

Abstract
Voltage-gated ion channels are crucial for regulation of electric activity of excitable tissues such as nerve cells, and play important roles in many diseases. During activation, the charged S4 segment in the voltage sensor domain translates across a hydrophobic core forming a barrier for the gating charges. This barrier is critical for channel function, and a conserved phenylalanine in segment S2 has previously been identified to be highly sensitive to substitutions. Here, we have studied the kinetics of Kv1-type potassium channels (Shaker and Kv1.2/2.1 chimera) through site-directed mutagenesis, electrophysiology, and molecular simulations. The F290L mutation in Shaker (F233L in Kv1.2/2.1) accelerates channel closure by at least a factor 50, although opening is unaffected. Free energy profiles with the hydrophobic neighbors of F233 mutated to alanine indicate that the open state with the fourth arginine in S4 above the hydrophobic core is destabilized by ∼17 kJ/mol compared to the first closed intermediate. This significantly lowers the barrier of the first deactivation step, although the last step of activation is unaffected. Simulations of wild-type F233 show that the phenyl ring always rotates toward the extracellular side both for activation and deactivation, which appears to help stabilize a well-defined open state.
Introduction
Voltage-gated potassium (Kv) channels are membrane proteins that form ion-conducting pores highly selective for K+ ions and transport these across the membrane (1). Because they play a fundamental role in the generation and propagation of the nerve impulse and in cell homeostasis, they are of high pharmaceutical relevance. Structurally, they are composed of a central pore for ion permeation formed by four subunits. Each subunit contains six transmembrane segments (S1–S6), where the first four helices form the voltage-sensor domain (VSD) that contains charged residues important for channel function.
The VSD undergoes conformational changes in response to variations in the membrane potential due to positively charged residues in the S4 helix being translated across the membrane (Fig. 1 A) (2–5). There is one up state (i.e., S4 closer to the extracellular side) that is referred to as active and a number of intermediate states with S4 positioned further down toward the intracellular side. The stable down state is referred to as a resting state, and it is only when all four S4 helices are activated that the channel can be opened (6,7). Currently, there are only crystal structures of the open state available (8,9) and exact atomic details about the gating process and kinetics are still lacking. In the crystal structure of the Kv1.2/2.1 chimera used in this work, the pore is open and the voltage sensor is in the up state.
Figure 1.
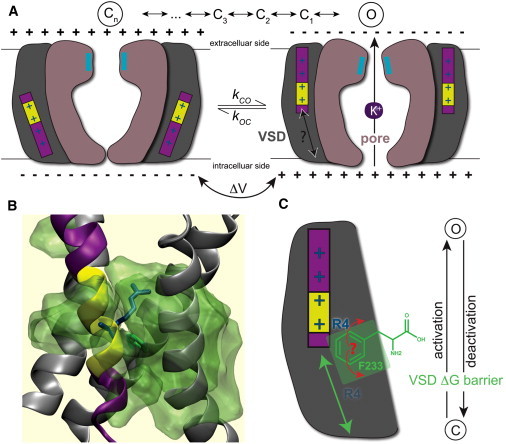
How different is activation from deactivation for the VSD S4 translation? (A) The translation of the charged S4 helix leads to a conformational change in the pore domain. The (de)activation comprises several intermediate states corresponding to S4 charges crossing a hydrophobic core, likely aided by a local 310-helix (yellow) segment sliding along the sequence as S4 translates. (B) Simulation system (white lipid chains, blue/red head-groups, and blue water) with the central hydrophobic core of the VSD is highlighted in transparent green. The R4 (R299) gating charge and F233 lock are drawn in blue, green sticks, respectively. (C) The phenyl ring has to rotate for the gating charges to cross, but is it merely pushed away in the direction of S4 translation, or can it help explain differences between activation and deactivation?
One distinguishes between the process of VSD activation accomplished through an upward/extracellular motion of S4 and VSD deactivation where the helix moves in a downward/intracellular direction. In the middle of the VSD there is a water-excluding portion formed by several hydrophobic residues (Fig. 1 B) that make a thin aperture for the S4 helix with its gating charges to move through. This hydrophobic core is ∼5 Å thick and clearly separates two cavities in contact with the extra- and intracellular regions, respectively (9,10). Channel deactivation likely involves several intermediate conformations of the VSD (11,12). Long et al. suggested the Kv1.2/2.1 chimera’s F233 play a role as a hydrophobic plug because it is a very conserved residue of the VSD and this might indicate a functional role in gating. This well-conserved aromatic residue is located in the middle of the S2 segment somewhat toward the intercellular side of the hydrophobic region (13). The importance of this residue for activation was first identified from conductance versus voltage G(V) relations as a function of mutations (14). More recently, Tao et al. (15) have shown that a rigid, cyclic (but not necessarily aromatic) side chain in the F233 position is important for gating, and that it interacts sequentially with all four gating charges. Recently, Lacroix and Bezanilla (16) showed through gating current measurements of several F233 mutants that a hydrophobic residue in this position primarily controls the transfer of the fourth gating charge (R4) across F233, whereas the movement of R1–R3 is not affected.
In this study, we have investigated the coupling between gating kinetics and the molecular-level dynamics of the F233 side chain. In particular, we are interested in the question whether activation and deactivation is the same process just occurring in opposite directions, or whether there are important differences (Fig. 1 C). Similar to Lacroix, we rely on voltage-clamp experiments for the experimental studies of (de)activation, but then use molecular dynamics (MD) simulations and free energy calculations to determine the related free energy barriers and F233 conformations on a molecular level. We have chosen to specifically focus on the translation of the fourth gating charge, R4 (R299 in Kv1.2/2.1), across the barrier, both because this last step of the activation (or first of deactivation) appears to be most influenced by F233 mutations, and because we have good models of the first intermediate state (C1) that gives us a reasonably accurate pathway between the two conformations (12).
Notably, a substitution of the F233 side chain to a tryptophan leads to a pronounced open state stabilization indicated by a left shift (i.e., more negative voltages required for deactivation) of 40 mV in G(V) (15). One possible explanation for this could be cation-π interactions. Aromatic side chains could catalyze the transmembrane passage of S4 charges through this type of interaction and F233 has been suggested to assist the S4 movement via this mechanism based on refined structural data of the Kv1.2 channel (13). Pless et al. (17) have also investigated tryptophan at position 233 (290 in Shaker) and found that the electronegative surface potential possibly contributes to an interaction with K5 (K302 in Kv1.2/2.1, or K374 in Shaker).
The transfer of the gating charges across the cell membrane is quite unfavorable due to the low dielectric environment of the lipid bilayer. To circumvent this problem the gating charges are not exposed to the hydrophobic part of the lipid bilayer but largely reside in the aqueous crevices. In previous studies (18,19) we showed that position 233 in the hydrophobic core is central for the free energy barrier for S4 translation. The rigid ring appears to determine the main barrier by enabling a switch-like transition between the open and resting states. Substitutions on this residue can modulate the deactivation of the voltage sensor and in this way influence the open propensity of the channel (19). The phenyl ring appears to be a lock or seal altering between two preferred positions; it either has a largely horizontal orientation with respect to the membrane plane where it effectively blocks the narrowest part of the voltage sensor cavity completely, or it rotates away to a largely vertical orientation where the R4 (R299) arginine side chain has the possibility to pass it (19).
The free energy barrier resulting from the arginines having to pass F233 has been shown to be quite high (18) (in particular, if the S4 helix is constrained to its cystal structure state), and likely a determining factor both for conformational changes on the molecular level and possibly the experimentally observed activation and deactivation kinetics. One possibility to overcome this barrier might be a secondary structure transition of the mostly α-helical S4 segment to partly adopt 310-helix structure (18,20–23). This would stretch the helix and also improve the salt bridge patterns between the gating charges and the glutamatic acids before and after the barrier. As suggested from recent models (12), this could even be limited to a short 310-helix region that slides in sequence during S4 helix translation (Fig. 1 A).
MD simulations allow us to compare F233’s behavior during the two opposite directions of R4 crossing the barrier, i.e., the first step of deactivation versus the last step of activation. For the modeling and simulation approaches we used the currently highest-resolution Kv1.2/2.1 structure (9) while the experimental studies are carried out on the Shaker K+ channel, primarily because the channel can be highly expressed in Xenopus oocytes, and it has very high sequence similarity to Kv1.2/2.1. Their voltage sensors should be largely functionally equivalent, though there are minor differences in sequence. For consistency the Kv1.2/2.1 numbering is used throughout the text, although the Kv1.2/2.1 chimera’s F233 corresponds to F290 in Shaker.
We found that the F233L mutation destabilizes the open state but does not appear to affect the activation. On the molecular level, the phenyl ring of F233 is consistently rotating upward regardless of the S4 translation direction in the simulations. The reason for this is likely the residue’s stabilization by hydrophobic residues above the ring, without any corresponding region below it. This appears to support a model where the gating charge transfer does not merely push the F233 side chain away, but the residue primarily stabilizes the open state of the channel and keeps it open longer. This could aid in giving potassium channels a slower deactivation response than they would have without this residue, which in turn could be a contributing factor to allow potassium ions to continue to flow out from the cell for a period of time and hyperpolarize the membrane even as the potential is becoming more negative (which would cause a more rapid response channel to close and cutoff the ion flux).
Methods
Electrophysiological experiments
For electrophysiological experiments, we expressed the Shaker H4 channel (24) incapable of fast inactivation (referred to as wild-type (WT) (25)) in Xenopus laevis oocytes. The F233L mutation was introduced as previously described (26). Oocyte preparation, cRNA synthesis and injection, and oocyte storage followed the procedure described previously (27). Animal experiments were approved by the local Animal Care and Use Committee at Linköping University. Ion currents were measured 3–6 days after cRNA injection using the two-electrode voltage-clamp technique as previously described (26). In brief, the holding voltage was set to −80 mV and ion currents measured at voltages between −80 and +75 mV (+50 mV for WT) in 5 mV increments. All experiments were performed at room temperature (∼20°C). The control solution contained in mM: 88 NaCl, 1 KCl, 15 HEPES, 0.4 CaCl2, and 0.8 MgCl2. pH was adjusted to 7.4 with NaOH yielding a final sodium concentration of 100 mM. Recordings of activation and deactivation kinetics were performed in a high-potassium solution where the NaCl in the control solution was replaced with KCl and pH set with KOH, yielding a final potassium concentration of 100 mM.
Electrophysiological analysis
The potassium conductance was calculated as
| (1) |
where is the steady-state current at the end of a 100-ms pulse, V the absolute membrane voltage, and the reversal potential for the K channel (set to −80 mV). V1/2 for WT and F233L were determined by fitting a single Boltzmann curve to the conductance data
| (2) |
where A is the amplitude, the midpoint, and s the slope of the curve. The activation and deactivation time constants were quantified by fitting an exponential curve to experimental data. Data are expressed as mean ± standard error.
In silico mutagenesis of hydrophobic residues in the VSD core
The F233L mutation was introduced using the PyMol Molecular Graphics System, version 1.2r3pre (DeLano Scientific, San Francisco, CA, http://www.pymol.org) mutagenesis plugin. In another system several residues (V172, I173, C229, I230, I231, and I263) in the hydrophobic core were mutated to alanine to assess their influence on the barrier. Each mutation was followed by steepest descent energy minimization for 1000 steps.
Simulation model of a voltage sensor domain
Helices S1 through S4 from the Kv1.2/2.1 chimera (PDB ID 2R9R) (9) were used to model an isolated voltage sensor (9). The simulation system was setup as previously described (18), with the Amber99SB-ILDN force field (29). All simulations were carried out with Gromacs 4.5.3 (30) using 5 fs time steps with virtual interaction sites for hydrogens. The final simulation system is presented in Fig. 1 A.
Steered MD and potential of mean force (PMF) calculations
In a previous study (12), models of several metastable intermediate states between open and resting states of the channel were generated from experimental constraints, resulting in both an open (O) state and a sequence of successively more closed conformations (at least C1, C2, C3). To investigate the difference between activation and deactivation, steered MD trajectories for the last step of activation (C1 → O) and the first step of deactivation (O → C1) were used to compare the processes (18,19). To enable equilibrium free energy calculations, frames with the S4 helix translation position (along its axis) spaced 0.2 Å apart were also selected from the trajectories. PMF curves were calculated by using umbrella sampling on the relative distance between the charged side chains in S4 (R0-R6) and the reference group formed by helices S1–S3. The umbrella harmonic force constant was set to 10,000 kJ/mol/nm2 and the reference distance in each window was kept constant during the run. Each umbrella point was run for 50 ns to ensure relaxation. The first 20 ns were discarded and the last 30 ns used for production. Standard errors were estimated by splitting the data into three parts and using jackknife statistics. PMF curves were calculated from the ensemble of simulations using the Gromacs g_wham program (30) based on the Kumar et al. weighted histogram analysis method (31).
Calculation of hydrophobic interactions and the F233 ring’s accessible surface area
Lennard-Jones van der Waals interactions were calculated for each hydrophobic residue in the core region against the F233 phenyl ring. In addition, the accessible surface area was computed using the Gromacs g_sas program (30), and the solvent accessible surface for all hydrophobic residues in contact with the F233 phenyl ring calculated both for WT and mutants simulations.
Results
The Kv1.2/2.1 F233L mutant requires more positive voltages for channel opening
To address the question whether the activation and deactivation processes have different characteristics, we started with functional recording of their kinetics for the Kv1.2/2.1 F233 (Shaker F290) WT channel in comparison to the F233L (Shaker F290L) mutant. Leucine was chosen as the mutant because it has been shown computationally that this single point mutation reduces the free energy barrier significantly (19). Fig. 2 A shows a representative current family for the F233L mutant in control solution, displaying WT-like currents. However, the voltage dependence of the channel is significantly affected by the F233L mutation (Fig. 2 B). for F233L is +23.2 ± 2.4 mV (n = 13) compared to −20.4 ± 0.5 mV (n = 5) for WT.
Figure 2.
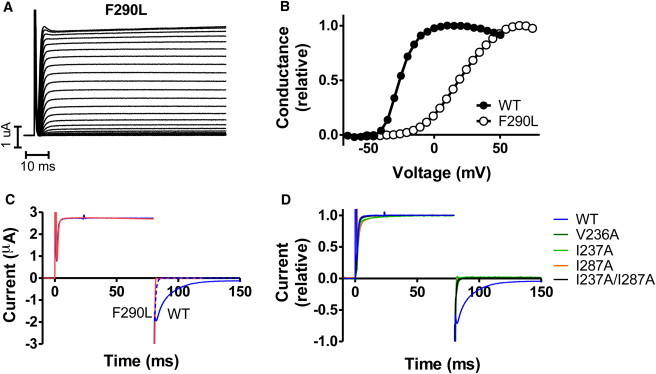
The F233L mutation destabilizes the open state. (A) Representative current family of the Kv1.2/2.1 F233L mutation (Shaker F290L) in control solution. The holding voltage was set to −80 mV and test pulses ranging from −80 to +75 mV in 5 mV increments. (B) Representative curves for WT and F233L (Shaker F290L). The F233L mutation shifts the channel voltage dependence toward more positive voltages. (C) Representative currents for channel activation (stepping to +50 mV from a holding voltage of −80 mV) and deactivation (stepping to −50 mV from an activation voltage of +50 mV) for WT (blue), and Shaker F290L (red). Legend residue numbers refer to Shaker. Experiments were performed in a high-potassium solution (see Methods). The activation is not affected by the F233L mutation, although channel deactivation is speeded up dramatically. The dashed blue curve is WT closure speeded up by a factor of 10. F290L is clearly faster. Panel D displays the same condition as in C but relative currents for mutations in the hydrophobic ring surrounding Shaker F290 (Kv1.2/2.1 F233).
In the simplest possible gating model with single closed (C) and open (O) states
| (3) |
a shift of the can be explained either by a stabilization of the closed state C (smaller rate constant ), and/or by a destabilization of the open state O (larger ). If the rate constants are calculated as
| (4) |
and
| (5) |
respectively, where and are the rates and voltages when and are equal, whereas and are the valencies associated with the respective transitions , the quotient of the factors by which the closing and opening rates are then affected can easily be calculated as
| (6) |
If mV and (close to WT data for the relation) then . This means that either the opening is slowed down very significantly, or the closing is similarly speeded up by the same amount, or a combination of the two effects. Kinetic simulations (see the Supporting Material) indicate that a slightly smaller factor is required if several closed states are included in the model and only the closing transition is affected.
Activation and deactivation kinetics appear to have different characteristics
To explore whether the opening is slowed down or the closing is accelerated we measured the time constants for opening and closing. Fig. 2 C shows that the opening at +50 mV for WT (blue) and F233L (red) overlap perfectly. It is noteworthy that these two recordings have almost identical steady-state amplitudes, which simplifies a direct comparison. The opening time constant is 1.0 ± 0.1 ms (n = 3) for WT and 1.1 ± 0.2 ms (n = 3) for F223L. After the opening at +50 mV the membrane voltage is stepped to −50 mV to close the channel. Because this experiment is carried out in the high-potassium solution the instantaneous current at −50 mV is expected to be of the same size but of opposite sign. The closure at −50 mV for WT (blue line) has a time constant of 10.1 ± 0.9 ms (n = 3). In sharp contrast, the closure of F233L is as fast as or faster than the time resolution of the experimental setup; that is, the closure is at least 20 times faster, probably more, for F233L than for WT. This suggests F233 is only rate limiting for channel closure and that the F233L mutation could destabilize the open state. These results indicate that the activation and deactivation processes might have important differences on the molecular level.
The F233 phenyl ring rotates upward for both activation and deactivation
To understand the reason for this more complex behavior and why the F233L mutation only speeds up channel deactivation we probed channel gating further through MD and free energy calculation approaches. Both we and others have previously suggested F233 as an important part of the barrier (9,19). A prominent feature in the simulations is how the F233 side chain rotates away to make room for each arginine that flips its side chain across the hydrophobic core (19). This local flipping of the crossing arginine in combination with the F233 rotation appears to be supported by the recent gating simulation of Jensen et al. (32). To investigate whether F233 has something to do with the activation versus deactivation differences, we have studied the ring position by measuring the distance between the center of mass of the phenyl ring relative to the F233 Cα atom. Interestingly, both in the activation (C1 → O) and deactivation (O → C1) steered simulations the F233 side chain rotates toward the extracellular side (or upward in the normal way of illustrating the channel) (Fig. 3 A). Both for activation and deactivation, the ring is displaced up to 3.5 Å when R4 crosses the barrier. The average relative center of mass-Cα position along the z axis when the lock is open is +3 Å with the ring having an almost vertical orientation, in contrast to the closed-lock position that has an average close to 0 Å, and the ring is virtually horizontal in that case (Fig. 3 A). It is noteworthy that conformations with the F233 ring pointing to the intracellular side hardly occur, even when the S4 helix is moving in this direction. It is only at the very beginning when R4 starts to push on the ring that we see a short transient displacement of −1 Å, but the lock rapidly moves back to a stable conformation without the arginine having passed. These results support the idea that the F233 side-chain dynamics is the source of the experimentally determined differences between activation and deactivation kinetics. Replacing the phenylalanine with a tryptophan has been shown to lead to an even higher energy barrier (15,19), and this is confirmed in simulations where we observe similar orientations as for the phenylalanine during R4 crossing, but in this case R4 has to perform an even longer flip compared to the WT (Fig. 3 C).
Figure 3.
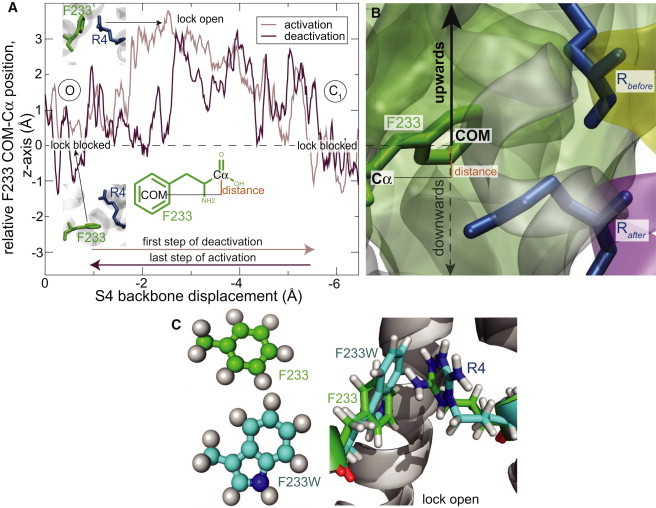
F233 rotates extracellularly for both activation and deactivation. (A) z-position of F233 side chain relative to Cα, averaged over 1 ns. The ring rotates upward largely along the same path for both activation and deactivation rather than merely being pushed in the translation direction. This could explain the gating kinetics differences, because the ring needs room to move upward during deactivation, whereas it can be pushed away under activation. Insets show the ring orientation for the indicated blocked/open positions. (B) Snapshot of the initial F233 orientation. The upward rotation is preferred both when R4 approaches the hydrophobic zone from above (deactivation, Rbefore) and below (activation, Rafter). (C) The F233W side chain exhibits similar rotation behavior as WT, rotating to the extracellular side in both cases. The main difference is the R4 side chain having to make a longer flip to find room when crossing F233W.
Nonpolar interactions to F233 are crucial for stabilizing the phenyl lock in the blocked orientation
To understand why the phenyl ring is predominantly stabilized in the horizontal direction, and how an extracellular direction rotation could affect deactivation more than activation, we analyzed the interactions between F233 and other hydrophobic residues in the core of the VSD. Ideally, one would like to mimic the experimental results with a free energy profile of F233L, but this is a small change combined with a model of the C1 state and a transition pathway derived from simulations on much shorter timescales than the experiments. Instead, we attempt to create a test with a stronger effect by identifying seven residues (V172, I173, C229, I230, I231, A262, and I263) with strong van der Waals interactions to the F233 phenyl ring (Fig. 4 A). All these residues show interaction energies in the range of 5–15 kJ/mol that they lose when the phenylalanine ring rotates into the open-lock orientation (cf. Fig. 4, A and B). For instance, the strongest interaction is 15 kJ/mol with I230 when the ring is in its blocked orientation and still 10 kJ/mol when the ring is rotated away to open the lock. The residues found are all located in the S1–S3 helices, close to F233 and form a semicircle-like arrangement above and around the phenyl ring as visualized in Fig. 4, C and D. This location above the ring appears to be an important factor to make the upward rotation more favorable.
Figure 4.
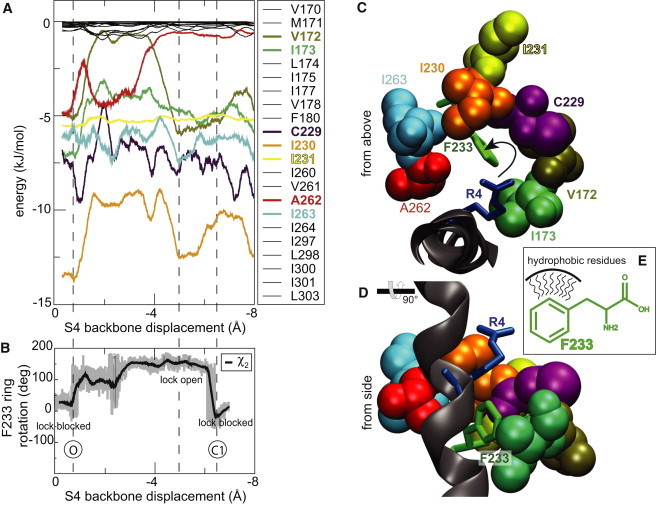
F233 is stabilized by van der Waals interactions to its neighbors. (A) Interaction energies between F233 and neighbor residues, with important residues marked in bold. Several neighbors appear to stabilize the blocked orientation of the lock. I230 shows the strongest interactions with energies approaching 15 kJ/mol. The F233 ring starts to rotate after 1 Å of translation, which is also where the interactions are reduced. After R4 has crossed (4–5 Å) the ring rotates back and regains its interactions. (B) Corresponding phenyl ring rotation around χ2 as a function of S4 translation. The hydrophobic residues in a semicircle above (C) and around (D) F233 help shield/stabilize the side chain (E) and favor the extracellular rotation of the phenyl ring.
To measure whether the hydrophobic residues stabilize the phenyl ring we conducted a control simulation where all seven hydrophobic residues identified were substituted for alanine that is less hydrophobic. After repeating the free energy calculation for this mutant, the result is a drop in the free energy barrier for the O → C1 transition of ∼15 kJ/mol. Because the difference between the barrier peak and C1 state is roughly maintained, we interpret this primarily as a destabilization of the open state since favorable interactions are lost, which agrees very well with the electrophysiology experiments. Fig. 5 A displays the profiles adjusted to match in the C1 state. This is obviously an arbitrary relative choice; it could also be interpreted as relative stabilization both of the barrier peak and C1 states. Part of the effect appears to come from more water entering the hydrophobic core; the solvent-accessible surface (Fig. 5 B) for these residues almost doubles with R4 crossing the barrier. Both the less hydrophobic core region in the VSD as well as the increased solvent accessibility is likely to contribute to the relative destabilization of the open state. Comparing with the Van der Waals energies (i.e., enthalpies), part of this might be explained from the I230 and A262 interaction differences between O and C1, but a large part is likely due to entropy effects where the residues might pack better in the O state where all gating charges have moved above the hydrophobic core.
Figure 5.
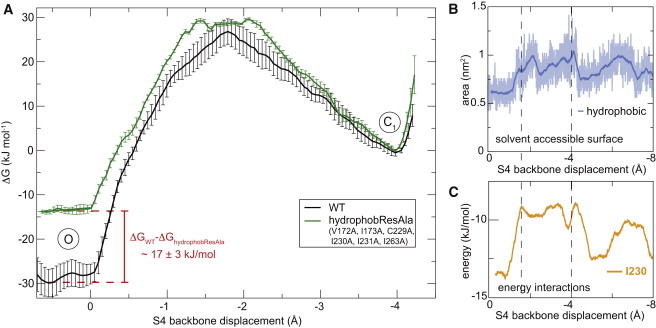
Perturbing interactions between F233 and hydrophobic neighbors destabilizes the open state. To control whether the interactions between F233 and its hydrophobic neighbors stabilize the open state, a control simulation was performed with these residues mutated to alanine. (A) The resulting PMF shows that the mutant has the open state relatively destabilized by roughly 17 kJ/mol, and a correspondingly lower deactivation barrier to C1. This agrees well with the experimental F233L gating kinetics shift also being explained by open state destabilization. (B) As the phenyl ring rotates away to allow R4 to pass, the solvent accessible surface of the hydrophobic residues increases transiently, largely matching the shape of the change in van der Waals energy (C) (cf. Fig. 4A).
The computational results predict that mutations of residues in the hydrophobic core that F233 is interacting with will increase the deactivation rate but not the activation rate. To critically assess this, we experimentally introduced corresponding mutations in the Shaker K channel and measured opening and closing kinetics as well as the G(V). Three residues located close to the F233 side chain in the Kv1.2/2.1 chimera structure were selected and mutated to alanines: V236A (V172A in the Kv1.2/2.1 chimera nomenclature; in S1), I237A (I173A in Kv1.2/2.1; in S1), and I287A (I230A in Kv1.2/2.1; in S2). Two combinations of the three mutations were also tested: V236A/I287A and I237A/I287A. All mutants except V236A/I287A expressed well. In all cases, the expressing mutants shifted the G(V) in a positive direction along the voltage axis (Table 1). The opening kinetics after a +50 mV stepping was hardly affected, whereas closing kinetics became faster for all the selected mutations. The kinetics was 13 times faster for V236A and at least 20 times faster for the other three mutations where the closing speed is faster than the experimental resolution (Fig. 2 D, Table 1). Thus, the additional electrophysiological tests clearly support the conclusion from the MD simulations that Shaker F290 appears to be held in position by surrounding hydrophobic residues, and that the phenylalanine must move out of this position for the S4 helix to move between kinetic states.
Table 1.
Experimental G(V) shifts and kinetics data for new mutants
| V1/2 (mV) | (ms) | (ms) | ||||
|---|---|---|---|---|---|---|
| WT | −20.4 ± 0.5 | n = 5 | 1.0 ± 0.1 | n = 3 | 10.1 ± 0.9 | n = 3 |
| F290L | +23.2 ± 2.4 | n = 13 | 1.1 ± 0.2 | n = 3 | <0.5 | n = 3 |
| V236A | −2.6 ± 1.1 | n = 8 | 1.1 ± 0.1 | n = 8 | 0.8 ± 0.1 | n = 7 |
| I237A | +34.5 ± 5.1 | n = 4 | 1.6 ± 0.2 | n = 6 | <0.5 | n = 6 |
| I287A | +17.7 ± 4.4 | n = 3 | 1.8 ± 0.3 | n = 4 | <0.5 | n = 4 |
| V236A/I287A | no expression | |||||
| I237A/I287A | +18.4 ± 0.6 | n = 3 | 1.2 ± 0.1 | n = 3 | <0.5 | n = 3 |
All mutations shift G(V) in a positive direction, and there is very little or no effect on the opening kinetics compared to WT, whereas the closing is accelerated by more than one order of magnitude in all cases, and for several mutants it is too fast to measure, which corresponds to at least a 20-fold speedup.
Discussion
The experimental studies on Shaker F290L showed quite normal opening kinetics with a time constant around 1 ms, but highly accelerated closing compared to WT. In addition, the voltage-dependence of the open probability was shifted to higher voltages by 44 mV. Together this indicates a destabilization of the open state. This fits well with the recent measurements of gating currents by Lacroix et al. (16) who showed that only the backward (deactivation) transition of the final activation step was affected for a number of F290 mutants. The kinetic effects on ion currents are larger than the Lacroix results for gating currents; this might be explained from the ion currents primarily being sensitive to the last S4 opening transition (C1 to O), which fits the MD part of the work well, whereas gating currents report on all steps. Similarly, in MD simulations of K channel gating by Jensen et al. (32), R4 appears to have a larger latency and to be slower to cross Kv1.2/2.1 F233 compared to the other charges. Going beyond the phenyl ring, the present simulations and experiments show this is not merely an isolated effect from the side chain, but rather explained by its interactions with the surrounding hydrophobic residues. Interestingly, these interactions appear to be responsible for the aromatic ring virtually always rotating toward the extracellular side, both during activation and deactivation. Had the phenyl ring merely been an unspecific steric hindrance the naive model would have been for the S4 charges to always push the ring in the translation direction.
Removing the hydrophobic interactions between Kv1.2/2.1 F233 and its neighbors through mutations has large effects on gating and destabilizes the open state of the VSD relative to the C1 state, although it hardly changes the transition state free energy (the peak of the barrier) relative to C1. This supports a highly important role for the interactions between F233 and its hydrophobic neighbors to stabilize the open state, which fits well with studies by Campos et al. (33) who showed that Shaker I287H results in a proton current. The hydrophobic stabilization idea also agrees well with results of Lacroix (16) who found the speed of VSD deactivation to be linearly correlated with the hydrophobicity of substituted side chains in the phenylalanine position. In the present work, this residue is found to have the strongest interactions with the F233 ring, although it should be noted that the other residue proposed to be functionally important (I177) does not show any strong influence on the free energy barrier between the O and C1 states studied here.
In particular, our results suggest that the activated state (i.e., the upstage) of the voltage sensors is clearly stabilized by interactions between the F233 phenyl ring and its hydrophobic neighbors, and mutations of these hydrophobic residues specifically destabilize the open state and cause a dramatic acceleration in deactivation gating kinetics, although it appears to have no effect at all on activation kinetics. These differences in gating kinetics also seem to be supported by a model where the phenyl ring can be pushed away during (rapid) activation, whereas the deactivation process requires the same ring to search for a low free-energy conformation in the opposite direction to the motion of gating charges. These conformational changes might be similar to (e.g., protein folding processes), where the folding is slow due to the entropic character of the barrier, although the unfolding direction is enthalpic in nature and much faster.
Both the present results and Lacroix (16) support the conclusion that it is mainly the O state that is changed by the mutations, whereas C1 is largely unaffected (Fig. 5 A), and this is likely to hold also for the more closed C2 and C3 states, represented, e.g., by our previous models (12). There might still be limited effects on the free energy barrier between the more closed states from removing the aromatic ring, but a free energy landscape where the C2 and C3 states are of roughly the same depth as C1 would be compatible with the observed behavior that the voltage sensor is either relatively stable in the open state, or it moves all the way down to the resting state.
F233 with its interactions thus appears to be directly responsible for the slow deactivation, with the phenyl ring simply forcing the channel to stay open longer despite an increasing negative membrane potential. Considering this residue is virtually perfectly conserved in voltage-gated potassium channels, it is likely not a coincidence. It could even be important for the shape of the action potential; at the peak of the action potential the sodium channels inactivate, which causes the intracellular voltage to drop because cations ions are still leaving the cell through the open potassium channels. If these potassium channels had a rapid deactivation response to voltage the channels might very well close too fast rather than allowing ions to continue to flow out from the cell for a period of time and achieve the membrane hyperpolarization necessary to prepare for the next action potential. This type of selective (de-)stabilization of channel states might be important for several processes, for instance neurotoxins known to specifically bind to the resting state (34,35), or polyunsaturated fatty acid interactions that shift the G(V) curve in the opposite direction (4).
Acknowledgments
The authors thank P. Bjelkmar and Berk Hess for many helpful discussions.
This work was supported by grants from the European Research Council (209825), the Swedish Foundation for Strategic Research, the Swedish Research Council, the Swedish Heart-Lung Foundation, the Swedish Brain Foundation, the County Council of Östergötland, Queen Silvias Anniversary Foundation, King Gustaf V and Queen Victoria’s Freemasons Foundation, and the Swedish Society for Medical Research. Computer resources were provided through the Swedish e-Science Research Center and Swedish National Infrastructure for Computing (SNIC 020/11-41).
Supporting Material
References
- 1.Hille B. 3rd ed. Sinauer Associates; Sunderland, MA: 2001. Ionic Channels in Excitable Membranes. [Google Scholar]
- 2.Larsson H.P., Baker O.S., Isacoff E.Y. Transmembrane movement of the shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 3.Starace D.M., Stefani E., Bezanilla F. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 1997;19:1319–1327. doi: 10.1016/s0896-6273(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 4.Börjesson S.I., Hammarström S., Elinder F. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys. J. 2008;95:2242–2253. doi: 10.1529/biophysj.108.130757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahern C.A., Horn R. Focused electric field across the voltage sensor of potassium channels. Neuron. 2005;48:25–29. doi: 10.1016/j.neuron.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 6.Zagotta W.N., Hoshi T., Aldrich R.W. Shaker potassium channel gating. II: transitions in the activation pathway. J. Gen. Physiol. 1994;103:279–319. doi: 10.1085/jgp.103.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pathak M.M., Kurtz L., Isacoff E. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 2005;125:57–69. doi: 10.1085/jgp.200409197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 9.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 10.Shafrir Y., Durell S.R., Guy H.R. Models of voltage-dependent conformational changes in NaChBac channels. Biophys. J. 2008;95:3663–3676. doi: 10.1529/biophysj.108.135335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delemotte L., Tarek M., Treptow W. Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2011;108:6109–6114. doi: 10.1073/pnas.1102724108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henrion U., Renhorn J., Elinder F. Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc. Natl. Acad. Sci. USA. 2012;109:8552–8557. doi: 10.1073/pnas.1116938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X., Wang Q., Ma J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA. 2010;107:11352–11357. doi: 10.1073/pnas.1000142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li-Smerin Y., Hackos D.H., Swartz K.J. alpha-helical structural elements within the voltage-sensing domains of a K(+) channel. J. Gen. Physiol. 2000;115:33–50. doi: 10.1085/jgp.115.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao X., Lee A., MacKinnon R. A gating charge transfer center in voltage sensors. Science. 2010;328:67–73. doi: 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lacroix J.J., Bezanilla F. Control of a final gating charge transition by a hydrophobic residue in the S2 segment of a K+ channel voltage sensor. Proc. Natl. Acad. Sci. USA. 2011;108:6444–6449. doi: 10.1073/pnas.1103397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pless S.A., Galpin J.D., Ahern C.A. Contributions of counter-charge in a potassium channel voltage-sensor domain. Nat. Chem. Biol. 2011;7:617–623. doi: 10.1038/nchembio.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwaiger C.S., Bjelkmar P., Lindahl E. 310-helix conformation facilitates the transition of a voltage sensor S4 segment toward the down state. Biophys. J. 2011;100:1446–1454. doi: 10.1016/j.bpj.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwaiger C., Börjesson S., Lindahl E. The deactivation free energy barrier in a voltage sensor domain is altered by substitutions in the voltage sensor domain. PLoS ONE. 2012;7:e45880. doi: 10.1371/journal.pone.0045880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villalba-Galea C.A., Sandtner W., Bezanilla F. S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. USA. 2008;105:17600–17607. doi: 10.1073/pnas.0807387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bjelkmar P., Niemelä P.S., Lindahl E. Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLOS Comput. Biol. 2009;5:e1000289. doi: 10.1371/journal.pcbi.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalili-Araghi F., Jogini V., Schulten K. Calculation of the gating charge for the Kv1.2 voltage-activated potassium channel. Biophys. J. 2010;98:2189–2198. doi: 10.1016/j.bpj.2010.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schow E.V., Freites J.A., Tobias D.J. Down-state model of the voltage-sensing domain of a potassium channel. Biophys. J. 2010;98:2857–2866. doi: 10.1016/j.bpj.2010.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamb A., Tseng-Crank J., Tanouye M.A. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1988;1:421–430. doi: 10.1016/0896-6273(88)90192-4. [DOI] [PubMed] [Google Scholar]
- 25.Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 26.Börjesson S.I., Elinder F. An electrostatic potassium channel opener targeting the final voltage sensor transition. J. Gen. Physiol. 2011;137:563–577. doi: 10.1085/jgp.201110599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Börjesson S.I., Parkkari T., Elinder F. Electrostatic tuning of cellular excitability. Biophys. J. 2010;98:396–403. doi: 10.1016/j.bpj.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reference deleted in proof.
- 29.Lindorff-Larsen K., Piana S., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hess B., Kutzner C., Lindahl E. GROMACS 4.0: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S., Bouzida D., Rosenberg J. The weighted histogram analysis method for free-energy calculations on biomolecules. 1. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 32.Jensen M.Ø., Jogini V., Shaw D.E. Mechanism of voltage gating in potassium channels. Science. 2012;336:229–233. doi: 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- 33.Campos F.V., Chanda B., Bezanilla F. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc. Natl. Acad. Sci. USA. 2007;104:7904–7909. doi: 10.1073/pnas.0702638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swartz K.J., MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 1997;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- 35.Lee H.C., Wang J.M., Swartz K.J. Interaction between extracellular Hanatoxin and the resting conformation of the voltage-sensor paddle in Kv channels. Neuron. 2003;40:527–536. doi: 10.1016/s0896-6273(03)00636-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.