

Abstract
We present a quantitative experimental demonstration of solvent-mediated communication between noncontacting biopolymers. We show that changes in the activity of a solvent component brought about by a conformational change in one biopolymer can result in changes in the physical properties of a second noncontacting biopolymer present in solution. Specifically, we show that the release of protons on denaturation of a donor polymer (in this case, a four-stranded DNA tetraplex, iDNA) modulates the melting temperature of a noncontacting, acceptor polymer [in this case poly(A)]. In addition to such proton-mediated cross talk, we also demonstrate counterion-mediated cross talk between noncontacting biopolymers. Specifically, we show that counterion association/release on denaturation of native salmon sperm DNA (the donor polymer) can modulate the melting temperature of poly(dA)⋅poly(dT) (the acceptor polymer). Taken together, these two examples demonstrate how poly(A) and poly(dA)⋅poly(dT) can serve as molecular probes that report the pH and free salt concentrations in solution, respectively. Further, we demonstrate how such through-solvent dialogue between biopolymers that do not directly interact can be used to evaluate (in a model-free manner) association/dissociation reactions of solvent components (e.g., protons, sodium cations) with one of the two biopolymers. We propose that such through-solution dialogue is a general property of all biopolymers. As a result, such solvent-mediated cross talk should be considered when assessing reactions of multicomponent systems such as those that exist in essentially all biological processes.
Decades of investigations of individual biopolymers in solution have provided us with a wealth of information on the properties of these important molecules of life. These studies have allowed us to define baseline biophysical and biochemical properties for these molecules as well as to characterize their contact interactions with other ligands (refs. 1–7 and references therein). However, the in vivo biochemical milieu includes multiple components that not only influence one another directly through well studied contact interactions (e.g., binding) but also can influence one another indirectly by means of solvent perturbations induced by one component or event propagating/diffusing through the solvent to impact on another component/event in solution. Although such indirect influences (e.g., linkage thermodynamics, coupled equilibria) have been appreciated conceptually for some time (ref. 8 and references therein), few, if any, quantitative experimental demonstrations have been reported of cross talk between noncontacting biopolymers. In this paper, we describe what we believe to be the first quantitative demonstration of this phenomenon. We also comment on the need to consider such cross talk when attempting to understand the complex behaviors of multicomponent cellular systems.
Interactions between biopolymers and small solvent components (anions, cations, protons, etc.) strongly influence the conformational states of biopolymers, their thermal and thermodynamic stabilities, as well as their biological functions (refs. 1 and 3–5, and references therein). Two well known examples of such interactions are the strong dependence of the melting temperatures of nucleic acid duplexes on cation type and concentration (9, 10) as well as the dependence of the thermal and thermodynamic stabilities of proteins on pH (11). Many other examples of such effects can be found in the literature. The effector molecules (small solvent components such as H+, Na+, etc., including H2O) generally act by preferentially binding to, or interacting with, one of the potential conformational states of the biopolymer thereby favoring this state over other states (ref. 1 and references therein). Understanding the linkage between the activity (concentration) of the effector molecules and the stability of a particular conformational state of a macromolecule often allows one to gain insights into mechanisms for the in vitro and in vivo control of biological processes as well as the forces that govern the folding properties of the participating biopolymers (ref. 8 and references therein).
Preferential interactions between small effector molecules and biopolymers also can modulate interactions between macromolecules. A classic example is provided by the binding of the lac repressor protein to its DNA target (10, 12–16). The binding affinity for this protein–nucleic acid interaction strongly depends on the ionic strength of the solution. On binding, a large number of cations and water molecules from the binding surface of the target DNA and a number of anions and water molecules from the protein binding surface are displaced into the bulk solvent, thereby providing a significant entropic driving force for this interaction. Indeed, the importance of solvation at the interface between interacting macromolecules can, in large part, be considered in terms of the entropy gain achieved on release of bound water to bulk solvent. The magnitude of such an entropy gain significantly depends on the overall solvent conditions. The preferential interactions between the interacting macromolecules (e.g., lac repressor protein and DNA) and the solvent effector molecules (e.g., H+, Na+, etc.) strongly influence the binding equilibrium, thereby providing a practical way for controlling the overall binding event. Such coupling between effector molecule–biopolymer and biopolymer–biopolymer interactions generally can be described adequately through the application of linkage thermodynamics (ref. 8 and references therein).
As noted above, it has been recognized for a long time that biological processes/activities can be controlled by modulating the interactions between macromolecules by changing effector molecule concentrations (12). However, what has not been fully appreciated heretofore (at least experimentally) is that conformational changes in one macromolecule can be communicated to another noncontacting biopolymer through the action of solvent effector molecules. We demonstrate here that such communication can, indeed, occur and that it can be quite substantial. For this demonstration, we make use of several nucleic acid model systems. We show that the conformational transition of a donor polymer can be detected by a noncontacting acceptor polymer also present in solution. To aid in the analysis and interpretation of our results, we use here unequal concentrations of acceptor and donor polymers. However, there is no fundamental reason for such a disparity in concentrations. Here, we simply have used a low concentration of acceptor polymer to allow us to treat it as a convenient probe to determine the overall change in solute concentration on denaturation of the donor polymer.
To demonstrate the cross talk noted above, we have focused on nucleic acid systems, although the effect is general and should apply to all classes of biopolymers. The nucleic acids we have selected possess well documented properties and sensitivities to alterations in solvent conditions. Moreover, the melting temperatures (Tm) of nucleic acid structures can be determined with high precision by modern differential scanning calorimeters (DSCs; ref. 17), and their values provide a convenient measure of solvent conditions. In fact, recent advances in calorimetric instrumentation (18, 19) allow modern DSCs to detect very precisely the melting temperature of vanishingly small concentrations of a reporter polymer in the presence of a large excess of background signal generated by a donor polymer (17). Such a measurement is not possible when using more conventional optical methods. The principles we describe here are general and can be used to study a myriad of other systems and processes, including the modulation in vitro of protein-folding events and states (ref. 2 and references therein). In this connection, it is worth noting that the acknowledged high biopolymer concentrations inside cells (20) and the reduced intracellular water activity (21) represent in vivo conditions that would encourage such solvent-mediated communication between noncontacting biopolymers.
In the sections that follow, we describe the systems we have selected and the measurements we have performed.
Materials and Methods
Materials.
Salmon sperm DNA, poly(A), and poly(dA)⋅poly(dT) were purchased from Amersham Pharmacia and used as supplied. Oligonucleotides d(TC5) and d[(C5T3)3C5], which form inter- and intramolecular tetraplexes, respectively, were synthesized by conventional phosphoramidite chemistry and purified by conventional DMT-on and DMT-off reverse-phase HPLC (where DMT refers to the dimethoxytrityl group protecting oligonucleotides during synthesis). Extinction coefficients for the oligonucleotides were determined by phosphate assay (22) in denaturing conditions (60°C) and were ɛ260 of d(TC5) = 39,500 M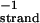 ⋅cm−1 and ɛ260 of d[(C5T3)3C5] = 227,000 M
⋅cm−1 and ɛ260 of d[(C5T3)3C5] = 227,000 M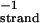 ⋅cm−1. The following extinction coefficients for the polymers were provided by the manufacturer or taken from the literature: ɛ260 of salmon sperm DNA = 13,800 M
⋅cm−1. The following extinction coefficients for the polymers were provided by the manufacturer or taken from the literature: ɛ260 of salmon sperm DNA = 13,800 M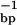 ⋅cm−1, ɛ260 of poly(A) = 9,800 M
⋅cm−1, ɛ260 of poly(A) = 9,800 M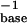 ⋅cm−1, and ɛ260 of poly(dA)⋅poly(dT) = 12,000 M
⋅cm−1, and ɛ260 of poly(dA)⋅poly(dT) = 12,000 M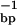 ⋅cm−1. Oligonucleotides were ion-exchanged into the desired buffer with the help of Sephadex G-25 spin columns. All polymer samples were dialyzed exhaustively against at least two changes of the desired buffer.
⋅cm−1. Oligonucleotides were ion-exchanged into the desired buffer with the help of Sephadex G-25 spin columns. All polymer samples were dialyzed exhaustively against at least two changes of the desired buffer.
Buffers.
The 10 mM Na+ buffer for the studies with poly(A) and d(TC5) was prepared by mixing appropriate quantities of 4 mM disodium succinate/1 mM Na2EDTA buffer with 4 mM succinic acid/1 mM Na2EDTA/8 mM NaCl buffer to give the desired pH. The 12 mM Na+ (pH 6.8) buffer for the studies with salmon sperm DNA and poly(dA)⋅poly(dT) was prepared by mixing appropriate quantities of 10 mM sodium cacodylate/1 mM Na2EDTA buffer with 10 mM cacodylic acid/1 mM Na2EDTA/10 mM NaCl buffer to give the desired pH of 6.8. Buffers of higher ionic strength were prepared by including the necessary amount of additional NaCl.
Methods.
All melting experiments were performed by using one of two NanoDSC II DSCs (Calorimetry Sciences, Provo, UT) with either cylindrical or capillary cells of nominal cell volume of 0.3 ml. Identical results were obtained with both instruments. Mixed donor and acceptor samples were used to fill the sample cell; acceptor samples alone were placed in the reference cell. A constant heating scan rate of 1 K/min was used. Melting temperatures were determined from the midpoint of the transition of the excess heat capacity curves. To avoid evaporative losses, samples were not degassed as is common practice. Nucleic acid concentrations were determined spectroscopically with a Varian model 300 Spectrophotometer.
Results
pH Modulated Communication Between Noncontacting Biopolymers.
To demonstrate that changes in the conformational state of one biopolymer can be communicated to a second noncontacting biopolymer, we use as a first example the interactions between the DNA oligomer d(TC5) and the RNA polymer poly(A).
d(TC5) Properties.
At low pH and low ionic strength, d(TC5) adopts a stable secondary structure, a four-stranded DNA tetraplex (iDNA; refs. 23–25). This structural motif is one in which one parallel-stranded d(TC5) duplex interacts with another d(TC5) duplex through intercalation between the base pairs (23–26). This tetraplex has been shown to be the preferred structure of cytosine-rich oligonucleotides at low pH (27, 28). The feature of the d(TC5) tetraplex that is of particular interest to us is the requirement for cytosine protonation to form the tetraplex. Specifically, about half of the cytosines in the tetraplex must be protonated to form the parallel C⋅C+ base-pair array that represents the repeating unit of this structure (23, 28). On a per-base level, the d(TC5) tetraplex is one of the most highly protonated nucleic acid structures known. As such, the d(TC5) tetraplex can serve either as a donor or as an acceptor of protons depending on solution conditions. Denaturation of the tetraplex at pH values above the pKa of free cytosine results in a net release of bound protons into the bulk buffer. Denaturation of the tetraplex at pH values below the pKa value of free cytosines (about pH 4.3–4.5) results in a net uptake of protons from the bulk buffer (28). Based on the design and solution conditions of our experiment, we use the d(TC5) tetraplex as a proton donor.
Poly(A) Properties.
The RNA poly(A) represents the other nucleic acid component in our system. Poly(A) forms a pH-dependent, parallel-stranded RNA double helix in which a protonated adenine of one RNA strand forms hydrogen bonds to a protonated adenine of a second RNA strand (29–32). Hydrogen bonding occurs through Hoogsteen interactions between the N7/NH26 positions of one adenine and the N7/NH26 position of the second adenine. Protonation is believed to occur at the N1 positions of both adenines. In contrast to the d(TC5) structure, the protonation site in the poly(A) complex is not involved directly in hydrogen bonding. Of particular interest here is the strong pH and ionic strength dependence of the melting temperature (Tm) of the acid poly(A) helix. Linear shifts in melting temperature by as much as 40 K per change in pH unit at a given ionic strength and Tm changes of 20 K for a 10-fold change in ionic strength at a given pH have been reported (31, 32). We have confirmed these results independently.¶ Therefore, the Tm of the acid poly(A) double helix provides a very sensitive measure or probe of the pH/ionic strength of a given solution. Furthermore, between pH 6.0 and pH 4.5 (i.e., near the pKa of free cytosine) the Tm of poly(A) is always higher than that of d(TC5) in low ionic strength buffer (10 mM Na+ buffer). We expect no direct interactions between d(TC5) and poly(A) under the solution conditions studied here. For all these reasons, poly(A) represents a very good proton acceptor for use in our study.
Solvent-Mediated Cross Talk Between d(TC5) and Poly(A).
In a low ionic strength buffer (10 mM Na+, pH 5.75) we combined an excess of the d(TC5) donor with the poly(A) acceptor and placed this solution into the sample cell of our DSC. The choice of this low ionic strength buffer was dictated by the fact that the d(TC5) tetraplex becomes unstable at an elevated sodium chloride concentration (28). For these experiments, the reference cell of the DSC was filled with an equal amount of poly(A). If the Tm of poly(A) remains unaffected by the presence of d(TC5), poly(A) in the sample and reference cells will melt at the same temperature, resulting in no measurable signal because of cancellation of the heats of denaturation. If d(TC5) influences the melting of poly(A) in some way, melting of poly(A) in the reference cell will occur at a different temperature from that of poly(A) in the sample cell, thereby precluding cancellation and resulting in the appearance of a positive transition in the sample cell and a negative transition in the reference cell. Inspection of the DSC melting profiles shown in Fig. 1 reveals that this behavior is exactly what we observe. Note the clear difference in melting temperature for poly(A) in the sample (positive peak) and the reference cells (negative peak). Depending on the concentration of d(TC5) (CT = 0.07 mMstrand → 0.89 mMstrand), the Tm of poly(A) in the sample cell is shifted to a higher temperature (relative to the reference cell) by as little as 1.7 K and by as much as 17 K. These ΔTm shifts correspond to apparent pH changes of 0.05 and 0.5 pH units, respectively. In fact, we have confirmed independently a decrease in pH of the sample solution after melting by direct measurement with a pH meter. This observation suggests a significant influence on poly(A) melting by the presence of the noncontacting d(TC5) solute. The magnitude of the observed Tm shift for poly(A) relative to the reference cell is independent of the heating rate (not shown), although the d(TC5) melting is rate dependent. The magnitude of the observed Tm shift is, however, proportional to the nominal pH of the bulk solution (Fig. 1), the concentration of the added d(TC5) (Fig. 2), and the total amount of buffer species present (Fig. 2). In the aggregate, these observations strongly suggest that the denaturation of the tetraplex causes the change in the melting temperature of poly(A), although no direct interactions between d(TC5) and poly(A) occur. We propose that the conformational transition of d(TC5) is communicated to poly(A) through the action of the released protons which alter the pH of the bulk solution. This experiment supports our hypothesis of “through-solution” communication between noncontacting biopolymers; in this case, differential proton binding represents the effector molecule that causes the noncontacting communication.
Figure 1.

Calorimetric melting profiles for d(TC5) donor and poly(A) acceptor in 10 mM Na+ buffer at three different pH values. The large positive transition in each of the three melting curves corresponds to the melting of d(TC5). The small negative peak corresponds to the melting of the poly(A) acid helix in the reference cell; the small positive peak at the higher temperature corresponds to the melting of the poly(A) acid helix in the sample cell. Black curve, pH 6.0, 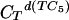 = 0.428 mMstrand, CTpoly(A) = 0.1169 mMbase; blue curve, pH 5.75,
= 0.428 mMstrand, CTpoly(A) = 0.1169 mMbase; blue curve, pH 5.75, 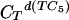 = 0.4146 mMstrand, CTpoly(A) = 0.0639 mMbase; red curve, pH 5.5,
= 0.4146 mMstrand, CTpoly(A) = 0.0639 mMbase; red curve, pH 5.5, 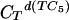 = 0.4247 mMstrand, CTpoly(A) = 0.068 mMbase.
= 0.4247 mMstrand, CTpoly(A) = 0.068 mMbase.
Figure 2.

The measured Tm shifts in two different buffer conditions for poly(A) in the sample cell relative to poly(A) in the reference cell as a function of d(TC5) concentration. The solution conditions are 10 mM Na+ buffer at pH 5.75.
Poly(A) as a Molecular Probe of pH and Protons.
We can use the proton-induced shift we measure in the Tm of the reporter (acceptor) poly(A) molecule in conjunction with the known Tm vs. pH reference curve for dilute poly(A) to determine the total number of protons released from d(TC5). In this analysis, the release of protons from the d(TC5) tetraplex causes a decrease in pH, which can be determined precisely from the Tm vs. pH reference curve for poly(A). Assuming a dTm/dpH of ≈40 K for poly(A) and a precision in Tm measurements of ±0.2 K, we can reliably measure pH changes as small as 0.005 pH unit by this approach. Thus, the poly(A) pH probe exhibits a sensitivity that significantly exceeds that commonly found with conventional pH probes.
We reasonably assume that d(TC5) concentration-dependent changes in proton concentration only cause shifts in the acid/base concentration of the buffer species, which, given the pH of the solution, can be estimated from the Henderson–Hasselbalch equation. Temperature-dependent changes in the pKa of buffer components can be ignored, as these should be identical to those of the poly(A) reference curve. Activity coefficients are ignored because of the low concentrations at which we work. In fact, to minimize the impact of the reporter (acceptor) poly(A) molecule on the overall solution conditions, it is present at a 50- to 100-fold lower concentration than our donor polymer [d(TC5)]. We also reasonably assume that changes in the degree of ionization of other solvent components, e.g., EDTA and poly(A), can be ignored, and that d(TC5) melting does not result in a net change in counterion release or uptake other than the release of protons. By using these reasonable assumptions, we calculate that, on average, 1.5 protons per d(TC5) monomer strand, or 6 protons per tetraplex, are released from d(TC5). The maximum formal value is 2.5 protons per monomer (10 protons per tetraplex). The observation that net proton release is less than the theoretical number of protons assumed to be associated with the d(TC5) structure (less than complete protonation of potential sites) is, as one would expect, because of end effects and/or negative cooperativity between adjacent cytosines, influences that have been reported for DNA triplexes (33–37). The point is that the sensitivity of the Tm of poly(A) to solution conditions makes it a sensitive probe for characterizing solution pH.
Solvent-Mediated Cross Talk Between Poly(A) and Intramolecular d(TC5): Reversing the Donor and Acceptor Systems.
To demonstrate that such through-solution communication between noncontacting biopolymers is general and not an artifact of our experimental design we have reversed the order of the donor and acceptor polymers in our experiments. In this mode, poly(A) now acts as the donor of protons, and a four-stranded DNA tetraplex (iDNA) acts as the acceptor. For this application, it was necessary to generate an intramolecular d(TC5) complex to increase its Tm above that of poly(A). To this end, we linked together the four d(TC5) strands through three all-thymine loops, thereby generating the desired intramolecular tetraplex d[(C5T3)3C5] (28, 38). This iDNA has an increased Tm although maintaining a pH profile very similar to that of the intermolecular d(TC5) tetraplex. The addition of increasing amounts of poly(A) to this intramolecular d[(C5T3)3C5] complex causes a Tm shift that is proportional to the poly(A) concentration added. This result is consistent with our previous experiment in which the addition of increasing amounts of d(TC5) caused a proportional Tm shift in poly(A). Note that in these experiments, the intramolecular d[(C5T3)3C5] oligomer is present at a concentration 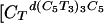 = 0.032 mMstrand] that is similar to that of the poly(A) polymer (CT = 0.03 mMbase → 2 mMbase), thereby limiting its use as a convenient pH probe under these conditions. We propose, therefore, that through-solution proton transfer has occurred from our donor poly(A) duplex to our acceptor, intramolecular d[(C5T3)3C5] complex. These experiments demonstrate that the through-solution interaction between noncontacting biopolymers represents a two-way communication in the strict sense because donor and acceptor polymers can interchange roles depending on solution conditions.
= 0.032 mMstrand] that is similar to that of the poly(A) polymer (CT = 0.03 mMbase → 2 mMbase), thereby limiting its use as a convenient pH probe under these conditions. We propose, therefore, that through-solution proton transfer has occurred from our donor poly(A) duplex to our acceptor, intramolecular d[(C5T3)3C5] complex. These experiments demonstrate that the through-solution interaction between noncontacting biopolymers represents a two-way communication in the strict sense because donor and acceptor polymers can interchange roles depending on solution conditions.
Counterion/Salt-Modulated Communication Between Biopolymers.
A second test of our hypothesis for solvent-mediated communication between noncontacting biopolymers involves condensed counterions rather than bound protons. For this demonstration, we used salmon sperm DNA as a counterion donor and poly(dA)⋅poly(dT) as a counterion acceptor. The association of a large number of counterions with native, duplex DNA to reduce the overall charge density of the polymer is well known (9, 10). On denaturation, a significant fraction of these counterions are released into solution, as the overall/combined charge density of the two single strands is less than that of the native duplex. Because denaturation of genomic DNA is largely irreversible on the time scale of our experiments (the kinetics for reassociation are very slow), denaturation of salmon sperm DNA can be used as a convenient donor of counterions. The properties of the poly(dA)⋅poly(dT) homopolymer duplex make it well suited to serve as our counterion acceptor. The increase in melting temperature of the poly(dA)⋅poly(dT) homopolymer that occurs on changes in the ionic strength is well documented (39–42). The Tm of poly(dA)⋅poly(dT) shifts by as much as 20 K per 10-fold increase in ionic strength. Furthermore, melting of poly(dA)⋅poly(dT) is reversible, and neither poly(dA), poly(dT), nor poly(dA)⋅poly(dT) is expected to interact with native or denatured salmon sperm DNA under the conditions used here. For all these reasons, poly(dA)⋅poly(dT) represents a good counterion acceptor.
Fig. 3 shows the changes we observed in Tm for poly(dA)⋅poly(dT) on addition of increasing amounts of native salmon sperm DNA (CT = 0.6 mMbp → 8.5 mMbp), as well as the changes that occurred on denaturation of the genomic DNA. In this experiment, the same amount of poly(dA)⋅poly(dT) (CT = 0.042 mMbp) was added to the reference solution and to the DNA sample. Interestingly, addition of native DNA already resulted in a noticeable Tm shift for poly(dA)⋅poly(dT) in the sample cell relative to the reference cell. The most likely explanation for this observation is that poly(dA)⋅poly(dT) senses the altered ionic strength brought about by the presence of the native donor DNA and its associated counterion cloud. Denaturation of the genomic DNA brings about a further and much larger shift in Tm which also is proportional to the concentration of the added donor DNA. Clearly, poly(dA)⋅poly(dT) responds to the conformational transition of the donor genomic DNA by a shift in its melting temperature. We attribute this observation to a change in solvent conditions induced by the release of counterions from the genomic DNA which are sensed by the poly(dA)⋅poly(dT) duplex as an increase in solvent salt concentration, thereby increasing the Tm of the duplex (9, 10).
Figure 3.

The measured Tm shifts for a trace amount of poly(dA)⋅poly(dT) resulting from the presence of native (circle) and denatured (square) salmon sperm DNA. The dashed lines represent the straight lines of best fit through the experimental data points. The solution conditions are 12 mM Na+/10 M cacodylate/1 mM Na2EDTA, pH 6.8.
We can use the observed Tm shift of poly(dA)⋅poly(dT) in conjunction with a Tm vs. log[Na+] reference/calibration curve to estimate the genomic DNA-induced change in solution ionic strength required to cause the observed increase in Tm for poly(dA)⋅poly(dT). As this change in apparent ionic strength is brought about by the excess salmon sperm DNA, it is most convenient to express the change as a percentage of the amount of DNA added. This analysis reveals that the observed Tm shift reflects an increase in solution ionic strength that corresponds to an amount of added NaCl equivalent to 5% of the amount of added native genomic DNA (per base). On denaturation of the genomic DNA, a further increase in Tm occurs that reflects an increase in solution ionic strength that corresponds to an additional amount of added NaCl equivalent to 9.5% of the added genomic DNA (per base). Counterion condensation theory, as developed by Manning (9) and Record et al. (10), suggests that only about 88% of the phosphates in the backbone of native DNA are neutralized by territorially and thermodynamically associated counterions, leaving free in solution about 12% of the counterions that neutralize the DNA (all charges have to be neutralized at all times). Our observed shift in Tm for poly(dA)⋅poly(dT) corresponds to a 5% increase in free counterion concentration, as calculated from our measured Tm dependences of poly(dA)⋅poly(dT) on NaCl concentration and on the concentration of added salmon sperm DNA. Efforts to incorporate this experimental result into existing polyelectrolyte counterion condensation treatments now will be possible (J.V., H.H.K., G. S. Manning, and K.J.B., unpublished data). Nevertheless, our result is in qualitative agreement with both the direction and magnitude of the value predicted by Manning/Record theory (9, 10). Application of linkage thermodynamics to counterion–polynucleotide interactions further suggests a release of about 0.17 Na+ ion per base (17%) on denaturation of native DNA. This result is again in qualitative agreement with the apparent additional increase in the ionic strength of the solution equivalent to 9.5% of the added amount of bulk genomic (donor) DNA. When one considers the molecular crowding effects that are an inherent part of any such experimental measurements, then the agreement between theory and experiment becomes even more impressive. These observations demonstrate the power of our technique to test theoretical predictions and speak well of our ability to measure with great precision small changes in ion concentration.
The response described above of our acceptor polymer, poly(dA)⋅poly(dT), to the presence of native DNA and its conformational change represents a further demonstration of through-solution communication between noncontacting biopolymers. For this example, the effector molecule is the cation concentration rather than the change in pH used in our previous example.
Conclusions
We have demonstrated that changes in the conformational state of one biopolymer can result in changes in the overall solution conditions, which, in turn, can result in changes in the physical properties of a second noncontacting biopolymer present in the same solution. In short, we have demonstrated through-solution communication between noncontacting biopolymers. On a practical level, our observations allow one to measure, in a model-free manner, the total concentrations of small solvent effector molecules in any given solution (e.g., inside liposomes, intact cells, and any change therein). On a more fundamental level, our observations allow one to test basic principles about the role of small solvent components in modulating biopolymer structure and stability. Our results suggest that the observed solvent-mediated communication between noncontacting biopolymers might represent a general feature by which conformational changes in a biopolymer (e.g., ligand binding or phosphorylation) can be communicated to other biopolymers without the requirement of direct contact between these biopolymers. As a result, such solvent-mediated cross talk should be considered when assessing reactions of multicomponent systems such as those that exist in essentially all biological processes.
Acknowledgments
We thank Dr. G. E. Plum (Rutgers University) for helpful discussions and critical reading of the manuscript. This work was supported by National Institutes of Health Grants GM23509, GM34469, and CA47995 (to K.J.B.).
Abbreviations
- Tm
melting temperature
- DSC
differential scanning calorimeter
- CT
total concentration of a given DNA polymer
Footnotes
Tm = −36.748 × pH + 551.55 (in K) (r = 0.99991) in 10 mM Na+ buffer and Tm = −40.57 × pH + 546.55 (in K) (r = 0.9999) in 100 mM Na+ buffer.
References
- 1.Bloomfield V A, Crothers D M, Tinoco I., Jr . Nucleic Acids. Structures, Properties, and Functions. Mill Valley, CA: Univ. Sci. Books; 2000. [Google Scholar]
- 2.Dobson C M, Sali A, Karplus M. Angew Chem Int Ed Engl. 1998;37:868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 3.Cantor C R, Schimmel P R. The Conformation of Biological Macromolecules. San Francisco: Freeman; 1980. [Google Scholar]
- 4.Cantor C R, Schimmel P R. Techniques for the Study of Biological Structure and Function. San Francisco: Freeman; 1980. [Google Scholar]
- 5.Cantor C R, Schimmel P R. The Behavior of Biological Macromolecules. San Francisco: Freeman; 1980. [Google Scholar]
- 6.Creighton T E. Protein Folding. New York: Freeman; 1992. [Google Scholar]
- 7.Creighton T E. Protein Function: A Practical Approach. New York: IRL; 1997. [Google Scholar]
- 8.Wyman J, Gill S J. Binding and Linkage. Functional Chemistry of Biological Macromolecules. Mill Valley, CA: Univ. Sci. Books; 1990. [Google Scholar]
- 9.Manning G S. Q Rev Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 10.Record M T, Jr, Anderson C F, Lohman T M. Q Rev Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 11.Privalov P L, Potekhin S A. Methods Enzymol. 1986;131:4–51. doi: 10.1016/0076-6879(86)31033-4. [DOI] [PubMed] [Google Scholar]
- 12.deHaseth P L, Lohman T M, Record M T., Jr Biochemistry. 1977;16:4783–4790. doi: 10.1021/bi00641a004. [DOI] [PubMed] [Google Scholar]
- 13.Record M T, Jr, Lohman M L, De Haseth P. J Mol Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 14.Record M T, Jr, deHaseth P L, Lohman T M. Biochemistry. 1977;16:4791–4796. doi: 10.1021/bi00641a005. [DOI] [PubMed] [Google Scholar]
- 15.Record M T, Jr, Anderson C F, Mills P, Mossing M, Roe J H. Adv Biophys. 1985;20:109–135. doi: 10.1016/0065-227x(85)90033-4. [DOI] [PubMed] [Google Scholar]
- 16.Record M T, Jr, Zhang W, Anderson C F. Adv Protein Chem. 1998;51:281–353. doi: 10.1016/s0065-3233(08)60655-5. [DOI] [PubMed] [Google Scholar]
- 17.Völker J, Blake R D, Delcourt S G, Breslauer K J. Biopolymers. 1999;50:303–318. doi: 10.1002/(SICI)1097-0282(199909)50:3<303::AID-BIP6>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 18.Privalov G, Kavina V, Freire E, Privalov P L. Anal Biochem. 1995;232:79–85. doi: 10.1006/abio.1995.9957. [DOI] [PubMed] [Google Scholar]
- 19.Privalov G P, Privalov P L. Methods Enzymol. 2000;323:31–62. doi: 10.1016/s0076-6879(00)23360-0. [DOI] [PubMed] [Google Scholar]
- 20.Daban J R. Biochemistry. 2000;39:3861–3866. doi: 10.1021/bi992628w. [DOI] [PubMed] [Google Scholar]
- 21.Record M T, Courtenay E S, Cayley D S, Guttman H J. Trends Biochem Sci. 1998;23:143–148. doi: 10.1016/s0968-0004(98)01196-7. [DOI] [PubMed] [Google Scholar]
- 22.Snell F D, Snell C T. Colorimetric Methods of Analysis, Including Some Turbidimetric and Nephelometric Methods. Huntington, NY: Krieger; 1972. [Google Scholar]
- 23.Gehring K, Leroy J L, Gueron M. Nature (London) 1993;363:561–565. doi: 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- 24.Leroy J L, Gehring K, Kettani A, Gueron M. Biochemistry. 1993;32:6019–6031. doi: 10.1021/bi00074a013. [DOI] [PubMed] [Google Scholar]
- 25.Leroy J L, Gueron M. Structure (London) 1995;3:101–120. doi: 10.1016/s0969-2126(01)00138-1. [DOI] [PubMed] [Google Scholar]
- 26.Berger I, Cai L, Chen L, Rich A. Biopolymers. 1997;44:257–267. doi: 10.1002/(SICI)1097-0282(1997)44:3<257::AID-BIP5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 27.Manzini G, Yathindra N, Xodo L E. Nucleic Acids Res. 1994;22:4634–4640. doi: 10.1093/nar/22.22.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mergny J-L, Lacroix L, Han X, Leroy J-L, Helene C. J Am Chem Soc. 1995;117:8887–8898. [Google Scholar]
- 29.Rich A. Nature (London) 1958;181:521–525. doi: 10.1038/181521a0. [DOI] [PubMed] [Google Scholar]
- 30.Rich A, Davis D R, Crick F H C, Watson J D. J Mol Biol. 1961;3:71–86. doi: 10.1016/s0022-2836(61)80009-0. [DOI] [PubMed] [Google Scholar]
- 31.Holcomb D N, Timasheff S N. Biopolymers. 1968;6:513–529. doi: 10.1002/bip.1968.360060407. [DOI] [PubMed] [Google Scholar]
- 32.Klump H, Ackerman T, Neumann E. Biopolymers. 1969;7:423–431. [Google Scholar]
- 33.Völker J, Klump H H. Biochemistry. 1994;33:13502–13508. doi: 10.1021/bi00249a039. [DOI] [PubMed] [Google Scholar]
- 34.Völker J, Osborne S E, Glick G D, Breslauer K J. Biochemistry. 1997;36:756–767. doi: 10.1021/bi962271l. [DOI] [PubMed] [Google Scholar]
- 35.Plum G E, Breslauer K J. J Mol Biol. 1995;248:679–695. doi: 10.1006/jmbi.1995.0251. [DOI] [PubMed] [Google Scholar]
- 36.Plum G E, Pilch D S, Singleton S F, Breslauer K J. Annu Rev Biophys Biomol Struct. 1995;24:319–350. doi: 10.1146/annurev.bb.24.060195.001535. [DOI] [PubMed] [Google Scholar]
- 37.Leitner D, Schroder W, Weisz K. Biochemistry. 2000;39:5886–5892. doi: 10.1021/bi992630n. [DOI] [PubMed] [Google Scholar]
- 38.Mergny J L, Lacroix L. Nucleic Acids Res. 1998;26:4797–4803. doi: 10.1093/nar/26.21.4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chamberlin M J. Fed Proc. 1965;24:1446–1457. [PubMed] [Google Scholar]
- 40.Riley M, Maling B, Chamberlin M J. J Mol Biol. 1966;20:359–389. doi: 10.1016/0022-2836(66)90069-6. [DOI] [PubMed] [Google Scholar]
- 41.Blake R D, Delcourt S G. Biopolymers. 1987;26:2009–2026. doi: 10.1002/bip.360261204. [DOI] [PubMed] [Google Scholar]
- 42.Blake R D, Delcourt S G. Biopolymers. 1990;29:393–405. doi: 10.1002/bip.360290211. [DOI] [PubMed] [Google Scholar]