

Abstract
Using both atomistic and coarse-grained (CG) models, we compute the three-dimensional stress field around a gramicidin A (gA) dimer in lipid bilayers that feature different degrees of negative hydrophobic mismatch. The general trends in the computed stress field are similar at the atomistic and CG levels, supporting the use of the CG model for analyzing the mechanical features of protein/lipid/water interfaces. The calculations reveal that the stress field near the protein-lipid interface exhibits a layered structure with both significant repulsive and attractive regions, with the magnitude of the stress reaching 1000 bar in certain regions. Analysis of density profiles and stress field distributions helps highlight the Trp residues at the protein/membrane/water interface as mechanical anchors, suggesting that similar analysis is useful for identifying tension sensors in other membrane proteins, especially membrane proteins involved in mechanosensation. This work fosters a connection between microscopic and continuum mechanics models for proteins in complex environments and makes it possible to test the validity of assumptions commonly made in continuum mechanics models for membrane mediated processes. For example, using the calculated stress field, we estimate the free energy of membrane deformation induced by the hydrophobic mismatch, and the results for regions beyond the annular lipids are in general consistent with relevant experimental data and previous theoretical estimates using elasticity theory. On the other hand, the assumptions of homogeneous material properties for the membrane and a bilayer thickness at the protein/lipid interface being independent of lipid type (e.g., tail length) appear to be oversimplified, highlighting the importance of annular lipids of membrane proteins. Finally, the stress field analysis makes it clear that the effect of even rather severe hydrophobic mismatch propagates to only about two to three lipid layers, thus putting a limit on the range of cooperativity between membrane proteins in crowded cellular membranes.
Introduction
Lipid membranes have traditionally been regarded as a passive medium (1) that hosts transmembrane proteins and forms a barrier that regulates the permeation of water and small solutes (2). Therefore, a simple theoretical framework (3–6) that replaces the membrane as a low dielectric slab (e.g., ) of finite thickness (20–30 Å) has been found useful for modeling of large transmembrane proteins (7–9), coupled folding/insertion of transmembrane helices (10), and predicting binding orientation of membrane proteins to lipid membranes (11,12).
On the other hand, it has become increasingly clear that for many membrane-mediated processes, membrane deformations of different scales are intimately involved. In addition to the striking examples of large-scale membrane fusion and remodeling (13), more local deformations, induced by the presence of charged residues (14), for example, have been implicated in the function of the voltage-sensing domain of ion channels (15). Moreover, it has been shown that the chemical and mechanical properties of lipid membranes may have a significant impact on the activities of many transmembrane proteins (16–18). The most remarkable example in this context is mechanosensitive channels, which are gated by tension or other forms of mechanical perturbation applied to the membrane (19,20).
To better understand how membrane properties influence the function of membrane proteins, it is essential to characterize the two interfaces that are present. The first is the water/membrane interface, which has been well studied and is relatively well understood (21). An important property associated with the water/membrane interface is the lateral pressure profile, which reveals heterogeneous stress distribution along the membrane normal direction (22,23). The pressure profiles of pure lipid bilayers have been calculated using both atomistic and coarse-grained simulations (23,24), and they provide a framework for connecting microscopic models to continuum mechanics models of membranes (25).
The second interface of relevance is the protein/membrane interface, which is of interest to mechanistic discussions because the stress field around proteins determines how they respond mechanically to the environment. However, few calculations at either the atomistic or coarse-grained levels have been reported so far, because these calculations are computationally demanding (see below). Even for the discussion of mechanosensitive channels, many previous studies have explored only the pressure profiles for pure lipid bilayers (23), thus assuming that the stress field in the vicinity of membrane proteins is similar to that in flat bilayers (22,26,27). Whether this is an appropriate assumption for the discussion of membrane-protein regulation remains to be clarified, since it has become fairly well accepted that lipid molecules surrounding a membrane protein (annular lipids) behave rather differently from those in the bulk (28).
One ideal system for characterizing the stress field appears to be the bacterial mechanosensitive channel of large conductance (MscL), which undergoes a significant structural transition in response to tension in the membrane and other agents of deformation (e.g., curvature generation induced by asymmetric insertion of cone-shaped lipids) (29–31). A direct characterization of the stress field around MscL, especially changes induced by perturbations in the membrane, can help highlight protein regions that act as tension sensors. Recently, Marrink and co-workers carried out precisely such calculations using coarse-grained molecular dynamics (MD) simulations (32). Although that study represents a major step forward, it provided only limited mechanistic insight, because the MscL surface is highly heterogeneous and the cylindrically averaged stress field included contributions from many different residue types at the protein/lipid interface; this complexity undermined the connection of the stress field to residue-specific contributions. Moreover, the authors did not address the issue of modulation of the stress field by hydrophobic mismatch, which has been shown to be important to the distribution, organization, and function of transmembrane proteins (17,33).
To overcome the technical difficulties associated with MscL, we have chosen to use gramicidin A (gA) as a model for the characterization of the stress field near a membrane protein. gA is a small protein that has a clearly defined hydrophobic belt and an aromatic girdle and is anchored to the water/membrane interface by four Trp residues near the C-terminus of each subunit in the form of a membrane-spanning dimer (34). It is an appropriate choice for our purpose because of the following considerations. First, gA is a well studied model system for illustrating the coupling between membrane properties and protein function (35–37), such as the impact of hydrophobic mismatch on ion conductance. Second, its small size allows simulations at both atomistic and coarse-grained levels, making it possible to explore the sensitivity of the calculated stress field to computational models. Third, the system exhibits a significant degree of cylindrical symmetry, which makes cylindrical averaging of the stress field meaningful and helps with convergence of calculations. In terms of its position in the membrane, its center of mass is consistently near the membrane midplane, and the gA dimer maintains the characteristic hydrophobic belts and aromatic girdles. Of more importance, the hydrophobic belt and the aromatic girdle compositions are distinct; only Trp residues are present in the aromatic girdle and only hydrophobic residues in the hydrophobic belt. This homogeneity guarantees that the stress field at a point in a cylindrical coordinate is largely dictated by the interaction between lipids and a single type of amino acid (e.g., Trp or Leu/Ala).
One limitation of using gA as the model system is that only negative mismatches (e.g., where the protein is shorter than the membrane thickness) can be studied, because gA tilts in the presence of a positive mismatch. Tilting of the protein prevents the use of a cylindrical coordinate and compromises the numerical convergence of stress field calculations. Nevertheless, as we show below, a great many new insights can be gleaned simply by studying the variation of the stress field with respect to different degrees of negative mismatch. Specifically, we analyze the stress field in the vicinity of a gA dimer, which helps define mechanical components in the protein as anchors (38,39); such analysis further supports the use of stress field calculations for identifying functional mechanical motifs, such as tension sensors, in membrane proteins. Second, the calculated variations in stress near the protein/membrane interface allow an estimate of the membrane deformation energy, and the results are comparable to estimates based on phenomenological models (40,41). Therefore, our calculations establish connections between particle-based models and (continuum) mechanical descriptions of the protein/membrane interface, paving the way for the development of more sophisticated continuum mechanical models for membrane proteins in complex environments (42–44).
In the next section, we first summarize computational models and methods of analysis. In the third section, we present results and discussion. Finally, we draw a few conclusions.
Computational Methods
All-atom and coarse-grained gA structures
A conducting gA channel is a homodimer of two single-stranded β-helical subunits. The sequence of a subunit is formyl-Val-Gly-Ala-Leu-Ala-Val-Val-Val-Trp-Leu-Trp-Leu-Trp-Leu-Trp-ethanolamine, where underlined residues are D-amino acids (45). Among the several dimeric structures of gA, PDB 1JNO (46) has been used for numerous computational studies (47–50). It has a formyl group at the N-terminus and ethanolamine at the C-terminus. The formyl groups can rotate freely in a monomeric gA, but they form strong hydrogen bonds with backbone nitrogen atoms for most of the simulations in a dimeric gA (Fig. 1 A). The structure of the gA dimer remains stable for the entire period of simulations and will not be discussed in detail, since our focus here is the stress field near the protein/membrane interface.
Figure 1.
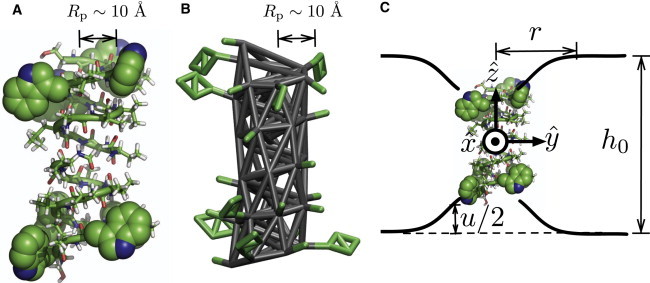
All-atom (A) and coarse-grained (B) structures of gramicidin A. (A) Anchoring Trp residues are shown in van der Waals representation and covalent bonds are shown in stick representation. (B) Elastic network model that models coarse-grained β-helix conformation is shown in gray sticks, with side chains in green sticks. (C) Schematic of bilayer deformation due to inclusion of a gA dimer. Throughout the report, we use a coordinate system with the origin at the center of mass of the gA dimer and the z axis normal to the membrane plane. Bilayer deformation at , , due to the gA dimer inclusion decreases monotonically as r increases over the scale of 20–30 Å depending on lipid chain length.
At the coarse grained (CG) level, the system is modeled using the MARTINI force field (24,51). Although we have developed an extension of the MARTINI model based on a careful consideration of electrostatics (52), we use the original MARTINI model here, because the system is not featured with highly charged species at the membrane/water interface. A CG gA dimer is built by using the seq2itp.pl script provided at the MARTINI website (53). The polar ethanolamine group at the C-terminus of gA is replaced by a Ser residue, because the functional group of ethanolamine is essentially a hydroxyl. A dimeric gA is modeled as a single chain by inserting an Ala residue in the space between two monomers (i.e., at the center of two formyl groups). Replacing the two formyl groups with an Ala residue is reasonable for our purpose, because formyl groups interact not with lipid hydrocarbon but mainly with protein atoms or water molecules in the channel, whereas we are interested only in protein-lipid interactions. The secondary structure of the gA dimer in a CG model is maintained using an elastic network with a harmonic constant of 25 kJ/mol·Å2, as suggested in the MARTINI force field (24) (Fig. 1 B). Compared to previous studies of protein-membrane interactions at the CG level, which have often treated the protein as a simple cylinder (e.g., de Meyer et al. (33)), the MARTINI model captures more chemical details.
Simulation overview
To study how the 3D stress field depends on hydrophobic mismatch, we carry out two all-atom (AA) simulations of a gA dimer in dimyristoylphosphatidylcholine (DMPC) and distearoylphosphatidylcholine (DSPC) bilayers and three CG simulations in DMPC, dipalmitoylphosphatidylcholine (DPPC), and DSPC (for a summary, see Table 1); the degree of mismatch increases in the order of DMPC, DPPC, and DSPC. Area/lipid is set to an experimental value at the relevant temperature: 65.4 Å2 at 323 K for DMPC, 63.3 Å2 at 323 K for DPPC, and 66.0 Å2 at 338 K for DSPC (54).
Table 1.
Simulation setups
| Model |
All-atom |
Coarse-grained |
|||
|---|---|---|---|---|---|
| Lipid | DMPC | DSPC | DMPC | DPPC | DSPC |
| No. of lipids | 72 | 72 | 288 | 288 | 288 |
| Simulation time (ns) | 100 | 220 | 600 | 600 | 600 |
AA systems are built as follows. First, a cylindrical hole with the size of a gA dimer is created in the center of a preequilibrated bilayer of 72 lipids by carrying out a short MD simulation (<1 ns) in the presence of a cylindrical harmonic potential (the GEO potential of the CHARMM package (55)). Then, the gA crystal structure is inserted into the hole and minimized in the absence of any restraining potential. Finally, the systems are equilibrated for ∼50 ns using Gromacs (56) before the production runs are carried out. For atomistic simulations, the CHARMM force field is used to describe all components of the system: CHARMM22 for proteins (57), CHARMM27r for lipids (58,59), the original TIP3P (60) for water, and CHARMM ion parameters (61).
CG systems are built as follows. First, the atomistic systems of gA dimer and lipid molecules, which are built as described above, are converted to the CG level according to the mapping scheme of MARTINI (Fig. 1 B). Then, the numbers of lipid molecules are increased from 72 to 288 by attaching a preequilibrated lipid bilayer. Those CG systems are equilibrated for at least 100 ns before the production runs.
For additional details of the AA and CG simulations, see the Supporting Material.
Stress field calculations
The local stress field as a function of position in 3D is calculated using the routine developed for our previous study (32,62). We divide the simulation box into rectangular blocks of ∼1 Å3 and label each block using three indices, . The local stress tensor for a block , , is defined by
| (1) |
where is the volume of a block; if particle i is inside the block and 0 otherwise; is the mass of particle i; is the velocity vector of particle i; ⊗ is a tensor product; is the force vector on particle i due to j; ; and is the localization function. The localization function, , is defined by the overlap length of the line connecting two particles i and j with a block (62). We use the Irving-Kirkwood contour, which is a straight line that connects two atoms; this is the common choice in most MD studies of stress profiles in bilayers (23,32,63). As discussed in Appendix A of Lindahl and Edholm (63), contributions from many-body force field terms (angle, dihedral) can always be expressed in pairwise terms; we use the same local virial computation algorithms as in the Gromacs package (56). Lateral and normal stress components, and , are defined by and , respectively.
In the production runs, positions and velocities are saved every 50 frames. Later, 3D local stress, , is obtained by recalculating kinetic and virial components from saved frames using Eq. 1. The stress tensor averaged over the system is consistent with the pressure coupling parameters, showing that our stress calculation algorithms are appropriate. For example, , , , , , and bar for the CG DSPC system. In general, off-diagnonal stress components are significantly smaller than diagonal components, regardless of the position (see Fig. S4 in the Supporting Material). Thus, we ignore the off-diagonal components in the following discussion. In the stress calculations, the coordinate system with origin at the center of the gA dimer is used as illustrated in Fig. 1 C. See the Supporting Material for additional details on the calculation of stress field.
Results and Discussions
In Fig. 2, density maps and lateral and normal stress fields computed using all-atom and coarse-grained simulations of DMPC and DSPC systems are plotted in rz coordinates by averaging 3D density and stress data over the azimuthal angle. Note that we use the terms attractive (repulsive), stretching (compressing), and negative (positive) stresses interchangeably throughout this work. For example, when a mechanical body in equilibrium is stretched (compressed) by surroundings, an attractive (repulsive) stress develops inside the body as a response. The signs are negative and positive for attractive and repulsive stresses, respectively. As discussed in the Supporting Material, the stress field is a result of balancing contributions from different interactions; i.e., individual stress contributions from bonded, electrostatic, and van der Waals interactions can exceed 10,000 bar, whereas the magnitude of the total stress is at most ∼1000 bar. Moreover, although the overall stress fields from AA and CG simulations are fairly consistent with each other (see below), individual components can differ substantially; this is not surprising, because the physical meaning of CG force field components does not necessarily correspond to that of the AA force field terms.
Figure 2.
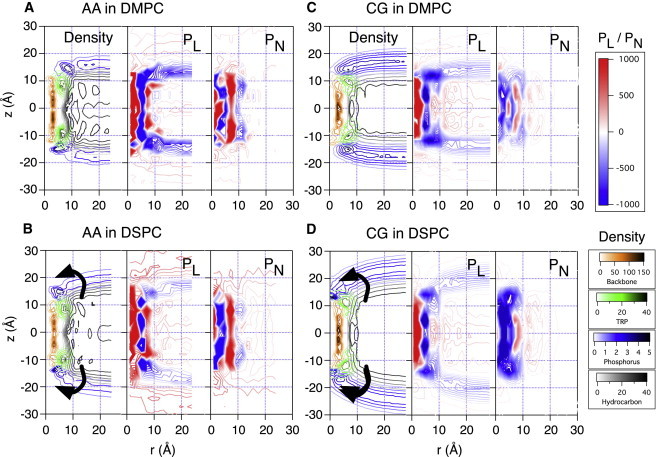
Number density map, lateral stress (), and normal stress () from AA DMPC (A), AA DSPC (B), CG DMPC (C), and CG DSPC (D) bilayers are shown. Number density is in nm−1 units and stress is in bar. As the membrane becomes thicker with lipids of longer tails, the curved surface of the water/membrane interface rotates with respect to the Trp anchors, as indicated by the arrows. The degree of hydrophobic mismatch is small for DMPC and significant (∼10 Å) for DSPC.
Last, we note that the CHARMM27r lipid force field is known to overestimate surface tension of a lipid bilayer (e.g., about 10–20 dyne/cm for DPPC) (59). This corresponds to the overall decrease in lateral stress by about 25–50 bar in the bilayer. Despite this shortcoming of the force field relative to the more recent version (64), the all-atom stress profiles calculated here serve well for a qualitative comparison with the CG result.
Stress inside a gA-dimer, r < ∼5 Å
Comparison of AA density map and stress profile (Fig. 2, A and B) reveals that the β-helix of a gA dimer can be characterized mechanically as a hollow cylinder of 5-Å radius and 20-Å height; its core region (mainly water channel at r < ∼3 Å) is highly repulsive (pL,pN > 1000 bar), whereas its backbone region (∼3 < r< 6 Å; colored in gold in the density maps) is highly attractive (pL,pN < −1000 bar) in all directions. As shown in Fig. S1 A, the β-helical structure is stabilized by electrostatic attractions due to hydrogen bonds that are almost parallel to the helical axis.
Because we use an elastic network for the CG proteins, the protein stress field from the CG simulations does not necessarily follow the same trends as the AA simulations. Nevertheless, the and trends in the CG model are qualitatively similar to those from AA calculations. One difference between AA and CG results is found in the protein responses to the disjoining force due to hydrophobic mismatch (65). The strong surface tension in the membrane/water interface is transferred to the body of a membrane protein, and thus membrane proteins in the presence of negative hydrophobic mismatch are subject to a strong disjoining force, , in the normal direction (17). In AA simulations, the protein response is focused mainly on the backbone region ( Å), because strong backbone hydrogen bonds are against stretching; accordingly, stretching stress in the normal direction is only observed in the backbone region (Fig. 2 B). By contrast, in the CG models, the protein response is scattered in space (Fig. 2 D), because the protein structure is maintained by the elastic network (Fig. 1 B). Conceptually, the disjoining force is the force exerted by the membrane to break a gA dimer into two monomers (65). However, it is important to note that the stress due to the disjoining force is not localized at the interface between two monomers but is distributed along the helical axis; i.e., the hydrophobic mismatch in principle perturbs not only the monomer-monomer contacts but also intramonomer interactions, although only the effect on the monomer-monomer contacts is directly measurable experimentally (36,65).
Using the stress field, we can quantify how strongly the hydrophobic mismatch stretches the embedded gA dimer. The mean disjoining forces from the CG simulations are estimated from the stress field inside the gA dimer, Å and Å. In this region, the mean normal stress, , equals approximately −356, −710, and −934 bar for DMPC, DPPC, and DSPC bilayers, respectively, and the mean lateral stress, , is 617, 555, and 573 bar for DMPC, DPPC, and DSPC bilayers, respectively. (Even though the convergence is relatively poor for the AA simulations, normal stress in the backbone region is clearly more attractive for gA in a DSPC bilayer than in a DMPC bilayer (Fig. 2, A and B)). Interestingly, only the magnitude of normal stress increases significantly as the degree of hydrophobic mismatch increases, whereas the lateral stress is more or less independent of the hydrophobic mismatch. Lundbæk et al. estimated that the disjoining force for the solvent-free gA/monoglyceride system is kcal/mol·Å/unit of hydrophobic mismatch (10 Å) (65); if we take Å2 as the effective cross-section area of gA, the calculated CG stress leads to a disjoining force in the range of 0.4–1.1 kcal/mol·Å, which is in reasonable agreement with the experimental estimate. Despite the significant disjoining force in a gA dimer, experiments showed that the gA structure is insensitive to the degree of hydrophobic mismatch for phospholipid acyl chain length ranging from 10 to 20 (66,67). Indeed, the structure of a gA dimer remains stable in AA simulations without any artificial restraints even with a DSPC bilayer (C18:0), which is significantly thicker than the gA dimer (∼10 Å hydrophobic mismatch).
Stress at the protein/lipid interface
At the protein/hydrocarbon interfaces, bulky Trp residues create a narrow band (aromatic girdles) of ∼10 Å in height, and the space between the two aromatic bands is filled with hydrocarbon (see density maps in Fig. 2). Comparisons of density maps and stress profiles show highly positive and due to repulsive van der Waals interactions between nonpolar residues and hydrocarbons in both AA and CG simulations ( Å, Å in Fig. 2, A–D). Although the magnitude of such repulsion is significantly larger than the normal stress in lipid hydrocarbons, the gA dimer remains stable in the membrane, because it is anchored by the Trp residues at the water/membrane interface (see below). At the first lipid shell, Å, highly attractive normal stress, , is observed in both AA and CG calculations although the attraction is more prominent in AA stress profiles. As shown in our accompanying article (68), C-C bonds of the annular lipid tails at Å are more tangential to the protein/lipid interface. These more ordered annular lipids create another interface between the ordered lipids and bulk lipids, and the attractive near Å is an indication of this lipid/lipid interface.
The manifestation of an ordered first lipid shell around a membrane protein is consistent with recent MD simulation studies, which showed that a membrane protein diffuses as a dynamic complex with annular lipids (28,69,70) or diffusion constants for the annular and bulk lipids are significantly different (37); experimental studies also indicate the existence of ordered annular lipids (17,28). It is noteworthy that a recent study using atomistic simulations of nonequilibrium compression/expansion suggested that the area compressibility modulus is increased by 22% due to the inclusion of a membrane protein (MscL) (71). Our observation of the sandwichlike stress distribution in the annular lipid region even with a mild degree of hydrophobic mismatch supports the existence of solidified lipids, as observed in that simulation study. For more discussion of the properties of annular lipids, see the accompanying article (68).
Trp residues at the water/membrane interface: mechanical anchors
Comparison of number density maps with the stress profile near the Trp residues in Fig. 2 clearly reveals a ring-shaped anchor to which the surface of the water/lipid interface is connected. As the membrane becomes thicker with lipids of longer tails, the curved surface of the water/membrane interface rotates with respect to the Trp anchors. The Trp density map is almost independent of lipid tail length, and the point at which the water/lipid interface meets Trp residues rotates, as indicated by the arrows in Fig. 2. Lateral stress in both AA and CG simulations along the Trp anchors is extremely negative, bar, which indicates a significant mechanical frustration between Trp residues and lipid headgroups. A recent experimental study (39) on the variation in the tilting angles of transmembrane helices upon systematically placing an anchor residue (Trp, Lys, Arg, or Gly) at each terminus revealed that Trp residues dictate the tilting angle. This suggests that Trp residues are more strongly anchored to the membrane/water interface than other residues. The extremely negative stress between Trp and the water/membrane interface found here is consistent with those experimental observations.
Stress in bulk lipids: Å
Inclusion of a gA dimer in a bilayer with unperturbed thickness, , larger than the height of the gA dimer causes a membrane deformation, , as illustrated schematically in Fig. 1 C. To elucidate and quantitatively in the vicinity of the inclusion, mean normal and lateral stresses as functions of z for a given r range, , are calculated by averaging and over :
| (2) |
where and is the number of data points at z and . For CG DMPC and CG DPPC systems, and are calculated for four different r ranges, , , , and Å. As shown in Fig. 3, A–C, the mean normal stress, , is almost flat for Å regardless of lipid type, indicating that the bilayers are unperturbed at Å. As r decreases, rises monotonically in the hydrocarbon region and reaches 50–100 bar at Å. Interestingly, normal compression ( bar) is observed only in the z range enclosed by the two minima in the lateral pressure . Thus, it is natural to define the effective membrane thickness, , as the distance between the two minima (i.e., the distance shown in Fig. 3). For unperturbed membranes, the distance is 27, 33, and 40 Å for DMPC, DPPC, and DSPC, respectively. Meanwhile, the unperturbed mean distance between phosphates in the two monolayers is 35, 41, and 48 Å for DMPC, DPPC, and DSPC, respectively. The two sets of distances are highly correlated (with a constant shift of 8 Å) for all bilayers and largely independent of membrane deformation , because depends significantly on the phosphate positions. In other words, the effective membrane thickness, , can be conveniently calculated by subtracting 8 Å from the phosphate-phosphate distance. In Fig. 4, A and B, effective monolayer deformation curves in AA and CG simulations are shown; these are converted into bilayer strain as a function of r in Fig. 4 C (see next subsection for details on the definition of strain). Note that strain is at most 10–15% at Å.
Figure 3.
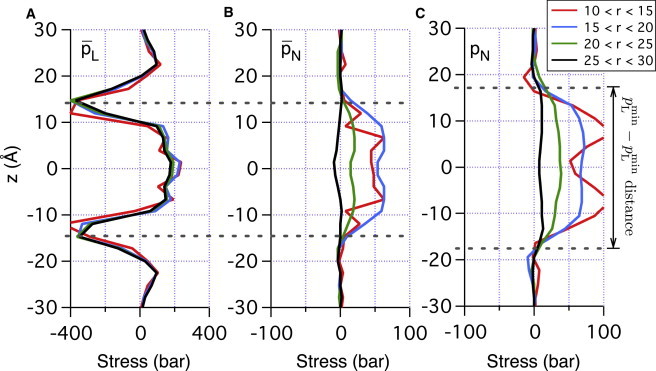
Mean lateral (A) and normal (B) stress in CG DMPC and mean normal stress in CG DPPC (C) system are shown as a function of normal displacement in four regions: Å, Å, Å, and Å. Note that only normal stress, , depends significantly on r.
Figure 4.
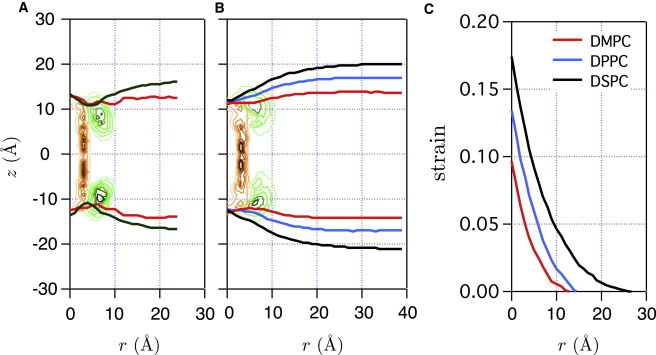
Effective monolayer deformation curves by a gA insertion for AA (A) and CG (B) simulations are obtained by shifting the mean phosphate curve of each monolayer by 4 Å toward the origin on the z axis. The mean phosphate curves are calculated from the number density maps of phosphate groups (Fig. 2). Effective leaflet thickness at r, , is defined by the distance between two effective monolayer deformation curves along the z axis. (C) Strain by bilayer deformations for CG simulations. See Estimation of Membrane Deformation Energy beyond the Annular Lipids for details on strain definition.
As shown in Fig. 3 A, has a lesser degree of dependence on r than does . This relatively weak dependence of on r indicates that lateral stress near a gA dimer is less significant. Indeed, more quantitative analysis in the next section shows that lateral compression accounts for only ∼30% of the total deformation energy for all the gA-bilayer systems studied here.
Estimation of membrane deformation energy beyond the annular lipids
The above discussion and recent study of Kim et al. (37) highlighted that the behaviors of annular lipids are complex and likely difficult to describe using a simple continuum elasticity theory. For regions beyond the annular lipids, however, it is worthwhile to explore the applicability of continuum elasticity models. Here, we demonstrate that the stress field at Å calculated in this study is consistent with the prediction from elasticity theory (40,41); we also estimate membrane deformation energy and compare it with previous theoretical estimations (40,41).
When a bilayer is deformed by from its unperturbed thickness in the z direction at (Fig. 1 C), area/lipid increases by from its unperturbed value due to the small volume compressibility of the bilayer (21,72). The normal and lateral strains are and , respectively. The associated membrane deformation energy is usually written in two different forms that can be used interchangeably, i.e., either
| (3) |
or
| (4) |
where is the bilayer area stretch moduli (20,21,40,41,73), because a constant volume of a lipid bilayer (, or, equivalently, ) is usually a valid assumption (72). This equality implies that the experimentally determined contains both lateral and normal components: . As discussed above (Fig. S4), the coupling between normal and lateral stresses is small. Therefore, we can decompose into the normal and lateral deformation energies, :
| (5) |
| (6) |
where and are the bilayer area stretch moduli in the normal and lateral directions, respectively.
Normal and lateral elastic stresses, and , respectively, can be obtained by taking the derivatives of and with respect to u:
| (7) |
Note that and , because we only consider negative hydrophobic mismatch in this study. To verify that our simulations agree with the continuum elasticity theory, we calculate mean normal and lateral stress averaged over z at , ,
| (8) |
where is the effective membrane thickness as defined above. In Fig. 5, A and B, and , respectively, are shown as functions of strain. For both DPPC and DSPC membranes, stresses depend linearly on strain when the latter is below 10–15%, which corresponds to the strain at Å (see Fig. 4 C). Thus, we see that the lipid bilayer obeys the elasticity theory at Å, and in Eq. 8 is equal to in Eq. 7 at Å. and can be estimated using the slopes in the elastic regime in Fig. 5, A and B, and the results are summarized in Table 2. The total bilayer-area stretch modulus, , is 35, 47, and 41 kcal/mol·nm2 for DMPC, DPPC, and DSPC, respectively. These values are in good agreement with experimental results showing that is ∼40 kcal/mol·nm2 for lipid bilayers with tail length ranging from 10 to 20 (72).
Figure 5.
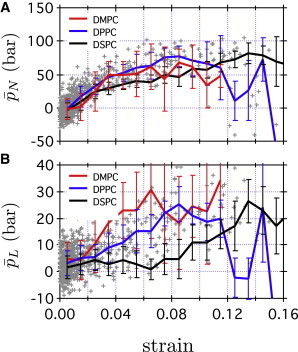
(A and B) Normal and lateral stresses, respectively, in CG simulations are shown as functions of strain . Gray crosses represent mean normal and lateral stresses in CG DPPC averaged over z at , , as defined in Eq. 8. Red, blue, and black lines indicate mean stresses at a given strain in CG DMPC, CG DPPC, and CG DSPC, respectively.
Table 2.
Summary of bilayer area stretch modulus and bilayer deformation energies from CG simulations (kcal/mol)
| DMPC | DPPC | DSPC | |
|---|---|---|---|
| 25 | 36 | 32 | |
| 10 | 11 | 9 | |
| 35 | 47 | 41 | |
| 0.8 | 0.9 | 1.7 | |
| 0.2 | 0.3 | 0.4 | |
| 1.0 | 1.2 | 2.1 |
Stretch modulus is given in kcal/mol·nm2 and deformation energy in kcal/mol.
The deformation free energy by gA has been estimated in seminal works by Huang (40) and Helfrich and Jakobsson (41) using phenomenological models based on the continuum elasticity theory. In these studies, they modeled a gA dimer as a cylinder of effective radius 10 Å and height 22 Å. By minimizing the phenomenological free-energy functional with the inclusion of a gA dimer setting the boundary condition (i.e., Å), they obtained the bilayer deformation, , and the bilayer-deformation free energy. Their phenomenological free-energy functional typically consists of three components: compression, splay, and surface tension. The compression term matches well with the normal compression energy in this study, whereas surface tension and splay contributions were regarded as insignificant (40,41). Specifically, the total deformation free energy of the phosphatidylcholine in an n-decane membrane (of unperturbed effective thickness 48 Å) by a model gA was kcal/mol according to Helfrich (41). Nielsen et al. also applied a similar analytic model with parameters for the gA/DMPC system; they obtained a total deformation energy of 2.4 kcal/mol and found that the compression term accounted for 70% of the total deformation energy (74).
By integrating in Eqs. 5 and 6 over x and y using , , and from the CG simulations as discussed above, we can estimate the bilayer-deformation energies, and , by a gA dimer where . As summarized in Table 2, computed deformation energies for DMPC, DPPC, and DSPC bilayers are 1.0, 1.2, and 2.1 kcal/mol, respectively. Compared to the previous analytic studies, our analysis predicts a lower total deformation energy. It is notable that the effective membrane thickness, , at the protein/lipid interface ( Å) in Fig. 4, A and B, is always larger than the effective height of gA ( Å), whereas all three analytic studies mentioned above (40,41,74) employed the boundary condition that the effective membrane thickness equals the effective gA height ( Å) at the protein/lipid interface. For example, the effective membrane thickness, , in our CG DMPC system changes from 25 Å at Å to 27 Å at large r, whereas the boundary condition in the analytic models is that Å at Å and 28.5 Å at large r (74). Therefore, previous analytic studies seem to overestimate the compression of the membrane in the vicinity of gA and, consequently, the bilayer compression energy induced by gA insertion. Uncertainty in the boundary condition of an elastic model at the membrane/protein interface has also been discussed in a recent study by Im and co-workers (37).
Conclusions
It has been increasingly recognized that mechanical properties of biomembranes can significantly modulate the functional characteristics of membrane proteins. To better understand the underlying physical basis and construct predictive models for such modulation, it is imperative to characterize the mechanical features of the protein/membrane interface. Since this is not easy to accomplish with current experimental techniques, we use both atomistic and CG simulations to compute stress field distribution around a membrane protein using the gA dimer as a model system. Moreover, we carry out such calculations with lipid bilayers of different thickness to probe how hydrophobic mismatch influences the stress field.
The calculations reveal that the stress field near the protein/lipid interface is rich in feature and exhibits a layered structure with both significant repulsive and attractive regions; the trends are largely consistent between atomistic and coarse-grained simulations, which supports the use of coarse-grained models for the mechanical analysis of more complex systems. The magnitude of the local stress field in a gA dimer induced by the hydrophobic mismatches is as high as bar, highlighting the mechanical stability of even simple membrane proteins. The stress field distributions with different bilayers help underline the Trp residues at the protein/membrane/water interface as mechanical anchors, suggesting that similar analysis is useful for identifying tension sensors in other membrane proteins, especially those involved in mechanosensation. Finally, the calculated stress field allows us to estimate free energy of deformation associated with hydrophobic mismatch, which is a fundamentally important quantity for the discussion of how mechanical properties of membrane modulate protein functions. Our stress analysis confirms that linear elasticity theory holds for the membrane region beyond the annular lipids. However, membrane deformations by the inclusion of a gA dimer in the presence of hydrophobic mismatch deviate from the general assumptions made by phenomenological models that the membrane thickness at the membrane/protein interface is independent of lipid type. We find that this in fact is lipid-dependent, due presumably to the balance between lipid mechanics and protein-lipid interactions.
At the conceptual level, the most significant aspect of this work is to foster a connection between microscopic and continuum mechanics models for proteins in complex environments. Although continuum mechanics models for pure membranes have been successful in explaining many fascinating behaviors of biomembranes (20,21,75,76), extension of such models to describe processes involving biopolymers with detailed structural features (42–44,77–81) is relatively new. To develop robust mechanical models that can describe the proper response of membrane proteins to mechanical perturbations, it is essential to have a thorough understanding of key features in the stress field distribution near membrane proteins and how such features vary with perturbation (e.g., different degrees of hydrophobic mismatch).
The magnitude and spatial range of heterogeneity in the calculated stress field provide concrete clues regarding the validity of assumptions commonly made in continuum mechanics models for membrane mediated processes. For example, despite commonly discussed annular lipids near membrane proteins, it is often assumed in continuum models that the membranes have homogeneous material properties. Our calculations show explicitly that the annular lipids indeed have very different mechanical properties compared to even the lipids in the second shell. Whether such differences make a significant contribution to the description of the process of interest depends on whether the distribution of these annular lipids is altered significantly. In the accompanying article (68), we study the potential of mean force (PMF) between two gA dimers in bilayers of different thickness. At separations that do not involve significant depletion of the annular lipids, the PMF from microscopic calculations can be understood well using a continuum model. At very short separations, however, the situation becomes more complex due to the necessary reorganization of annular lipids, and thus, the well depth of the PMF is not easy to predict correctly using a simple continuum model. Finally, another useful observation from the stress field analysis is that the effect of even rather severe hydrophobic mismatch (a gA dimer in DSPC with ∼10 Å mismatch) propagates to only about two to three lipid layers (i.e., ∼25 Å from the center of a gA dimer). This limits the range of cooperatively between membrane proteins in crowded cellular membranes (20,73).
Additional simulation details and analysis of the stress calculations are included in the Supporting Material.
Acknowledgments
Computational resources from the National Center for Supercomputing Applications at the University of Illinois and the Center of High Throughput Computing at UW-Madison are greatly appreciated.
This research was supported by the National Institutes of Health (R01-GM084028).
Supporting Material
References
- 1.Parsegian A. Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature. 1969;221:844–846. doi: 10.1038/221844a0. [DOI] [PubMed] [Google Scholar]
- 2.Finkelstein A. Water and nonelectrolyte permeability of lipid bilayer membranes. J. Gen. Physiol. 1976;68:127–135. doi: 10.1085/jgp.68.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiu D., Shenkin P.S., Still W.C. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J. Phys. Chem. A. 1997;101:3005–3014. [Google Scholar]
- 4.Dominy B.N., Brooks C.L., III Development of a generalized Born model parametrization for proteins and nucleic acids. J. Phys. Chem. B. 1999;103:3765–3773. [Google Scholar]
- 5.Tanizaki S., Feig M. A generalized Born formalism for heterogeneous dielectric environments: application to the implicit modeling of biological membranes. J. Chem. Phys. 2005;122:124706. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 6.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52:176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 7.Tanizaki S., Feig M. Molecular dynamics simulations of large integral membrane proteins with an implicit membrane model. J. Phys. Chem. B. 2006;110:548–556. doi: 10.1021/jp054694f. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh N., Prat-Resina X., Cui Q. Microscopic pKa analysis of Glu286 in cytochrome c oxidase (Rhodobacter sphaeroides): toward a calibrated molecular model. Biochemistry. 2009;48:2468–2485. doi: 10.1021/bi8021284. [DOI] [PubMed] [Google Scholar]
- 9.Roux B. Influence of the membrane potential on the free energy of an intrinsic protein. Biophys. J. 1997;73:2980–2989. doi: 10.1016/S0006-3495(97)78327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Im W., Brooks C.L., 3rd Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talasaz A.H., Nemat-Gorgani M., Davis R.W. Prediction of protein orientation upon immobilization on biological and nonbiological surfaces. Proc. Natl. Acad. Sci. USA. 2006;103:14773–14778. doi: 10.1073/pnas.0605841103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jajjar E., Mihajlovic M., Reuter N. Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins. 2008;71:1655–1669. doi: 10.1002/prot.21853. [DOI] [PubMed] [Google Scholar]
- 13.Doherty G.J., McMahon H.T. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu. Rev. Biophys. 2008;37:65–95. doi: 10.1146/annurev.biophys.37.032807.125912. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt N., Mishra A., Wong G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 15.Krepkiy D., Mihailescu M., Swartz K.J. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–479. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Israelachvili J.N. Refinement of the fluid-mosaic model of membrane structure. Biochim. Biophys. Acta. 1977;469:221–225. doi: 10.1016/0005-2736(77)90185-7. [DOI] [PubMed] [Google Scholar]
- 17.Andersen O.S., Koeppe R.E., 2nd Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt D., MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. USA. 2008;105:19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kung C. A possible unifying principle for mechanosensation. Nature. 2005;436:647–654. doi: 10.1038/nature03896. [DOI] [PubMed] [Google Scholar]
- 20.Phillips R., Ursell T., Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boal D. Cambridge University Press; Cambridge, United Kingdom: 2002. Mechanics of the Cell. [Google Scholar]
- 22.Cantor R.S. Lateral pressures in cell membranes: a mechanism for modulation of protein function. J. Phys. Chem. B. 1997;101:1723–1725. [Google Scholar]
- 23.Gullingsrud J., Schulten K. Gating of MscL studied by steered molecular dynamics. Biophys. J. 2003;85:2087–2099. doi: 10.1016/s0006-3495(03)74637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 25.Safran S.A. Addison-Wesley; New York: 1994. Statistical Thermodynamics of Surfaces, Interfaces, and Membranes. [Google Scholar]
- 26.Meyer G.R., Gullingsrud J., Martinac B. Molecular dynamics study of MscL interactions with a curved lipid bilayer. Biophys. J. 2006;91:1630–1637. doi: 10.1529/biophysj.106.080721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsh D. Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys. J. 2007;93:3884–3899. doi: 10.1529/biophysj.107.107938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Powl A.M., East J.M., Lee A.G. Importance of direct interactions with lipids for the function of the mechanosensitive channel MscL. Biochemistry. 2008;47:12175–12184. doi: 10.1021/bi801352a. [DOI] [PubMed] [Google Scholar]
- 29.Chang G., Spencer R.H., Rees D.C. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- 30.Steinbacher S., Bass R., Rees D.C. Structures of the prokaryotic mechanosensitive channels MscL and MscS. Curr. Top. Membr. 2007;58:1–24. [Google Scholar]
- 31.Perozo E., Kloda A., Martinac B. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 2002;9:696–703. doi: 10.1038/nsb827. [DOI] [PubMed] [Google Scholar]
- 32.Ollila O.H.S., Risselada H.J., Marrink S.J. 3D pressure field in lipid membranes and membrane-protein complexes. Phys. Rev. Lett. 2009;102:078101. doi: 10.1103/PhysRevLett.102.078101. [DOI] [PubMed] [Google Scholar]
- 33.de Meyer F.J., Venturoli M., Smit B. Molecular simulations of lipid-mediated protein-protein interactions. Biophys. J. 2008;95:1851–1865. doi: 10.1529/biophysj.107.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun H., Greathouse D.V., Koeppe R.E., 2nd The preference of tryptophan for membrane interfaces: insights from N-methylation of tryptophans in gramicidin channels. J. Biol. Chem. 2008;283:22233–22243. doi: 10.1074/jbc.M802074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lundbaek J.A., Collingwood S.A., Andersen O.S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. J. R. Soc. Interface. 2010;7:373–395. doi: 10.1098/rsif.2009.0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lundbaek J.A., Koeppe R.E., 2nd, Andersen O.S. Amphiphile regulation of ion channel function by changes in the bilayer spring constant. Proc. Natl. Acad. Sci. USA. 2010;107:15427–15430. doi: 10.1073/pnas.1007455107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim T., Lee K.I., Im W. Influence of hydrophobic mismatch on structures and dynamics of gramicidin A and lipid bilayers. Biophys. J. 2012;102:1551–1560. doi: 10.1016/j.bpj.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Planque M.R., Kruijtzer J.A., Killian J.A. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane α-helical peptides. J. Biol. Chem. 1999;274:20839–20846. doi: 10.1074/jbc.274.30.20839. [DOI] [PubMed] [Google Scholar]
- 39.Vostrikov V.V., Daily A.E., Koeppe R.E., 2nd Charged or aromatic anchor residue dependence of transmembrane peptide tilt. J. Biol. Chem. 2010;285:31723–31730. doi: 10.1074/jbc.M110.152470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang H.W. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 1986;50:1061–1070. doi: 10.1016/S0006-3495(86)83550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Helfrich P., Jakobsson E. Calculation of deformation energies and conformations in lipid membranes containing gramicidin channels. Biophys. J. 1990;57:1075–1084. doi: 10.1016/S0006-3495(90)82625-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Y., Cao G., Cui Q. A finite element framework for studying the mechanical response of macromolecules: application to the gating of the mechanosensitive channel MscL. Biophys. J. 2006;91:1248–1263. doi: 10.1529/biophysj.106.085985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X., Cui Q., Yethiraj A. Gating mechanisms of mechanosensitive channels of large conductance, I: a continuum mechanics-based hierarchical framework. Biophys. J. 2008;95:563–580. doi: 10.1529/biophysj.107.128488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mondal S., Khelashvili G., Weinstein H. Quantitative modeling of membrane deformations by multihelical membrane proteins: application to G-protein coupled receptors. Biophys. J. 2011;101:2092–2101. doi: 10.1016/j.bpj.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarges R., Witkop B. Garmicidin A: V. The structure of valine and isoleucine-gramicidin A. J. Am. Chem. Soc. 1965;87:2011–2020. doi: 10.1021/ja01087a027. [DOI] [PubMed] [Google Scholar]
- 46.Townsley L.E., Tucker W.A., Hinton J.F. Structures of gramicidins A, B, and C incorporated into sodium dodecyl sulfate micelles. Biochemistry. 2001;40:11676–11686. doi: 10.1021/bi010942w. [DOI] [PubMed] [Google Scholar]
- 47.Allen T.W., Andersen O.S., Roux B. Structure of gramicidin a in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR data. J. Am. Chem. Soc. 2003;125:9868–9877. doi: 10.1021/ja029317k. [DOI] [PubMed] [Google Scholar]
- 48.Allen T.W., Andersen O.S., Roux B. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 2004;101:117–122. doi: 10.1073/pnas.2635314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen T.W., Andersen O.S., Roux B. Ion permeation through a narrow channel: using gramicidin to ascertain all-atom molecular dynamics potential of mean force methodology and biomolecular force fields. Biophys. J. 2006;90:3447–3468. doi: 10.1529/biophysj.105.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allen T.W., Andersen O.S., Roux B. On the importance of atomic fluctuations, protein flexibility, and solvent in ion permeation. J. Gen. Physiol. 2004;124:679–690. doi: 10.1085/jgp.200409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Monticelli L., Kandasamy S., Marrink S.J. The MARTINI coarse grained force field: extension to proteins. J. Chem. Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 52.Wu Z., Cui Q., Yethiraj A. A new coarse-grained force field for membrane-peptide simulations. J. Chem. Theory Comput. 2011;11:3793–3802. doi: 10.1021/ct200593t. [DOI] [PubMed] [Google Scholar]
- 53.http://md.chem.rug.nl/cgmartini
- 54.Petrache H.I., Dodd S.W., Brown M.F. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 56.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 57.MacKerell A.D.J., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 58.Feller S.E., MacKerell A.D., Jr. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 2000;104:7510–7515. [Google Scholar]
- 59.Klauda J.B., Brooks B.R., Pastor R.W. An ab initio study on the torsional surface of alkanes and its effect on molecular simulations of alkanes and a DPPC bilayer. J. Phys. Chem. B. 2005;109:5300–5311. doi: 10.1021/jp0468096. [DOI] [PubMed] [Google Scholar]
- 60.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 61.Beglov D., Roux B. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J. Chem. Phys. 1994;100:9050–9063. [Google Scholar]
- 62.Yoo J., Cui Q. Curvature generation and pressure profile modulation in membrane by lysolipids: insights from coarse-grained simulations. Biophys. J. 2009;97:2267–2276. doi: 10.1016/j.bpj.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindahl E., Edholm O. Spatial and energetic-entropic decomposition of surface tension in lipid bilayers from molecular dynamics simulations. J. Chem. Phys. 2000;113:3882–3893. [Google Scholar]
- 64.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lundbaek J.A., Andersen O.S. Spring constants for channel-induced lipid bilayer deformations. Estimates using gramicidin channels. Biophys. J. 1999;76:889–895. doi: 10.1016/S0006-3495(99)77252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greathouse D.V., Hinton J.F., Koeppe R.E., 2nd Gramicidin A/short-chain phospholipid dispersions: chain length dependence of gramicidin conformation and lipid organization. Biochemistry. 1994;33:4291–4299. doi: 10.1021/bi00180a025. [DOI] [PubMed] [Google Scholar]
- 67.Wallace B.A., Veatch W.R., Blout E.R. Conformation of gramicidin A in phospholipid vesicles: circular dichroism studies of effects of ion binding, chemical modification, and lipid structure. Biochemistry. 1981;20:5754–5760. doi: 10.1021/bi00523a018. [DOI] [PubMed] [Google Scholar]
- 68.Yoo J., Cui Q. Membrane-mediated protein-protein interactions: a coarse-grained simulation analysis on gramicidin A. Biophys. J. 2012;104:117–127. doi: 10.1016/j.bpj.2012.11.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niemelä P.S., Miettinen M.S., Vattulainen I. Membrane proteins diffuse as dynamic complexes with lipids. J. Am. Chem. Soc. 2010;132:7574–7575. doi: 10.1021/ja101481b. [DOI] [PubMed] [Google Scholar]
- 70.Aponte-Santamaría C., Briones R., de Groot B.L. Molecular driving forces defining lipid positions around aquaporin-0. Proc. Natl. Acad. Sci. USA. 2012;109:9887–9892. doi: 10.1073/pnas.1121054109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeon J., Voth G.A. Gating of the mechanosensitive channel protein MscL: the interplay of membrane and protein. Biophys. J. 2008;94:3497–3511. doi: 10.1529/biophysj.107.109850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rawicz W., Olbrich K.C., Evans E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000;79:328–339. doi: 10.1016/S0006-3495(00)76295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ursell T., Huang K.C., Phillips R. Cooperative gating and spatial organization of membrane proteins through elastic interactions. PLOS Comput. Biol. 2007;3:e81. doi: 10.1371/journal.pcbi.0030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nielsen C., Goulian M., Andersen O.S. Energetics of inclusion-induced bilayer deformations. Biophys. J. 1998;74:1966–1983. doi: 10.1016/S0006-3495(98)77904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shibata Y., Hu J., Rapoport T.A. Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 2009;25:329–354. doi: 10.1146/annurev.cellbio.042308.113324. [DOI] [PubMed] [Google Scholar]
- 76.Ursell T.S., Klug W.S., Phillips R. Morphology and interaction between lipid domains. Proc. Natl. Acad. Sci. USA. 2009;106:13301–13306. doi: 10.1073/pnas.0903825106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang Y., Yoo J., Chen X. Gating mechanisms of mechanosensitive channels of large conductance, II: systematic study of conformational transitions. Biophys. J. 2008;95:581–596. doi: 10.1529/biophysj.107.128496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bathe M. A finite element framework for computation of protein normal modes and mechanical response. Proteins. 2008;70:1595–1609. doi: 10.1002/prot.21708. [DOI] [PubMed] [Google Scholar]
- 79.Choe S., Hecht K.A., Grabe M. A continuum method for determining membrane protein insertion energies and the problem of charged residues. J. Gen. Physiol. 2008;131:563–573. doi: 10.1085/jgp.200809959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma L., Yethiraj A., Cui Q. A computational framework for mechanical response of macromolecules: application to the salt concentration dependence of DNA bendability. Biophys. J. 2009;96:3543–3554. doi: 10.1016/j.bpj.2009.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roos W.H., Gibbons M.M., Wuite G.J. Squeezing protein shells: how continuum elastic models, molecular dynamics simulations, and experiments coalesce at the nanoscale. Biophys. J. 2010;99:1175–1181. doi: 10.1016/j.bpj.2010.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.