in samples containing Ca2+ inferred from the QCM-D experiment.
Figure 4.
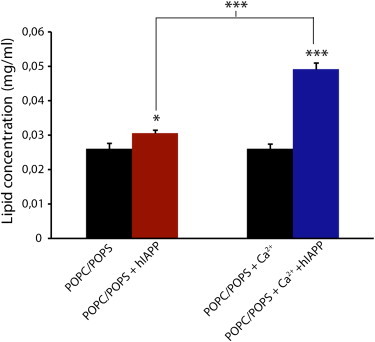
Membrane fragmentation through a detergent-like mechanism induced by hIAPP fibers. Lipid concentration in the supernatant after incubation with 15 μM hIAPP for 24 hr at 25°C with 1 mg/ml LUVs of POPC/POPS (molar ratio 7:3) in the presence or absence of 100 μM Ca2+, and centrifugation at 14,000 rpm as measured by the Stewart assay. Black bars represent controls in the absence of peptide. The presence of Ca2+ significantly enhances fragmentation of membranes induced by hIAPP fiber formation. All experiments were performed in 10 mM HEPES buffer, 100 mM NaCl, pH 7.4 at 25°C. Results are the average of three experiments. The error bars indicate the standard error of measurement.
AFM Reveals a Fiber-Dependent Defect on SLB Only in the Presence of Ca2+
The dye-leakage, membrane-sedimentation, QCM-D, and NMR experiments all indicate that two different types of hIAPP-membrane interactions exist depending on the presence of Ca2+, which in turn leads to two different mechanisms for membrane disruption. This difference can be directly visualized by AFM experiments. In particular, the detergent-like mechanism favored in samples containing Ca2+ should lead to the appearance of large defects in the integrity of the SLB that are possible to detect by AFM experiments.
AFM images of POPC/POPS (molar ratio 7:3) SLBs incubated with hIAPP for short (2 hr) and long (24 hr) durations with and without Ca2+ are shown in Fig. 5. Calcium causes a marked change in the morphology of hIAPP deposited on the bilayer after prolonged incubation times. In the presence of Ca2+, after 24 hr, hIAPP forms a rich population of large, long fiber-like structures parallel to the SLB surface, with a lateral size of 79.8 ± 12.9 nm and a height of ∼9.8 ± 3.3 nm (Fig. 5 D). In this case, damaged regions can also be observed in the SLB (Fig. 6 B), suggesting severe degradation of the membrane consistent with the removal of lipid from the mica surface, in agreement with the loss of mass detected in the QCM traces (Fig. 2 B). Such damaged regions in the membrane were not observed in the absence of calcium (Figs. 5 B and 6 A). Strikingly, long fiber-like species were also not detected in the absence of calcium after incubation for 24 hr (Fig. 5 B). Instead, a large number of smaller cylindrical structures (4.5 ± 1.6 nm high and 82.7 ± 14.5 nm wide) were detected apparently emerging from the SLB. This observation is surprising because we observed the fiber formation with ThT for the sample without Ca2+ (Fig. S4); however, the similarity of the ThT curves for hIAPP alone and hIAPP in the presence of LUV POPC/POPS (Fig. S4) suggests that in this case, hIAPP fibers are mainly formed in solution rather than on the bilayer surface, and therefore are not detected by AFM. As a control, AFM images were also acquired for hIAPP in the absence of SLB (Fig. S13), as well as for SLB films exposed to rIAPP solution. Images of the SLB incubated with rIAPP show the absence of fibers and defects in the SLB (Fig. S14).
Figure 5.

AFM images of hIAPP fibers with and without calcium ions. (A and B) Top: AFM images of POPC/POPS (7:3) SLBs after exposure to 2 μM hIAPP alone for 2 hr (A) and 24 hr (B). (C and D) Bottom: AFM images of POPC/POPS SLBs incubated with both 2 μM hIAPP and 100 μM Ca2+ ions for 2 hr (C) and 24 hr (D). All experiments were performed in 10 mM HEPES buffer, 100 mM NaCl, pH 7.4 at 25°C.
Figure 6.
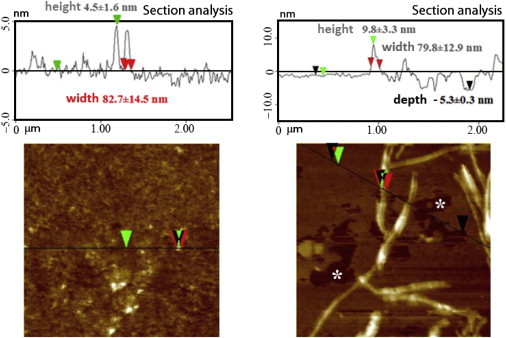
AFM section analysis. (A and B) Structures formed on POPC/POPS (7:3) SLBs after exposure to 2 μM hIAPP (A) or 2 μM hIAPP with 100 μM Ca2+ ions (B) for 24 hr. The zero value refers to the height of the membrane in the absence of hIAPP. Green and red arrows refer to height and width measurements, respectively. Defects on the SLB are evident only in the presence of calcium ions (marked with white asterisks on the AFM image and black arrows in the sectional analysis). All experiments were performed in 10 mM HEPES buffer, 100 mM NaCl, pH 7.4 at 25°C.
Discussion
The possible role of lipids at different stages of hIAPP aggregation, and the molecular details of hIAPP-associated membrane damage in T2DM are subjects of intense debate (18,52). It has been proposed that the interaction of IAPP with the membrane results in the loss of lipids from the target membrane (67). It has also been suggested that small-sized IAPP oligomers form ion-permeable structures when inserted into the membrane (8,13,64,68). Despite intensive investigation, our knowledge about the early events that trigger hIAPP fibril growth and/or membrane damage in the cell is still limited (16). This lack of knowledge mirrors our lack of understanding of the triggers that convert hIAPP, which is normally stored safely at high concentrations, into a potentially cytotoxic species in diabetic subjects (12). In this context, the dysregulation of Ca2+ concentration that occurs with aging (32,33), as well as the observation of significantly increased levels of cytosolic Ca2+ levels in β cells of transgenic mice overexpressing hIAPP, suggest a close relationship among Ca2+ dyshomeostasis, abnormal hIAPP–membrane interaction, and, eventually, islet cell toxicity (69).
In this study, we have shown that calcium ions play a significant role in affecting both mechanisms of hIAPP-induced membrane disruption (Fig. 7). NMR quenching experiments show that Ca2+ ions hinder the insertion of prefibrillar hIAPP species into negatively charged lysoPG micelles (Fig. 3). Although the NMR data clearly show binding of the hIAPP monomer to the micelle in the presence of calcium (Fig. S8), the cross-linking of lipid molecules caused by the chelation of calcium with the lipid headgroups likely prevents the penetration of hIAPP into the micelle (Fig. 3). Moreover, the QCM-D experiments show that for samples without Ca2+ ions, the addition of hIAPP causes an immediate increase in ΔD, which is correlated with the viscosity of the membrane (Fig. 2 A), suggesting that the binding of prefibrillar species of hIAPP in the absence of Ca2+ induces a rupture of lipid-lipid interactions resulting in insertion of hIAPP into the hydrophobic region of the bilayer. Changes in ΔD are more modest in the presence of Ca2+, indicating that hIAPP causes smaller changes in the membrane viscosity when Ca2+ is present. This interpretation is also supported by a close analysis of the ΔD depth profile, which reveals that hIAPP penetrates to a lesser degree into the bilayer in the presence of calcium (Fig. 2 B), in agreement with the NMR results. This difference in binding mode can be directly observed by AFM. In the absence of Ca2+, hIAPP forms cylindrical oligomers on the membrane, whereas in the presence of Ca2+ the dominant species is long fibers similar to those seen in solution (Fig. 6). Taken together, the QCM-D, AFM, and NMR results indicate that Ca2+ switches the binding mode of both prefibrillar and fibrillar hIAPP, causing a shallower insertion into the membrane with less disruption of the hydrophobic core of the bilayer.
Figure 7.
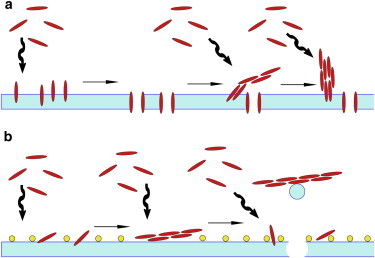
(A and B) Cartoon schematic of the possible interactions of hIAPP with lipid bilayers without Ca2+ (A) and with Ca2+ (B).
This difference in the membrane-binding mode of hIAPP is reflected in the membrane permeabilization induced by prefibrillar hIAPP species (Fig. 1 A and Fig. S3A). In the absence of calcium, hIAPP efficiently disrupts POPC/POPS membranes immediately after the addition of the peptide (Fig. S3A). The presence of Ca2+ ions seems to inhibit this phenomenon, possibly by increasing the rigidity of the bilayer (Fig. 2) and by shielding the negative charge on the membrane surface.
Whereas Ca2+ decreased membrane permeabilization by prefibrillar hIAPP, it increased membrane fragmentation during hIAPP fiber elongation. This is reflected in the decrease in the mass of the absorbed SLB in the QCM-D data at later time periods for samples containing Ca2+ ions (Fig. 2 B) than in samples without Ca2+, suggesting that Ca2+ enhances membrane disruption during amyloid formation. Direct evidence for this process was provided by dye-release assays performed for samples containing preformed hIAPP fibers (Fig. 1 B and Fig. S3B), which showed that calcium ions enhanced the release of the dye during the fiber elongation process, confirming a close correlation between fiber growth and membrane disruption in the presence of Ca2+. Moreover, lipid sedimentation assays revealed the appearance of small lipid aggregates after prolonged incubation of hIAPP with LUVs containing Ca2+ (Fig. 4), suggesting that the fiber growth induced the removal of lipids from the bilayer by a detergent-like mechanism. Direct evidence of a different interaction of the amyloid state of hIAPP with the bilayer in the presence of Ca2+ was provided by AFM. A large array of fibrils, together with irregular holes in the lipid matrix, is evident in the presence of Ca2+ (Fig. 6 B); however, similar defects in the membrane are not detected in AFM images of the Ca2+-free sample (Fig. 6 A). Similarly, the loss of mass at ∼950 min in the presence of Ca2+ detected by QCM may be correlated with the detachment of hIAPP from the SLB surface with the consequent extraction of a small amount of lipid from the bilayer through a detergent-like mechanism (54). Notably, the ΔD curves continue to increase linearly after the loss of mass, confirming that hIAPP continues to interact with the remaining SLB, and the remaining membrane is similar in structure to the original SLB. Similar behavior was previously observed in experiments with Aβ1-40, in which the membrane apparently resealed after fiber formation and membrane fragmentation (55). The reason for the increase in membrane disruption caused by fiber elongation is not straightforward, but may be related to the increase in negative (outward) curvature of the membrane upon Ca2+ binding (70), which has been linked to hIAPP membrane disruption and toxicity (15).
Conclusions
Our observations support the hypothesis that multiple environmental factors, such as a local increase of protein concentration and an unbalanced cytosolic influx-efflux of ions, are likely to play a critical role in the mechanism of IAPP interaction with the membrane (71). According to our results, Ca2+ ions may act as switches that hamper the insertion of monomeric IAPP into the membrane. Consequently, although hIAPP is not embedded in the lipid core, most likely due to the rigidifying influence of calcium on the bilayer, hIAPP-enriched microenvironments form in the proximity of the membrane surface and start to self-assemble into oligomers.
We are aware of the inherent difficulties of extrapolating results from models in vitro to the biological context, particularly for a system as complex as the Ca2+/hIAPP/membrane assembly. Nevertheless, it is worthwhile to mention that mediation of hIAPP-induced apoptosis by the disturbance of intracellular Ca2+ homeostasis has already been shown in human pancreatic tissues (72). The significance of what we have demonstrated here is that this alteration of the levels of Ca2+ ions can also lead to an enhancement of hIAPP-mediated membrane damage via fibrillogenesis. In this case, because membrane disruption is the source of Ca2+ dysregulation, a positive feedback loop can be created whereby the influx of Ca2+ into the cell by hIAPP feeds forward into the enhancement of membrane disruption by Ca2+, creating a cycle that leads eventually to the death of the cell. Our observations suggest that elevated Ca2+ levels in T2DM may play an additional role beyond a passive role as a product of hIAPP-induced membrane disruption, and that unregulated Ca2+ homeostasis may also play an active role in the control of pathophysiological hIAPP–membrane interactions.
Finally, we note that because hIAPP itself does not bind Ca2+ (36,73), calcium’s effect on membrane permeabilization by hIAPP is due to its influence on the physical properties of the membrane, not to a particular structural feature of hIAPP itself. This implies that the changes in membrane structure that occur upon binding of Ca2+ are likely to influence other membrane active peptides and proteins as well. Although the effect of Ca2+ on membrane active peptides has not been extensively studied, it is known to inhibit membrane permeabilization by the antimicrobial membrane-disrupting peptides alamethicin (74) and gramicidin (75), amyloidogenic oligomers of lysozyme (76), and at least one type of nonfibrillar channel produced by oligomerization of the amyloidogenic protein α-synuclein, which has been implicated in Parkinson’s disease (77). These findings are consistent with our observation that Ca2+ inhibits membrane permeabilization by prefibrillar hIAPP, which may involve a channel-like mechanism (8,78–81). To our knowledge, the enhancement of membrane fragmentation by Ca2+ has not been reported before. However, membrane fragmentation is frequently observed for many antimicrobial peptides (82) and has also been reported for other amyloidogenic proteins, such as Aβ1-40 (83,84) and α-synuclein (85,86). Because calcium dysregulation is intimately involved with apoptosis and many other cellular processes (87), future studies examining the generality of Ca2+’s effect on membrane active proteins are vital to understand the in vivo effects of these proteins.
Acknowledgments
We thank Dr. Subramanian Vivekanandan for help with NMR measurements.
This study was supported by research funds from the National Institutes of Health (DK078885 to A.R.) and the University of Catania.
Contributor Information
Ayyalusamy Ramamoorthy, Email: ramamoor@umich.edu.
Carmelo La Rosa, Email: clarosa@unict.it.
Supporting Material
References
- 1.Sipe J.D., Cohen A.S. Review: history of the amyloid fibril. J. Struct. Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 2.Moore C.X., Cooper G.J.S. Co-secretion of amylin and insulin from cultured islet β-cells: modulation by nutrient secretagogues, islet hormones and hypoglycemic agents. Biochem. Biophys. Res. Commun. 1991;179:1–9. doi: 10.1016/0006-291x(91)91325-7. [DOI] [PubMed] [Google Scholar]
- 3.Clark A., Cooper G.J., Turner R.C. Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet. 1987;2:231–234. doi: 10.1016/s0140-6736(87)90825-7. [DOI] [PubMed] [Google Scholar]
- 4.Westermark P., Wilander E., Johnson K.H. Islet amyloid polypeptide-like immunoreactivity in the islet B cells of type 2 (non-insulin-dependent) diabetic and non-diabetic individuals. Diabetologia. 1987;30:887–892. doi: 10.1007/BF00274799. [DOI] [PubMed] [Google Scholar]
- 5.Betsholtz C., Svensson V., Westermark P. Islet amyloid polypeptide (IAPP):cDNA cloning and identification of an amyloidogenic region associated with the species-specific occurrence of age-related diabetes mellitus. Exp. Cell Res. 1989;183:484–493. doi: 10.1016/0014-4827(89)90407-2. [DOI] [PubMed] [Google Scholar]
- 6.Higham C.E., Jaikaran E.T., Clark A. Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Lett. 2000;470:55–60. doi: 10.1016/s0014-5793(00)01287-4. [DOI] [PubMed] [Google Scholar]
- 7.Lorenzo A., Razzaboni B., Yankner B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 8.Mirzabekov T.A., Lin M.C., Kagan B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 9.Haataja L., Gurlo T., Butler P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriarty D.F., Raleigh D.P. Effects of sequential proline substitutions on amyloid formation by human amylin20-29. Biochemistry. 1999;38:1811–1818. doi: 10.1021/bi981658g. [DOI] [PubMed] [Google Scholar]
- 11.Westermark P., Engström U., Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westermark P., Andersson A., Westermark G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 13.Anguiano M., Nowak R.J., Lansbury P.T., Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41:11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 14.Konarkowska B., Aitken J.F., Cooper G.J. The aggregation potential of human amylin determines its cytotoxicity towards islet β-cells. FEBS J. 2006;273:3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 15.Smith P.E.S., Brender J.R., Ramamoorthy A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: the case of islet amyloid polypeptide. J. Am. Chem. Soc. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brender J.R., Salamekh S., Ramamoorthy A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc. Chem. Res. 2012;45:454–462. doi: 10.1021/ar200189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hebda J.A., Miranker A.D. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annu Rev Biophys. 2009;38:125–152. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 18.Last N.B., Rhoades E., Miranker A.D. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc. Natl. Acad. Sci. USA. 2011;108:9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lashuel H.A., Lansbury P.T., Jr. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 20.Scalisi S., Sciacca M.F., La Rosa C. Self-assembling pathway of HiApp fibrils within lipid bilayers. ChemBioChem. 2010;11:1856–1859. doi: 10.1002/cbic.201000090. [DOI] [PubMed] [Google Scholar]
- 21.Sparr E., Engel M.F.M., Killian J.A. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577:117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 22.Engel M.F., Khemtémourian L., Höppener J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green J.D., Kreplak L., Aebi U. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J. Mol. Biol. 2004;342:877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 24.Brender J.R., Lee E.L., Gafni A. Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophys. J. 2011;100:685–692. doi: 10.1016/j.bpj.2010.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jayasinghe S.A., Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 26.Gehman J.D., O’Brien C.C., Separovic F. Metal effects on the membrane interactions of amyloid-β peptides. Eur. Biophys. J. 2008;37:333–344. doi: 10.1007/s00249-007-0251-2. [DOI] [PubMed] [Google Scholar]
- 27.Fraser S.P., Suh Y.H., Djamgoz M.B.A. Ionic effects of the Alzheimer’s disease β-amyloid precursor protein and its metabolic fragments. Trends Neurosci. 1997;20:67–72. doi: 10.1016/s0166-2236(96)10079-5. [DOI] [PubMed] [Google Scholar]
- 28.Mattson M.P., Cheng B., Rydel R.E. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawahara M., Kuroda Y., Rojas E. Alzheimer’s β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- 30.Resnick L.M. Cellular calcium and magnesium metabolism in the pathophysiology and treatment of hypertension and related metabolic disorders. Am. J. Med. 1992;93(2A):11S–20S. doi: 10.1016/0002-9343(92)90290-r. [DOI] [PubMed] [Google Scholar]
- 31.Barbagallo M., Gupta R.K., Resnick L.M. Cellular ions in NIDDM: relation of calcium to hyperglycemia and cardiac mass. Diabetes Care. 1996;19:1393–1398. doi: 10.2337/diacare.19.12.1393. [DOI] [PubMed] [Google Scholar]
- 32.Squier T.C., Bigelow D.J. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci. 2000;5:D504–D526. doi: 10.2741/squier. [DOI] [PubMed] [Google Scholar]
- 33.Nitahara J.A., Cheng W., Anversa P. Intracellular calcium, DNase activity and myocyte apoptosis in aging Fischer 344 rats. J. Mol. Cell. Cardiol. 1998;30:519–535. doi: 10.1006/jmcc.1997.0616. [DOI] [PubMed] [Google Scholar]
- 34.Gerke V., Creutz C.E., Moss S.E. Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 35.Rescher U., Gerke V. Annexins—unique membrane binding proteins with diverse functions. J. Cell Sci. 2004;117:2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- 36.Sciacca M.F.M., Pappalardo M., La Rosa C. Calcium-activated membrane interaction of the islet amyloid polypeptide: implications in the pathogenesis of type II diabetes mellitus. Arch. Biochem. Biophys. 2008;477:291–298. doi: 10.1016/j.abb.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 37.Grasso D., Grasso G., Sóvágó I. Environmental effects on a prion’s helix II domain: copper(II) and membrane interactions with PrP180-193 and its analogues. Chemistry. 2005;12:537–547. doi: 10.1002/chem.200500534. [DOI] [PubMed] [Google Scholar]
- 38.Grasso D., Milardi D., Rizzarelli E. DSC study of the interaction of the prion peptide PrP106–126 with artificial membranes. New J. Chem. 2001;25:1543–1548. [Google Scholar]
- 39.Milardi D., Sciacca M.F.M., La Rosa C. The role of aromatic side-chains in amyloid growth and membrane interaction of the islet amyloid polypeptide fragment LANFLVH. Eur. Biophys. J. 2011;40:1–12. doi: 10.1007/s00249-010-0623-x. [DOI] [PubMed] [Google Scholar]
- 40.Stewart J.C. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980;104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 41.Merchant T.E., Glonek T. 31P NMR of tissue phospholipids: competition for Mg2+, Ca2+, Na+ and K+ cations. Lipids. 1992;27:551–559. doi: 10.1007/BF02536139. [DOI] [PubMed] [Google Scholar]
- 42.Nanga R.P.R., Brender J.R., Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schanda P., Forge V., Brutscher B. Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2007;104:11257–11262. doi: 10.1073/pnas.0702069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roux M., Bloom M. Calcium binding by phosphatidylserine headgroups. Deuterium NMR study. Biophys. J. 1991;60:38–44. doi: 10.1016/S0006-3495(91)82028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boettcher J.M., Davis-Harrison R.L., Rienstra C.M. Atomic view of calcium-induced clustering of phosphatidylserine in mixed lipid bilayers. Biochemistry. 2011;50:2264–2273. doi: 10.1021/bi1013694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vernier P.T., Ziegler M.J., Dimova R. Calcium binding and head group dipole angle in phosphatidylserine-phosphatidylcholine bilayers. Langmuir. 2009;25:1020–1027. doi: 10.1021/la8025057. [DOI] [PubMed] [Google Scholar]
- 47.Knight J.D., Miranker A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 48.Düzgüneş N., Nir S., Papahadjopoulos D. Calcium-induced and magnesium-induced fusion of mixed phosphatidylserine-phosphatidylcholine vesicles—effect of ion binding. J. Membrane Biol. 1981;59:115–125. doi: 10.1007/BF01875709. [DOI] [PubMed] [Google Scholar]
- 49.McLaughlin S., Mulrine N., McLaughlin A. Adsorption of divalent cations to bilayer membranes containing phosphatidylserine. J. Gen. Physiol. 1981;77:445–473. doi: 10.1085/jgp.77.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martín-Molina A., Rodríguez-Beas C., Faraudo J. Effect of calcium and magnesium on phosphatidylserine membranes: experiments and all-atomic simulations. Biophys. J. 2012;102:2095–2103. doi: 10.1016/j.bpj.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jan A., Adolfsson O., Lashuel H.A. Aβ42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Aβ42 species. J. Biol. Chem. 2011;286:8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brender J.R., Dürr U.H.N., Ramamoorthy A. Membrane fragmentation by an amyloidogenic fragment of human islet amyloid polypeptide detected by solid-state NMR spectroscopy of membrane nanotubes. Biochim. Biophys. Acta. 2007;1768:2026–2029. doi: 10.1016/j.bbamem.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasahara K., Morigaki K., Hamada D. Binding of islet amyloid polypeptide to supported lipid bilayers and amyloid aggregation at the membranes. Biochemistry. 2012;51:6908–6919. doi: 10.1021/bi300542g. [DOI] [PubMed] [Google Scholar]
- 54.Sciacca M.F.M., Brender J.R., Ramamoorthy A. Phosphatidylethanolamine enhances amyloid fiber-dependent membrane fragmentation. Biochemistry. 2012;51:7676–7684. doi: 10.1021/bi3009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sciacca M.F.M., Kotler S.A., Ramamoorthy A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sauerbrey G. Verwendung von schwingquarzen zur wagung dunner schichten und zur mikrowagung. Z. Phys. 1959;155:206–222. [Google Scholar]
- 57.Feiler A.A., Sahlholm A., Caldwell K.D. Adsorption and viscoelastic properties of fractionated mucin (BSM) and bovine serum albumin (BSA) studied with quartz crystal microbalance (QCM-D) J. Colloid Interface Sci. 2007;315:475–481. doi: 10.1016/j.jcis.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 58.Richter R., Mukhopadhyay A., Brisson A. Pathways of lipid vesicle deposition on solid surfaces: a combined QCM-D and AFM study. Biophys. J. 2003;85:3035–3047. doi: 10.1016/S0006-3495(03)74722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sasahara K., Hall D., Hamada D. Effect of lipid type on the binding of lipid vesicles to islet amyloid polypeptide amyloid fibrils. Biochemistry. 2010;49:3040–3048. doi: 10.1021/bi9019252. [DOI] [PubMed] [Google Scholar]
- 60.Brender J.R., Hartman K., Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nanga R.P.R., Brender J.R., Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP(1-19) in micelles by NMR spectroscopy. Biochemistry. 2008;47:12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glabe C.G., Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66(2, Suppl 1):S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- 63.Janson J., Ashley R.H., Butler P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 64.Lashuel H.A., Hartley D., Lansbury P.T., Jr. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 65.Wahlgren M., Arnebrant T., Lundstrom I. The adsorption of lysozyme to hydrophilic silicon-oxide surfaces—comparison between experimental data and models for adsorption kinetics. J. Colloid Interface Sci. 1995;175:506–514. [Google Scholar]
- 66.Patil S.M., Xu S.H., Alexandrescu A.T. Dynamic α-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balali-Mood K., Ashley R.H., Bradshaw J.P. Neutron diffraction reveals sequence-specific membrane insertion of pre-fibrillar islet amyloid polypeptide and inhibition by rifampicin. FEBS Lett. 2005;579:1143–1148. doi: 10.1016/j.febslet.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 68.Porat Y., Kolusheva S., Gazit E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry. 2003;42:10971–10977. doi: 10.1021/bi034889i. [DOI] [PubMed] [Google Scholar]
- 69.Parkash J., Chaudhry M.A., Rhoten W.B. Intracellular calcium ion response to glucose in β-cells of calbindin-D28k nullmutant mice and in βHC13 cells overexpressing calbindin-D28k. Endocrine. 2002;18:221–229. doi: 10.1385/ENDO:18:3:221. [DOI] [PubMed] [Google Scholar]
- 70.Taheri-Araghi S., Ha B.Y. Electrostatic bending of lipid membranes: how are lipid and electrostatic properties interrelated? Langmuir. 2010;26:14737–14746. doi: 10.1021/la102052r. [DOI] [PubMed] [Google Scholar]
- 71.Calloni G., Lendel C., Chiti F. Structure and dynamics of a partially folded protein are decoupled from its mechanism of aggregation. J. Am. Chem. Soc. 2008;130:13040–13050. doi: 10.1021/ja8029224. [DOI] [PubMed] [Google Scholar]
- 72.Huang C.J., Gurlo T., Butler P.C. Calcium-activated calpain-2 is a mediator of β cell dysfunction and apoptosis in type 2 diabetes. J. Biol. Chem. 2010;285:339–348. doi: 10.1074/jbc.M109.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brender J.R., Hartman K., Ramamoorthy A. Role of zinc in human islet amyloid polypeptide aggregation. J. Am. Chem. Soc. 2010;132:8973–8983. doi: 10.1021/ja1007867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hall J.E., Cahalan M.D. Calcium-induced inactivation of alamethicin in asymmetric lipid bilayers. J. Gen. Physiol. 1982;79:387–409. doi: 10.1085/jgp.79.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gambale F., Menini A., Rauch G. Effects of calcium on the gramicidin A single channel in phosphatidylserine membranes. Screening and blocking. Eur. Biophys. J. 1987;14:369–374. doi: 10.1007/BF00262322. [DOI] [PubMed] [Google Scholar]
- 76.Meratan A.A., Ghasemi A., Nemat-Gorgani M. Membrane integrity and amyloid cytotoxicity: a model study involving mitochondria and lysozyme fibrillation products. J. Mol. Biol. 2011;409:826–838. doi: 10.1016/j.jmb.2011.04.045. [DOI] [PubMed] [Google Scholar]
- 77.Zakharov S.D., Hulleman J.D., Cramer W.A. Helical α-synuclein forms highly conductive ion channels. Biochemistry. 2007;46:14369–14379. doi: 10.1021/bi701275p. [DOI] [PubMed] [Google Scholar]
- 78.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kagan B.L., Thundimadathil J. Amyloid peptide pores and the β sheet conformation. Adv. Exp. Med. Biol. 2010;677:150–167. doi: 10.1007/978-1-4419-6327-7_13. [DOI] [PubMed] [Google Scholar]
- 80.Kagan B.L., Jang H., Nussinov R. Antimicrobial properties of amyloid peptides. Mol. Pharm. 2012;9:708–717. doi: 10.1021/mp200419b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirakura Y., Kagan B.L. Amyloid peptide channels are inhibited by Congo Red and blocked by zinc. Biophys. J. 1999;76 A443–A443. [Google Scholar]
- 82.Bechinger B., Lohner K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 83.Nakazawa Y., Suzuki Y., Asakura T. The interaction of amyloid Aβ (1-40) with lipid bilayers and ganglioside as studied by 31P solid-state NMR. Chem. Phys. Lipids. 2009;158:54–60. doi: 10.1016/j.chemphyslip.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Michikawa M., Gong J.S., Yanagisawa K. A novel action of alzheimer’s amyloid β-protein (Aβ): oligomeric Aβ promotes lipid release. J. Neurosci. 2001;21:7226–7235. doi: 10.1523/JNEUROSCI.21-18-07226.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reynolds N.P., Soragni A., Seeger S. Mechanism of membrane interaction and disruption by α-synuclein. J. Am. Chem. Soc. 2011;133:19366–19375. doi: 10.1021/ja2029848. [DOI] [PubMed] [Google Scholar]
- 86.van Rooijen B.D., Claessens M.M.A.E., Subramaniam V. Membrane interactions of oligomeric α-synuclein: potential role in Parkinson’s disease. Curr. Protein Pept. Sci. 2010;11:334–342. doi: 10.2174/138920310791330659. [DOI] [PubMed] [Google Scholar]
- 87.Mattson M.P., Chan S.L. Calcium orchestrates apoptosis. Nat. Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.