

Abstract
Studies of initial activities of carbon monoxide dehydrogenase (CODH) from Rhodospirillum rubrum show that CODH is mostly inactive at redox potentials higher than −300 mV. Initial activities measured at a wide range of redox potentials (0–500 mV) fit a function corresponding to the Nernst equation with a midpoint potential of −316 mV. Previously, extensive EPR studies of CODH have suggested that CODH has three distinct redox states: (i) a spin-coupled state at −60 to −300 mV that gives rise to an EPR signal termed Cred1; (ii) uncoupled states at <−320 mV in the absence of CO2 referred to as Cunc; and (iii) another spin-coupled state at <−320 mV in the presence of CO2 that gives rise to an EPR signal termed Cred2B. Because there is no initial CODH activity at potentials that give rise to Cred1, the state (Cred1) is not involved in the catalytic mechanism of this enzyme. At potentials more positive than −380 mV, CODH recovers its full activity over time when incubated with CO. This reductant-dependent conversion of CODH from an inactive to an active form is referred to hereafter as “autocatalysis.” Analyses of the autocatalytic activation process of CODH suggest that the autocatalysis is initiated by a small fraction of activated CODH; the small fraction of active CODH catalyzes CO oxidation and consequently lowers the redox potential of the assay system. This process is accelerated with time because of accumulation of the active enzyme.
Carbon monoxide dehydrogenase (CODH) from Rhodospirillum rubrum is a nickel-iron-sulfur enzyme that carries out the reversible oxidation of CO to CO2 (1–4). Kinetic analyses have demonstrated that Ni is required for CODH activity (3, 5, 6). Previous spectroscopic studies have indicated that CODH contains the following iron-sulfur clusters: (i) a B-cluster with properties typical for an all-cysteinyl-liganded [Fe4S4] cluster; and (ii) an Ni-containing C-cluster that we have proposed to contain an [FeNi] binuclear subcluster (proposed subcluster of C-cluster) component bridged to or interacting with a slow-relaxing (EPR properties) Fe-containing subcluster component hereafter referred to as FeSC (7). Previously, the C-cluster was thought to contain a single Ni atom bridged to an [Fe4S4] cluster (8). The B-cluster is thought to mediate electron transfer from the C-cluster to an electron acceptor, CooF (9, 10). EPR spectroscopic studies of CODH from R. rubrum have revealed that the two C-subclusters ([FeNi] cluster and FeSC) and the B-cluster have different redox-active states, and that spin-couplings among the redox clusters result in unusual EPR signals at various redox states (10). Three distinct redox states of the [FeNi] subcluster have been proposed: [Fe2+−Ni2+]4+, [Fe3+−Ni2+−H−]4+, and [Fe2+−Ni2+−H−]3+ (10). The Fe-containing subcluster has been proposed to have two distinct oxidation states, FeSC2+ and FeSC1+, separated by one electron. The B-cluster is believed to have two redox states, [Fe4S4]B2+ and [Fe4S4]B1+ (8, 10). In addition, CODH contains a third cluster, the D-cluster. This cluster is an all-cysteinyl-liganded Fe4S4 cluster that bridges the two subunits of the CODH dimer (data not shown). The redox events and role of this cluster are not known at this time.
The redox-dependent EPR signals of the metal clusters and the corresponding spin states are suggested to be (10) (i) at the potential range of −60 to −300 mV, a gave = 1.87 signal is observed and is termed Cred1. The Cred1 EPR signal is suggested to result from a spin coupling of [Fe3+−Ni2+−H−]4+ with FeSC1+. (ii) By lowering the potential below −316 mV, the [FeNi] cluster (proposed subcluster of C-cluster) is reduced further by one electron to a diamagnetic state. This state is assigned to an uncoupled state (Cunc) containing one paramagnetic cluster, FeSC1+, with two diamagnetic species, [Fe2+−Ni2+−H−]3+ and [Fe4S4]B2+. (iii) Addition of excess CO2 to the uncoupled state produces an EPR signal that is designated as Cred2B. Cred2B is suggested to result from an antiferromagnetic spin coupling of [Fe2+−Ni2+]4+ with FeSC1+. (iv) Further lowering of reduction potential to ≲−400 mV produces yet another EPR coupling signal, termed Cred2A. The Cred2A signal is proposed to result from a spin interaction of FeSC1+ with [Fe4S4]B1+.
The measurement of CODH activity under oxidizing conditions produces various degrees of initial activities during the period termed “lag phase” (11). Full CODH activity is observed by extending the assay period, or prereducing the assay system. Typically, the prereduction has been performed by addition of a small amount of sodium dithionite [DTH, E′ = ≃−500 mV at pH 7.5 vs. standard hydrogen electrode (SHE)] (12). Accordingly, most previously reported in vitro CODH activities were measured under reduced or partially reduced conditions to avoid the lag phase. Without an understanding of the lag phase of CODH activity, the measurement of CODH activity at various redox states was not feasible. Consequently, the activities of CODH at each defined redox state were not understood fully.
In this study, analyses of initial activities of CODH reveal that the lag phase represents a redox-dependent conversion of an inactive form of CODH to an active form. This phenomenon can be interpreted as autocatalytic activation (13), and indicates that CO oxidation by a small fraction of active CODH lowers the redox potential of the system, resulting in accumulation of active CODH. This analysis also indicates that most of the CODH at redox potentials >−300 mV is catalytically inactive.
Materials and Methods
Buffers and Experimental Conditions.
Mops [3-(N-morpholino)propanesulfonic acid] buffer (pH 7.5; United States Biochemical) was used in all protein purifications and assays of CODH activity. Unless otherwise indicated, all buffers were purged of oxygen.
Redox Buffers.
The redox dyes used in the system for monitoring reduction of dyes were (i) one-electron dyes, benzyl viologen (E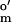 = −350 mV; Sigma) and methyl viologen (MV; E
= −350 mV; Sigma) and methyl viologen (MV; E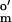 = −457 mV; Aldrich); and (ii) two-electron dyes (used in the presence of 0.01 μM MV), indigo carmine (E
= −457 mV; Aldrich); and (ii) two-electron dyes (used in the presence of 0.01 μM MV), indigo carmine (E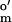 = −125 mV vs. SHE; Sigma), 2-hydroxy-1,4-naphthoquinone (E
= −125 mV vs. SHE; Sigma), 2-hydroxy-1,4-naphthoquinone (E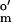 = −145 mV; Aldrich), phenosafranin (E
= −145 mV; Aldrich), phenosafranin (E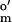 = −252 mV, Aldrich), and neutral red (E
= −252 mV, Aldrich), and neutral red (E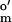 = −325 mV; Sigma). The same dyes, except MV, were used to poise the assay system for monitoring the reduction of [Fe-S] clusters (UV-visible observable iron-sulfur clusters) of CODH.
= −325 mV; Sigma). The same dyes, except MV, were used to poise the assay system for monitoring the reduction of [Fe-S] clusters (UV-visible observable iron-sulfur clusters) of CODH.
Growth of R. rubrum and Purification of CODH.
Wild-type strain (UR2) R. rubrum was cultured as described (1, 6, 14). CODH was purified as described for the wild-type enzyme (15).
Preparation of Oxidized CODH.
As-isolated CODH (in 100 mM Mops buffer containing 2 mM DTH) was loaded onto a DE-52 column (0.5 cm × 2 cm, diethylaminoethyl cellulose; Whatman) and washed with 100 mM Mops buffer. A 100 mM solution of oxidized thionin (E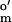 = +64 mV vs. SHE) in 100 mM Mops buffer was passed over the column-bound CODH and was removed subsequently by washing the column with 100 mM Mops buffer. Thionin-oxidized CODH was eluted by using 100 mM Mops buffer containing 200 mM NaCl, and the eluent was passed down a Sephadex G-25 column (0.5 cm × 20 cm; Amersham Pharmacia) to remove NaCl. The Sephadex G-25 column was preequilibrated with 100 mM Mops buffer before use.
= +64 mV vs. SHE) in 100 mM Mops buffer was passed over the column-bound CODH and was removed subsequently by washing the column with 100 mM Mops buffer. Thionin-oxidized CODH was eluted by using 100 mM Mops buffer containing 200 mM NaCl, and the eluent was passed down a Sephadex G-25 column (0.5 cm × 20 cm; Amersham Pharmacia) to remove NaCl. The Sephadex G-25 column was preequilibrated with 100 mM Mops buffer before use.
Protein Assays.
Protein assays were performed in the absence of DTH. Protein content was measured by using the bicinchoninic acid method (16). BSA (grade A; Sigma) was standardized to a carbonic anhydrase reference.
Monitoring Redox-Dependent Activation of CODH.
The activation process of CODH was monitored by reduction of MV and [Fe-S] clusters of CODH in the presence of CO. The determination of CO concentration in the system was performed by a hemoglobin-binding assay (1, 4). Spectra were recorded on a Shimadzu model 1605 dual-beam spectrophotometer.
Determination of the Activation Process of CODH.
(i) Monitoring reduction of MV (A). The absorbance change at 578 nm caused by the reduction of MV during activation of CODH as a function of time (t) was converted to CODH activity as shown in Eq. 1,
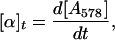 |
1 |
where [α]t represents CODH activity at any given time.
(ii) Monitoring reduction of [Fe-S] clusters. CODH activity was correlated directly to the reduction of [Fe-S] clusters (A418) as indicated in Eq. 2.
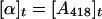 |
2 |
Converting Activity to a Fraction of Active CODH.
CODH activity at any given time ([α]t) can be converted to a fraction of active enzyme (γ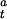 ) as shown in Eq. 3, where Vmax is the maximum CO oxidation activity and [CODH
) as shown in Eq. 3, where Vmax is the maximum CO oxidation activity and [CODH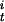 ] and [CODH
] and [CODH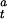 ] represent the fractions of inactive and active forms of CODH, respectively, at the given time t.
] represent the fractions of inactive and active forms of CODH, respectively, at the given time t.
 |
3 |
Analysis of the Fraction of Active CODH as a Function of Time.
The redox-dependent activation of CODH can be expressed as in Eq. 4.
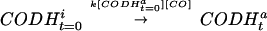 |
4 |
This process requires a small portion of active CODH at assay time 0 ([CODH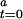 ]) that acts as an “initiator.” Furthermore, inactive CODH is converted to active CODH with a second-order rate constant of the activation process (k) in the presence of [CO]. Thus, Eq. 4 represents the reaction as a second-order autocatalytic reaction (13). Because [CO] >> [CODH
]) that acts as an “initiator.” Furthermore, inactive CODH is converted to active CODH with a second-order rate constant of the activation process (k) in the presence of [CO]. Thus, Eq. 4 represents the reaction as a second-order autocatalytic reaction (13). Because [CO] >> [CODH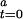 ], the kinetic parameter can be simplified to a pseudo-first-order rate constant, k′ = k[CODH
], the kinetic parameter can be simplified to a pseudo-first-order rate constant, k′ = k[CODH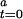 ]. From Eqs. 3 and 4, a function of the kinetic parameters can be expressed as shown in Eq. 5, where [CODH
]. From Eqs. 3 and 4, a function of the kinetic parameters can be expressed as shown in Eq. 5, where [CODH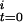 ] represents the initial fraction of inactive CODH at assay time 0.
] represents the initial fraction of inactive CODH at assay time 0.
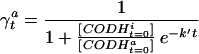 |
5 |
The pseudo-first-order assumption, [CO] >> [CODHi], is valid at all times for the process monitored by MV reduction. However, because a relatively large amount of enzyme is required to monitor the reduction of [Fe-S] clusters, the concentration of CO is decreased significantly with time. Thus, the pseudo-first-order assumption for the process monitoring the reduction of [Fe-S] clusters is valid only during the initial stage of the reaction. Therefore, only the data from the early stages of the assays of reduced [Fe-S] clusters were fitted to Eq. 5.
Measurement of Initial CODH Activities at Various Redox Potentials.
Activity assays of CODH, poised at a wide range of redox potentials (−50 to −500 mV), were performed either by monitoring the dye reduction (0.1 mM dye) by CODH or by monitoring the reduction of [Fe-S] clusters of CODH (0.01 mM), as a function of time from 5.0 to 5.1 sec. The short initial period, 5 sec, was required to prepare the assay for each experiment. Redox buffer systems were poised by addition of DTH. Redox potentials were calculated from the fraction of dye in the reduced and oxidized states. Note that the concentration of the used redox dye (0.1 mM, as described above) was in excess compared with the CODH concentrations used in both the dye-reduction assay (0.5 μM CODH, described in the legends for Figs. 1 and 2) and the [Fe-S] cluster-reduction assay (0.01 mM CODH, as described above). Therefore, the redox potentials of the prepoised assay solutions virtually were unchanged for 0–10 sec.
Figure 1.

Redox-dependent activation of CODH. (A) The autocatalytic activation process of CODH was monitored by the increase in A578 (absorbance of reduced MV) caused by accumulation of reduced MV. The assays were conducted by using anaerobically sealed assay cuvettes (1.5-ml size). Oxidized CODH (0.5 μM, final conc.) was added to an assay mixture containing 10 mM oxidized MV in 100 mM Mops, with variable CO concentrations, and in the absence of reductant. CO concentrations in the cuvettes were (a) 226 ± 11.8, (b) 185 ± 16.1, (c) 179 ± 14.9, and (d) 156 ± 8.2 μM. The progress of CODH activation was monitored by MV reduction at A578 as a function of time (5–200 sec). (A Inset) Transformation of these data using Eqs. 1 and 3 yields fractions of active CODH (thick lines) as a function of time at variable CO concentrations. The transformed data were fitted to Eq. 5 (thin lines) by using GRAPHPAD PRISM software. (B) Oxidized CODH (0.01 mM, final conc.) was added to a solution containing 100 mM Mops with various CO concentrations, in the absence of reductant or MV. The activation process of CODH was monitored by the decrease in A418 (absorbance of oxidized [Fe-S] clusters) caused by the accumulation of reduced [Fe-S] clusters of CODH. The data were transformed to fractions of active enzyme (thick lines) by using Eqs. 2 and 3. The transformed data were fitted to Eq. 5 (thin lines) by using GRAPHPAD PRISM software. Applied CO concentrations were (a) 591 ± 22.7, (b) 488 ± 17.3, (c) 441 ± 15.3, (d) 359 ± 10.5, and (e) 148 ± 19.2 μM.
Figure 2.

Determination of the initial activity of CODH at various redox potentials. The measurement of the initial activity of CODH from 5.0 to 5.1 sec at various redox potentials was performed by addition of oxidized enzyme (0.5 μM for reduction of MV; 0.01 mM for reduction of [Fe-S] clusters, final conc.) to an assay mixture poised at the desired redox potentials, as described in Materials and Methods. Initial-activity data were obtained by monitoring reduction of redox dye or reduction of [Fe-S] clusters. Initial activities were converted to a fraction of active enzyme at the given time (t = 5 sec) by using Eqs. 1, 2, and 3. The fraction of active enzyme at t = 5 sec was transformed to a fraction of active enzyme at t = 0 sec (quantified fraction of active enzyme at the initial stage of reaction) by Eq. 6. The transformed data determined from the two methods, monitored by reduction of MV (○) and of [Fe-S] clusters (●), at any given redox potential were similar. The sequentially transformed data were fitted to the Nernst equation by using GRAPHPAD PRISM software (solid line). The value of E′m for the transition of inactive to active enzyme is −316 ± 8.4 mV, and the number of electrons involved in this transition is 1.0 ± 0.2.
Transformation of Initial Activity to Initial Fraction of Active CODH.
The measured initial activities at various redox potentials, defined as the velocity from 5.0 to 5.1 sec, were converted to fractions of active enzyme (γ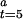 ). A rearrangement of Eq. 5 gives a formula (Eq. 6) by which γ
). A rearrangement of Eq. 5 gives a formula (Eq. 6) by which γ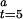 can be converted to a fraction of active enzyme at time 0, γ
can be converted to a fraction of active enzyme at time 0, γ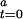 , given the second-order autocatalytic rate constant k, [CO], and the constant T = 5 seconds.
, given the second-order autocatalytic rate constant k, [CO], and the constant T = 5 seconds.
 |
6 |
Data Analyses.
Analyses of data were performed by using GRAPHPAD PRISM 3.00 software (GraphPad, San Diego).
Results
Analysis of the Activation Process of CODH.
Fully oxidized CODH has no detectable initial CO oxidation activity (Fig. 1A, t = 0). However, the activity is recovered at prolonged assay times. The activation process of CODH depends on time and on the concentration of CO. The addition of a small amount of a strong reductant (i.e., DTH or titanium citrate) during the activation process drastically increases the fraction of active CODH, and the addition of reductant in stoichiometric excess to CODH results in an instantaneous recovery of maximum activity (data not shown). Thus, the activation process is redox-dependent, which suggests that the activation of CODH corresponds to a change of redox state as a function of time. The redox-dependent activation of CODH at each CO concentration can be transformed as shown in Eq. 5. CODH stored with excess thionin over 2 h does not show any activation, because the autocatalytic process requires an initiator. The oxidation of CO by the initiator CODH results in a lowering of redox potential and consequently activates the rest of inactive CODH. For the MV reduction assays (Fig. 1A), the pseudo-first-order rate constants (k′) determined at each CO concentration were replotted against CO concentrations to derive the second-order rate constant for the activation process of CODH: k = 272 ± 3.8 s−1M−1 [CO]. The constant k is independent of the equilibrium constants from saturation kinetics (i.e., Km for CO), because these parameters represent distinctly different processes. Therefore, a CO-saturated condition is not required for the determination of the k value.
The later stages of data curves (plateau regions in Fig. 1A Inset) represent fully activated CODH with various amounts of CO within an appropriate time. Because the maximum CO oxidation catalysis of fully activated CODH can be achieved only under CO-saturating conditions (>200 μM CO; ref. 6), the CO oxidation turnover rates under the non-CO-saturating conditions are smaller than CO oxidation turnover rates at the CO-saturating condition. Data presented in the Fig. 1A Inset represent rates of CODH activity at various CO concentrations standardized against the maximum rates of CODH activity under CO-saturating conditions. Therefore, the fraction of activated CODH observed at low concentrations of CO seems smaller than the fraction of activated CODH at relatively high concentrations of CO.
Fig. 1B shows the time course of autocatalysis without any electron mediator (i.e., no benzyl viologen or MV) as measured by the reduction of the [Fe-S] clusters (0.01 mM CODH) at the CO-saturating condition. The data were fitted to Eq. 5, and k was determined to be 231 ± 11.5 s−1M−1 [CO]. As indicated in Materials and Methods, data-fitting was performed only at the early stages of the process; fitted curves (thin lines) at the later stage of each reaction were skewed from the data curve (thick lines) because of the violation of the pseudo-first-order assumption.
A statistical analysis, a two-sample t test, shows that the k values obtained by the two assay methods (reduction of MV and [Fe-S] clusters) are the same within a 99% confidence interval. The average k value for the rate of activation of CODH is 252 ± 14.4 s−1M−1 [CO].
The activation process of CODH in the absence of viologens (MV and benzyl viologen) was relatively slow (data not shown). However, there is no effect of viologens on the activation process when concentrations of CODH are higher than 0.01 mM in the assay system, as analyzed above. These results suggest that the redox-dependent autocatalysis process is apparently independent of the presence of electron mediators in the assay system when the CODH concentration is higher than 0.01 mM.
Determination of the Redox-Active Species of CODH.
Fig. 2 shows the redox dependence of initial CODH activity. Nernst analysis shows that the transition of inactive to active CODH occurs with an Em (midpoint potential) = −316 ± 8.4 mV, and that the number of electrons (n) involved in this transition is 1.0 ± 0.2. Note that in the redox range from 0 to −300 mV, most of the CODH is catalytically inactive.
Discussions
Redox Dependence of CODH Activity.
The analysis of the redox-dependent activation of CODH indicates that CODH can exist in both active and inactive states. Data from reduction of MV and [Fe-S] clusters fit a simple, two-component, second-order autocatalysis model with R2 > 0.9998 (Fig. 1 A and B), indicating that the distribution of enzyme can be treated as binomial (i.e., inactive and active CODH). Based on the data (Fig. 2) and on previous EPR spectroscopic studies (10), it is evident that the Cred1 state of CODH that occurs at potential >−300 mV is not involved in the CO oxidation catalysis of CODH. Furthermore, at potentials <−380 mV, most of CODH is fully active, and these potentials correspond to those producing the following EPR signals: (i) the Cunc state in the absence of CO2; and (ii) the Cred2B state in the presence of CO2 (10). Because the conversion of inactive to active enzyme involves one electron (n = 1 process), Cunc (active state) is one electron more reduced than the Cred1 state (inactive state), an observation consistent with the previous spin assignment (10).
Implications for the Mechanism of CODH.
These analyses, in conjunction with the previous spectroscopic results, indicate that the Cred2B and Cunc states are involved in the catalytic mechanism of CODH, but that the Cred1 state of CODH is not involved. This conclusion is significant to the mechanism of the CODH system, as it precludes a role for the Cred1 state of the enzyme in the catalytic mechanism of CO oxidation by CODH. This analysis is inconsistent with the model proposed for the mechanism of Clostridium thermoaceticum (12). We suggest that the results presented in figure 4 of ref. 12 are consistent with an autocatalytic activation of C. thermoaceticum CODH. Further, it is important to note that the activation process of CODH is an in vitro kinetic artifact, with respect to CO oxidation, and is induced by the active-seed enzyme in the assay system.
Acknowledgments
We thank Drs. W. W. Cleland, C. R. Staples, B. G. Fox, and T. C. Brunold for discussions and insightful comments. We also thank Dr. R. K. Thauer for many useful discussions. The work described here was supported by Department of Energy Basic Energy Sciences Program Grant DE-FG02-87ER13691 (to P.W.L.).
Abbreviations
- CODH
carbon monoxide dehydrogenase
- MV
methyl viologen
- DTH
sodium dithionite
- FeSC
proposed Fe-containing subcluster of C-cluster
- SHE
standard hydrogen electrode
- [Fe-S] clusters
UV-visible observable iron-sulfur clusters
References
- 1.Bonam D, Murrell S A, Ludden P W. J Bacteriol. 1984;159:693–699. doi: 10.1128/jb.159.2.693-699.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonam D, Ludden P W. J Biol Chem. 1987;262:2980–2987. [PubMed] [Google Scholar]
- 3.Ensign S A, Bonam D, Ludden P W. Biochemistry. 1989;28:4968–4973. doi: 10.1021/bi00438a010. [DOI] [PubMed] [Google Scholar]
- 4.Ensign S A. Biochemistry. 1995;34:5372–5381. doi: 10.1021/bi00016a008. [DOI] [PubMed] [Google Scholar]
- 5.Bonam D, McKenna M C, Stephens P J, Ludden P W. Proc Natl Acad Sci USA. 1988;85:31–35. doi: 10.1073/pnas.85.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ensign S A, Hyman M R, Ludden P W. Biochemistry. 1989;28:4973–4979. doi: 10.1021/bi00438a011. [DOI] [PubMed] [Google Scholar]
- 7.Staples C R, Heo J, Spangler N J, Kerby R L, Roberts G P, Ludden P W. J Am Chem Soc. 1999;121:11034–11044. [Google Scholar]
- 8.Hu Z, Spangler N J, Anderson M E, Xia J, Ludden P W, Lindahl P A, Münck E. J Am Chem Soc. 1996;118:830–845. [Google Scholar]
- 9.Ensign S A, Ludden P W. J Biol Chem. 1991;266:18395–18403. [PubMed] [Google Scholar]
- 10.Heo J, Staples C R, Telser J, Ludden P W. J Am Chem Soc. 1999;121:11045–11057. [Google Scholar]
- 11.Lu W P, Jablonski P E, Rasche M, Ferry J G, Ragsdale S W. J Biol Chem. 1994;269:9736–9742. [PubMed] [Google Scholar]
- 12.Seravalli J, Kumar M, Lu W-P, Ragsdale S W. Biochemistry. 1997;36:11241–11251. doi: 10.1021/bi970590m. [DOI] [PubMed] [Google Scholar]
- 13.Frost A A, Pearson R G. Kinetics and Mechanism. New York: Wiley; 1953. pp. 9–26. [Google Scholar]
- 14.Ludden P W, Roberts G P, Kerby R L, Spangler N, Fox J, Shelver D, He Y, Watt R. In: Microbial Growth on C1 Compounds. Lidstrom M E, Tabita F R, editors. San Diego: Academic; 1996. pp. 183–190. [Google Scholar]
- 15.Heo J, Staples C R, Halbleib C M, Ludden P W. Biochemistry. 2000;39:7956–7963. doi: 10.1021/bi992958g. [DOI] [PubMed] [Google Scholar]
- 16.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]