

Abstract
Increased acid excretion may promote renal injury. To evaluate this in African Americans with hypertensive nephrosclerosis, we studied the association between the net endogenous acid production and progression of kidney disease in 632 patients in the AASK trial. Protein and potassium intakes were estimated from 24-hour urea nitrogen and potassium excretion, and used to estimate net endogenous acid production, averaged over 2 years, approximating routine intake. The link between net endogenous acid production and the I125iothalamate glomerular filtration rate (iGFR) and time to end stage renal disease or doubling of serum creatinine was analyzed using mixed models and Cox proportional hazards regressions. The trend in higher net endogenous acid production was significantly associated with a faster decline in iGFR over a median of 3.2 years. After adjustment for age, body mass index, baseline iGFR, urine protein to creatinine ratio and randomized treatment group, the trend in higher net endogenous acid production remained significantly associated with a faster decline in iGFR at a rate 1.01 mL/min/1.73 m2 per year faster in the highest to the lowest quartile. However, in time to event analyses over a median of 7.7 years, the adjusted hazard ratio (1.10) for composite renal events per 25 mEq/day higher net endogenous acid production was not significant. Hence, our findings implicate endogenous acid production as a potential modifiable risk factor for progressive kidney disease.
Introduction
Chronic kidney disease (CKD) is a major public health problem affecting 13% of the US population.(1) Increased risk of morbidity and mortality are evident even among those with only mild decreases in kidney function.(2) Preventive strategies which are low cost, low risk and scalable are needed to address the epidemic of kidney disease.
Metabolic acidosis, a consequence of decreased renal acid excretion, is a modifiable risk factor for CKD progression.(3-5) In CKD, overall acid excretion is impaired with increased per nephron acid excretion to compensate for nephron loss.(6, 7) In turn, increased acid excretion by the nephron may promote tubulointerstitial injury and contribute to disease progression.(8, 9) Alkali supplements can lower acid excretion and slow disease progression.(4, 10) Manipulation of the net endogenous acid production through diet may be an additional strategy to decrease renal acid excretion that may be more amenable to wide implementation as a public health initiative.
Net endogenous acid production is determined by the balance of fixed acid and alkali precursors in the diet. Fixed acid in the diet is derived largely from protein intake and alkali from organic anions, such as citrate and acetate, which are naturally bound to cations, such as potassium.(11) For this reason, net endogenous acid production can be estimated from the ratio of protein and potassium in the diet.(12-14) In this study, we estimate net endogenous acid production in this manner and evaluate its association with CKD progression in a cohort of African Americans with CKD.
Results
Six hundred thirty two participants from the African American Study of Kidney Disease and Hypertension (AASK) trial and cohort study were included in this analysis. Reasons for exclusion are summarized in Figure 1. Median age was 55 years (range 22 to 70 years). Median I125iothalamate glomerular filtration rate (iGFR) was 48.6 mL/min/1.73 m2 (interquartile range 36.6 to 58.5 mL/min/1.73 m2). Median estimated net endogenous acid production was 72.8 mEq/d (interquartile range 57.2 to 89.5 mEq/d). Median estimated protein intake was 64.9 g/d (interquartile range 53.1 to 76.3 g/d) and median estimated potassium intake was 43.5 mEq/d (interquartile range 33.8 to 55.9 mEq/d). The baseline characteristics of the study population stratified by quartiles of net endogenous acid production are presented in Table 1. Higher net endogenous acid production was associated with current smoking, lower income, and lower serum bicarbonate. Differences in protein intake across categories of NEAP were smaller than differences in potassium intake.
Figure 1. Summary of reasons for participant exclusion from study population.
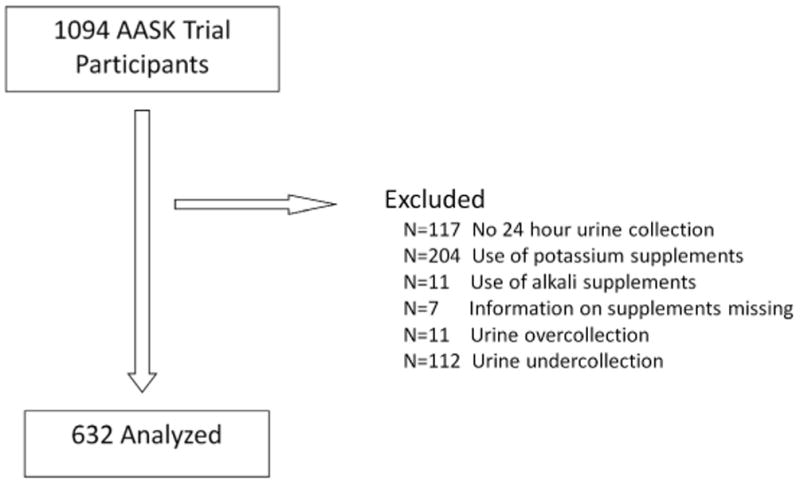
Table 1. Baseline characteristics of the study population by quartiles of net endogenous acid production (NEAP).
| Quartiles of NEAP (mEq/day) | |||||
|---|---|---|---|---|---|
| 1 (18.2 – 57.1) | 2 (57.2 – 72.8) | 3 (72.9 – 89.5) | 4 (89.6 – 232.5) | ||
| Characteristic: Mean ± SD or n (%) | (n=158) | (n=158) | (n=158) | (n=158) | p-value† |
| Age (years) | 55 ± 10 | 56 ± 10 | 54 ± 11 | 54 ± 11 | 0.41 |
| Female sex (%) | 63 (39.9) | 56 (35.4) | 69 (43.7) | 51 (32.3) | 0.17 |
| History of heart disease (%) | 84 (53.2) | 75 (47.5) | 79 (50.0) | 87 (55.1) | 0.54 |
| Smoking (%) | 0.008 | ||||
| Never | 73 (46.2) | 63 (39.9) | 62 (39.2) | 66 (41.8) | |
| Former | 55 (34.8) | 55 (34.8) | 43 (27.2) | 35 (22.2) | |
| Current | 30 (19.0) | 40 (25.3) | 53 (33.5) | 57 (36.1) | |
| Total Income (%)* | 0.005 | ||||
| <$15,000 | 65 (41.1) | 66 (41.8) | 74 (46.8) | 84 (53.2) | |
| ≥$15,000 | 68 (43.0) | 72 (45.6) | 50 (31.7) | 42 (26.6) | |
| Body mass index (kg/m2) | 30.3 ± 6.4 | 30.3 ± 6.2 | 29.3 ± 5.5 | 29.5 ± 6.4 | 0.10 |
| Body mass index (%) | 0.07 | ||||
| <25 kg/m2 | 25 (15.8) | 34 (21.5) | 36 (22.8) | 45 (28.5) | |
| 25-30 kg/m2 | 67 (42.4) | 52 (32.9) | 61 (38.6) | 45 (28.5) | |
| >30 kg/m2 | 66 (41.8) | 72 (45.6) | 61 (38.6) | 68 (43.0) | |
| Randomized to low BP goal (%) | 80 (50.6) | 86 (54.4) | 72 (45.6) | 69 (43.7) | 0.21 |
| Randomized drug (%) | 0.58 | ||||
| Ramipril | 57 (36.1) | 68 (43.0) | 64 (40.5) | 61 (38.6) | |
| Metoprolol | 65 (41.1) | 55 (34.8) | 68 (43.0) | 67 (42.4) | |
| Amlodipine | 36 (22.8) | 35 (22.2) | 26 (16.5) | 30 (19.0) | |
| Serum phosphorus (mg/dL) | 3.5 ± 0.6 | 3.5 ± 0.6 | 3.6 ± 0.7 | 3.5 ± 0.6 | 0.55 |
| Serum bicarbonate (mEq/L) | 25.7 ± 2.9 | 25.7 ± 2.8 | 25.0 ± 2.9 | 24.6 ± 3.3 | <0.001 |
| Serum potassium (mEq/L) | 4.1 ± 0.5 | 4.1 ± 0.6 | 4.1 ± 0.5 | 4.2 ± 0.5 | 0.07 |
| Serum potassium > 5.0 mEq/L | 7 (4.4) | 6 (3.8) | 9 (5.7) | 7 (4.4) | 0.88 |
| Urine protein/creatinine (%) | 0.29 | ||||
| <0.22 | 104 (66.7) | 118 (74.7) | 108 (68.8) | 116 (73.9) | |
| ≥0.22 and <1.0 | 37 (23.7) | 27 (17.1) | 27 (17.2) | 29 (18.5) | |
| ≥1.0 | 15 (9.6) | 13 (8.2) | 22 (14.0) | 12 (7.6) | |
| iGFR (mL/min/1.73 m2) | 46.5 ± 13.4 | 48.1 ± 13.7 | 46.4 ± 13.9 | 48.1 ± 13.1 | 0.52 |
| Estimated protein intake (g/d)‡ | 64.6 ± 19.1 | 64.6 ± 16.4 | 66.3 ± 16.6 | 68.4 ± 20.4 | 0.04 |
| Estimated potassium intake (mEq/d)‡ | 65.4 ± 20.3 | 47.8 ± 12.8 | 40.5 ± 10.2 | 32.4 ± 10.7 | <0.001 |
Column % do not total 100 due to participants who declined to report income
p-value is p-trend by univariate linear regression (continuous variables) or Pearson's χ2 (categorical variables)
Estimated from 24 hour urine collections between 12 and 36 months post-randomization in the AASK trial phase
iGFR: I125 iothalamate glomerular filtration rate; BP: blood pressure
I125iothalamate GFR slope (iGFR) analysis
The slope of iGFR was estimated from all available iGFR measurements performed every 6 months during the AASK trial. A mean of 6.9 iGFR measurements were available per individual to estimate iGFR slope over a median of 3.2 years.
The association of dietary intake and iGFR slope over follow up in the trial phase is presented in Table 2. In unadjusted models, there was a faster rate of decline in iGFR with higher quartiles of net endogenous acid production (p-trend=0.05) with an absolute difference in iGFR slope of -0.86 mL/min/1.73m2/year (95% CI -1.72 to -0.01; p= 0.05) in the highest compared to the lowest quartile. After adjustment for age, body mass index, randomized treatment group, baseline iGFR, and urine protein to creatinine ratio, higher quartiles of net endogenous acid production remained associated with a higher rate of decline (i.e. more negative slope) in iGFR over follow up (p-trend=0.01). Higher quartiles of net endogenous acid production remained associated with a higher rate of decline in iGFR (p-trend=0.02) after further adjustment for serum bicarbonate.
Table 2.
I125 iothalamate glomerular filtration rate (iGFR) slopes (mL/min/1.73m2/year) by quartiles of estimated net endogenous acid production (NEAP), dietary protein intake, and dietary potassium intake.
| iGFR slope (mL/min/1.73 m2/year) | |||||||
|---|---|---|---|---|---|---|---|
| Unadjusted | Adjusted* | Bicarbonate Adjusted† | |||||
|
| |||||||
| Quartiles | Absolute slope (95% CI) | Difference from Q1 (95% CI) | P-value | Difference from Q1 (95% CI) | P-value | Difference from Q1 (95% CI) | P-value |
|
| |||||||
| Estimated NEAP | |||||||
|
| |||||||
| 1 | −1.45 (−2.02, −0.88) | Ref | -- | Ref | -- | Ref | -- |
| 2 | −2.11 (−2.75, −1.46) | −0.59 (−1.44, 0.26) | 0.17 | −0.69 (−1.46, 0.09) | 0.08 | −0.69 (−1.45, 0.08) | 0.08 |
| 3 | −2.25 (−2.85, −1.65) | −0.76 (−1.61, 0.10) | 0.08 | −0.85 (−1.63, −0.08) | 0.03 | −0.82 (−1.59, −0.04) | 0.04 |
| 4 | −2.34 (−2.93, −1.76) | −0.86 (−1.72, −0.01) | 0.05 | −1.01 (−1.79, −0.23) | 0.01 | −0.94 (−1.72, −0.16) | 0.02 |
| p-trend | 0.05 | 0.01 | 0.02 | ||||
|
| |||||||
| Estimated Protein Intake | |||||||
|
| |||||||
| 1 | −1.99 (−2.58, −1.39) | Ref | -- | Ref | -- | Ref | -- |
| 2 | −1.24 (−1.82, −0.66) | 0.66 (−0.21, 1.54) | 0.14 | 0.60 (−0.23, 1.42) | 0.16 | 0.54 (−0.28, 1.36) | 0.20 |
| 3 | −2.58 (−3.17, −1.98) | −0.57 (−1.43, 0.29) | 0.19 | −0.42 (−1.25, 0.42) | 0.33 | −0.45 (−1.28, 0.38) | 0.29 |
| 4 | −2.26 (−2.88, −1.64) | −0.28 (−1.13, 0.57) | 0.52 | −0.05 (−0.90, 0.80) | 0.91 | −0.04 (−0.89, 0.80) | 0.92 |
| p-trend | 0.13 | 0.37 | 0.39 | ||||
|
| |||||||
| Estimated Potassium Intake | |||||||
|
| |||||||
| 1 | −2.26 (−2.85, −1.66) | Ref | -- | Ref | -- | Ref | -- |
| 2 | −2.00 (−2.53, −1.47) | 0.15 (−0.73, 1.02) | 0.74 | 0.27 (−0.55, 1.08) | 0.52 | 0.26 (−0.55, 1.07) | 0.53 |
| 3 | −1.88 (−2.51, −1.25) | 0.38 (−0.50, 1.26) | 0.40 | 0.62 (−0.20, 1.45) | 0.14 | 0.57 (−0.25, 1.39) | 0.17 |
| 4 | −2.05 (−2.70, −1.40) | 0.24 (−0.63, 1.11) | 0.59 | 0.71 (−0.13, 1.54) | 0.10 | 0.65 (−0.18, 1.49) | 0.13 |
| p-trend | 0.51 | 0.07 | 0.09 | ||||
Slopes adjusted for randomized blood pressure and drug groups and categories of age, proteinuria, baseline GFR and body mass index.
Adjusted for above plus quartiles of serum bicarbonate
CI: confidence interval; Q1: quartile 1
Estimated protein intake was not associated with iGFR slope in unadjusted or adjusted models (p-trend=0.13 and 0.37, respectively). Estimated potassium intake was not associated with the rate of iGFR decline in unadjusted models (p-trend=0.51), but higher potassium intake was marginally associated with a slower rate of iGFR decline after adjustment (p-trend=0.07). Adjusted differences in iGFR slope across quartiles of dietary intake are summarized in Figure 2.
Figure 2. Adjusted* difference in l125iothalamate glomerular filtration rate (iGFR) slope (mL/min/1.73m2/yr) by quartiles of net endogenous acid production (NEAP), protein and potassium intake.
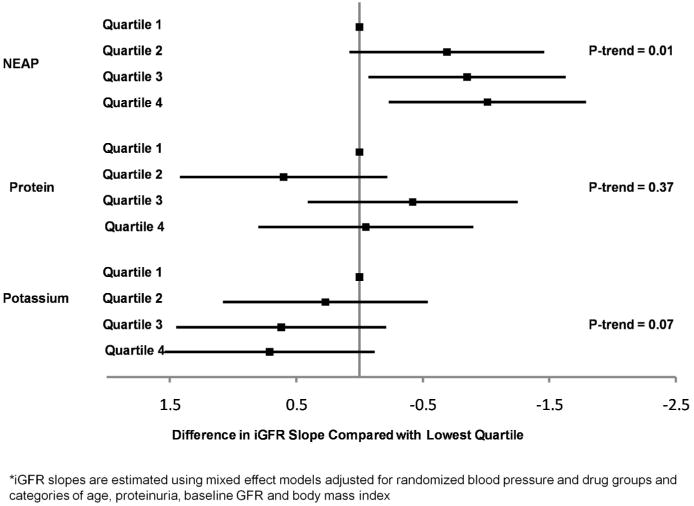
The association between net endogenous acid production and iGFR slope was similar for participants with mild/moderate CKD (iGFR > 45 mL/min/1.73m2) versus more advanced CKD (iGFR ≤ 45 mL/min/1.73m2) (p-interaction = 0.69; Table 3). There was some evidence that the association between net endogenous acid production and iGFR slope may differ for those with proteinuria, defined as urine protein to creatinine ratio (UPCR) ≥ 0.22, versus not (UPCR <0.22). Among participants with UPCR <0.22, higher quartiles of net endogenous acid production were associated with a higher rate of decline in iGFR in a graded fashion (p-trend<0.01), but not among participants with UPCR ≥ 0.22 (p-trend =0.84). The interaction between quartiles of net endogenous acid production and UPCR was not statistically significant (p-interaction = 0.19). Finally, the association between quartiles of net endogenous acid production and iGFR slope was present among a more restricted subgroup of participants with serum bicarbonate above current clinical practice targets (≥22 mEq/L; n=564). In this subgroup, participants in the highest quartile of net endogenous acid production had an absolute difference in iGFR slope of -0.97 mL/min/1.73m2/year (95% CI -1.86 to -0.09; p=0.03) compared with the lowest quartile, with a trend across quartiles (p-trend=0.03).
Table 3.
Adjusted* difference in I125iothalamate glomerular filtration rate (iGFR) slope (mL/min/1.73 m2/year) compared to lowest quartile associated with quartiles of estimated net endogenous acid production (NEAP) stratified by urine protein to creatinine ratio (UPCR) and severity of kidney disease.
| Difference in iGFR slope (mL/min/1.73 m2/year) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Urine protein to creatinine ratio | Baseline iGFR | |||||||
|
|
||||||||
| < 0.22 (n=444) | ≥ 0.22 (n=182) | ≤ 45 mL/min/1.732 (n=257) | > 45 mL/min/1.732 (n=369) | |||||
| Quartiles | Difference from Q1 (95% CI) | P-value | Difference from Q1 (95% CI) | P-value | Difference from Q1 (95% CI) | P-value | Difference from Q1 (95% CI) | P-value |
|
| ||||||||
| 1 | Ref | -- | Ref | -- | Ref | -- | Ref | -- |
| 2 | −0.73 (−1.63, 0.18) | 0.12 | 0.09 (−1.46, 1.64) | 0.91 | −0.54 (−1.71, 0.63) | 0.36 | −0.67 (−1.69, 0.35) | 0.20 |
| 3 | −0.88 (−1.78, 0.03) | 0.06 | −0.30 (−1.78, 1.17) | 0.69 | −0.77 (−1.92, 0.39) | 0.19 | −0.83 (−1.86, 0.21) | 0.12 |
| 4 | −1.29 (−2.18, −0.40) | <0.01 | −0.01 (−1.64, 1.62) | 0.99 | −1.68 (−2.89, −0.47) | <0.01 | −0.69 (−1.70, 0.31) | 0.18 |
| P- trend | <0.01 | 0.84 | <0.01 | 0.18 | ||||
| P-interaction | 0.19 | 0.69 | ||||||
Slopes adjusted for randomized blood pressure and drug groups and categories of age, proteinuria, baseline GFR and body mass index.
Q1: quartile 1; CI: confidence interval
Note: adjusted slope models include 626/632 participants (>99%), due to missing covariate information in 6 participants
Time to renal event analysis
There were 229 renal events (ESRD or doubling of serum creatinine from trial baseline) over long term follow-up from 12 months post-randomization through the trial and cohort phases. Seventy participants died prior to reaching the composite renal endpoint. Among those with UPCR < 0.22, there were 101 renal events compared with 128 renal events among those with UPCR ≥ 0.22. The adjusted association between quartiles of net endogenous acid production and composite renal events is presented overall and stratified by categories of UPCR in Table 4. There was not a statistically significant association between higher net endogenous acid production and composite renal events overall (p = 0.17). Among those with UPCR <0.22, higher net endogenous acid production was associated with composite renal events (p=0.05), although the interaction was not statistically significant (p=0.32).
Table 4.
Adjusted* hazard ratios for composite end stage renal disease (ESRD) or doubling of serum creatinine† associated with quartiles of net endogenous acid production (NEAP) and stratified by urine protein to creatinine ratio (UPCR)
| Adjusted Hazard Ratio | ||||||
|---|---|---|---|---|---|---|
| Overall (n=625) |
UPCR < 0.22 (n=444) |
UPCR ≥ 0.22 (n=181) |
||||
|
| ||||||
| Quartiles of NEAP | HR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value |
|
| ||||||
| 1 | 1.0 | -- | 1.0 | -- | 1.0 | -- |
| 2 | 0.95 (0.64, 1.41) | 0.81 | 1.02 (0.58, 1.79) | 0.95 | 0.77 (0.43, 1.37) | 0.37 |
| 3 | 1.06 (0.73, 1.55) | 0.76 | 1.06 (0.61, 1.87) | 0.83 | 1.04 (0.62, 1.73) | 0.89 |
| 4 | 1.22 (0.82, 1.83) | 0.33 | 1.38 (0.77, 2.48) | 0.28 | 1.26 (0.72, 2.18) | 0.42 |
| Continuous (per 25 mEq/d) | 1.10 (0.96, 1.26) | 0.17 | 1.22 (1.00, 1.49) | 0.05 | 1.07 (0.90, 1.27) | 0.46 |
Adjusted for age, sex, baseline GFR, proteinuria, body mass index, income, randomized blood pressure and drug assignment, and study phase.
Death is treated as a competing risk25
p-interaction for NEAP and categories of proteinuria =0.32
HR: hazard ratio; CI: confidence interval
Note: adjusted survival models include 625/632 participants (99%), due to missing covariate information in 7 participants
Sensitivity analyses
Analyses using estimates of net endogenous acid production obtained from all urine collections, without exclusion based on the total creatinine production, and using estimates obtained from urine collections averaged between trial baseline and 24 months post-randomization, were unchanged from the primary analyses. Additionally, in time to event analyses, results were similar if death was included as a component of the composite endpoint in lieu of being treated as a competing risk.
Discussion
In this study of African Americans with hypertensive CKD, a dietary pattern resulting in higher net endogenous acid production was associated with a faster rate of CKD progression. To date, several observational and randomized studies have implicated low serum bicarbonate levels as a modifiable risk factor for CKD progression.(3-5) Lowering net endogenous acid production of the diet may raise serum bicarbonate (13) and may be an alternative strategy to target abnormal acid base homeostasis in CKD without large sodium loads.
The association between net endogenous acid production and CKD progression was independent of serum bicarbonate and similar in a subset achieving target serum bicarbonate levels (15), suggesting that the risk associated with net endogenous acid production may not be due to its effect on serum bicarbonate per se. We acknowledge that serum bicarbonate concentration can be prone to measurement error and it is possible that we were not able to fully account for the effect of serum bicarbonate in these models.(3) However, several additional studies support mechanisms of renal injury due to acidic diets that are independent of serum bicarbonate concentration. Animal studies have shown that diets generating high net endogenous acid production may accelerate renal injury by increasing demand for renal ammonium excretion (8), and by promoting intracellular acidosis, both of which develop before clinically overt metabolic acidosis.(16, 17) Other human studies have demonstrated that modulation of net endogenous acid production through administration of fixed acid or alkali, alters the rate of GFR decline and pro-fibrotic mediators in the urine such as endothelin and aldosterone without clinically relevant changes in serum bicarbonate.(10, 18)
In our analyses, we did not observe strong evidence that the association between net endogenous acid production and CKD progression varies by baseline CKD stage. In both the slope and time to event analyses, we did observe that the association between net endogenous acid production and CKD progression was stronger among those without proteinuria. We would interpret these findings cautiously given that there was not a statistically significant test of interaction and statistical significance within one subgroup could be related, in part, to a larger sample size. It is important to recognize, however, that these analyses were highly stratified and had limited power to test these interactions. Animal studies suggest that renal acid loading specifically promotes tubulointerstitial injury, a pattern characterized by less proteinuria.(8, 19) Although the non-proteinuric group is at lower risk of ESRD, they remain at elevated risk for mortality associated in a graded fashion with decreased GFR.(20)
Point estimates of the association between net endogenous acid production and time to ESRD or doubling of serum creatinine were consistent with the findings from slope analyses, but did not confirm these findings statistically. Our power in these models was limited and the confidence intervals did not exclude clinically important associations. Additionally, less than half of the events occurred in the large subpopulation of participants without proteinuria, the group in whom net endogenous acid production appeared to be a stronger risk factor in slope analyses.
In this population the net endogenous acid production was more strongly influenced by variability in potassium, rather than protein, intake. It is important to note that the net endogenous acid production considers the balance of protein and potassium intake in the diet and was more strongly associated with GFR decline than either protein or potassium intake alone. Current clinical guidelines recommend restricted protein intake in CKD.(21) Although our study does not refute these guidelines, it suggests that balance of protein intake with natural sources of alkali, such as fruits and vegetables, may be more important. We were not able to definitively evaluate the safety of such diets in patients with CKD in this study although the observed prevalence of hyperkalemia was low.
This study has several limitations. The AASK study population was highly selected, including only African Americans with hypertensive kidney disease. For this reason, it is difficult to generalize these results to other racial groups with different dietary patterns or those with etiologies of kidney disease other than hypertension. We do not have direct measures of diet in this study. We are unable to account for dietary factors other than total protein and potassium intake which may affect endogenous acid production, including the differential impact of protein from plant versus animal sources and intake of additives in processed foods.
This study also has several important strengths. We have multiple measures of the net endogenous acid production through frequent 24 hour urine collections, frequent measures of iGFR over long term follow up, and careful collection of patient characteristics, including detailed medication histories. Additionally, we also have event data over a long follow up period across both the AASK trial and cohort phases, allowing consideration of both GFR slope and time to event analyses.
In conclusion, we have observed that higher net endogenous acid production is associated with a faster rate of decline in GFR among African Americans with hypertensive kidney disease. This association may be stronger among patients without proteinuria. These results should be interpreted cautiously in light of results from time to event models which are consistent with, but do not confirm statistically, the findings seen in models of GFR slope. Our findings implicate net endogenous acid production as a potentially modifiable risk factor for progressive kidney disease worthy of further study.
Methods
Study Population
The AASK trial was a multicenter, 2 × 3 factorial, randomized, controlled trial of intensive vs. standard blood pressure control, using one of three primary anti-hypertensive agents (ramipril, metoprolol or amlodipine), in self-identified African Americans with hypertensive nephrosclerosis.(22) Participants without ESRD at the end of the trial were offered enrollment in an observational cohort phase.(23, 24) This analysis includes 632 participants from AASK with eligible urine collections between 12 and 36 months in the trial. Urine collections were eligible if the 24 hour total creatinine excretion was within 30% of expected (22.1 mg/kg in men; 17.2 mg/kg in women), indicating a complete collection (25) and excluded if the participant was taking potassium or alkali supplements. The protocol and procedures were approved by the institutional review board of each center and all participants provided written informed consent.
Data collection
Twenty four hour urine collections were performed every 6 months throughout the trial and annually in the cohort. Urines were analyzed at a central laboratory for urea nitrogen, potassium, sodium and creatinine. Ideal body weight (IBW) was calculated using previously published equations.(26) Dietary protein intake was estimated from 24 hour urine urea nitrogen (UUN) excretion using the Maroni equation [protein intake = 6.25 *(UUN + 0.031*IBW) – urinary protein (g/d) if daily urine protein excretion ≥ 5 g].(27) Dietary potassium intake was estimated as the total 24 hour urine potassium excretion.(28) Net endogenous acid production was estimated from these intakes as previously described: net endogenous acid production (mEq/d) = −10.2 + 54.5 [protein intake (g/d) ÷ potassium intake (mEq/d)].(12) Estimates derived from urines collected between 12 and 36 months post-randomization were averaged to provide a measure of habitual dietary intake. Urines from the initial 12 months were not used due to frequent medication titrations during this time which may have altered steady state potassium excretion. Three hundred ten participants (49%) had 3 to 6 measurements available, 137 participants (22%) had 2 measurements and 185 participants (29%) had one measurement. Intake was also estimated from eligible urines in the cohort phase and averaged over the first 24 months for use in time dependent Cox models.
I125iothalamate GFR was collected twice at baseline, then at 3, 6 and every 6 months thereafter during the AASK trial phase. Serum creatinine was used to estimate glomerular filtration rate (eGFR) using a study specific equation in the cohort phase.(23, 29) UPCR was evaluated in spot urine samples at trial and cohort baseline. Renal events occurring over combined follow up in the trial and cohort phases were defined as ESRD (initiation of dialysis or renal transplantation) or doubling of serum creatinine from trial baseline.
Statistical Analysis
Baseline characteristics of the study population were examined across quartiles of net endogenous acid production using linear regression (continuous variables) or Pearson's χ2 (categorical variables). Values from trial baseline were used for these analyses, except for serum phosphorus and bicarbonate, for which values from 12 months post-randomization were used. UPCR and iGFR from trial baseline were selected given differential effects of the randomized drugs on proteinuria and acute effects on iGFR.
The association between net endogenous acid production and the rate of change of iGFR from 12 months post-randomization to the end of the trial was evaluated using linear mixed models with maximum likelihood estimation (median follow-up 3.2 years). Covariates hypothesized to contribute to CKD progression were included in adjusted models if they were associated with iGFR slope in univariate analyses (p<0.10) or significantly improved adjusted model fit. Models were adjusted for age (in quartiles), randomized group, body mass index (categorized as < 25 kg/m2; 25-30 kg/m2; > 30 kg/m2), baseline iGFR (categorized as >60 mL/min/1.73 m2; 46-60 mL/min/1.73 m2; 31-45 mL/min/1.73 m2; and ≤ 30 mL/min/1.73 m2) and proteinuria (categorized as baseline UPCR < 0.22, ≥0.22 and < 1, or ≥ 1). A subsequential adjustment was then made for serum bicarbonate (categorized in quartiles). Secondary analyses evaluated the association of estimated protein and potassium intake with iGFR slope.
The association between net endogenous acid production and the relative risk of composite renal events from 12 months post-randomization through follow-up in the cohort (median of 7.7 years) was tested using time-dependent Cox proportional hazards models with death treated as a competing risk.(30) Net endogenous acid production was analyzed as time-varying with updated exposure classification at cohort baseline. Models were adjusted for age, sex, categories of income, baseline GFR (time-varying: baseline iGFR for trial phase; baseline eGFR for cohort phase), log UPCR (time varying), categories of body mass index (time varying), study phase (trial vs. cohort), and randomized blood pressure and antihypertensive drug assignment. Secondary analyses evaluated the association of estimated protein and potassium intake with renal events. Sensitivity analyses evaluated a composite endpoint of death and renal events.
Models were stratified by categories of baseline GFR (i.e. iGFR >45 mL/min/1.73 m2 vs. ≤45 mL/min/1.73 m2), proteinuria (UPCR <0.22 vs. ≥0.22), and serum bicarbonate <22 or ≥22 mEq/L. The cutpoint for proteinuria was pre-specified and has been used as a standard threshold for all AASK analyses.(31, 32) Cutpoints for GFR and serum bicarbonate are clinically relevant cutpoints, consistent with prior literature and current practice guidelines.(33) Interactions were tested between net endogenous acid production and both baseline GFR and proteinuria using interaction terms in adjusted models. Sensitivity analyses were performed using estimates of net endogenous acid production obtained from all urines without exclusion based on the total creatinine, and using urine collections from trial baseline to 24 months post-randomization instead of 12 to 36 months.
All analyses were performed using STATA Special Edition 11.0 (College Station, Texas, 2009). Hypotheses were tested using a two-sided type 1 error rate of 0.05.
Acknowledgments
Funding sources: NIH, National Kidney Foundation of Maryland
We would like to acknowledge the time and commitment of the participants, investigators and staff of the AASK study. AASK was supported by grants to each clinical center and the coordinating center from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). In addition, AASK was supported by the Office of Research in Minority Health (now the National Center on Minority Health and Health Disparities, NCMHD) and the following institutional grants from the National Institutes of Health: M01 RR-00080, M01 RR-00071, M0100032, P20-RR11145, M01 RR00827, M01 RR00052, 2P20 RR11104, RR029887, and DK 2818-02. King Pharmaceuticals Pfizer Inc, AstraZeneca Pharmaceuticals, Glaxo Smith Kline, Forest Laboratories, Pharmacia and Upjohn donated antihypertensive medications. This work does not necessarily reflect the opinions of the AASK study or the NIDDK. JJS was supported by National Institute of Diabetes and Digestive and Kidney Diseases grant T32 DK 00732-14 and 5KL2RR025006 from the National Center for Research Resources, a component of the NIH and NIH Roadmap for Medical Research, as well as the National Kidney Foundation of Maryland. RSP is supported by grant 5R01DK072367-03 from the National Institute of Diabetes, Digestive and Kidney Diseases. BCA was supported in part by grant R21DK078218 from the National Institute of Diabetes and Digestive and Kidney Diseases. CAMA was supported by K01 HL092595-02 from the National Heart Lung and Blood Institute.
This work was presented as an oral presentation at the American Society of Nephrology's annual meeting on November 12, 2011 in Philadelphia, PA.
Footnotes
Disclosure: The authors have no relevant disclosures to report.
References
- 1.Coresh J, Selvin E, Stevens LA, Manzi J, et al. Prevalence of Chronic Kidney Disease in the United States. JAMA. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 2.van der Velde M, Matsushita K, Coresh J, Astor BC, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int. 2011;79:1341–1352. doi: 10.1038/ki.2010.536. [DOI] [PubMed] [Google Scholar]
- 3.Raphael KL, Wei G, Baird BC, Greene T, et al. Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney Int. 2010;79:356–362. doi: 10.1038/ki.2010.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate Supplementation Slows Progression of CKD and Improves Nutritional Status. J Am Soc Nephrol. 2009;20:2075–2084. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah SN, Abramowitz M, Hostetter TH, Melamed ML. Serum Bicarbonate Levels and the Progression of Kidney Disease: A Cohort Study. Am J Kidney Dis. 2009;54:270–277. doi: 10.1053/j.ajkd.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorhout-Mees EJ, Machado M, Slatopolsky E, Klahr S, et al. The functional adaptation of the diseased kidney : III. Ammonium excretion. J Clin Invest. 1966;45:289–296. doi: 10.1172/JCI105342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodman AD, L J, Lennon EJ, Relman AS. Production, Excretion and Net Balance of Fixed Acid in Patients with Renal Acidosis. J Clin Invest. 1965;44:495–506. doi: 10.1172/JCI105163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nath KA, H M, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats: Interactions of dietary acid load, ammonia, and complement component-C3. J Clin Invest. 1985;76:667–675. doi: 10.1172/JCI112020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phisitkul S, Hacker C, Simoni J, Tran RM, et al. Dietary protein causes a decline in the glomerular filtration rate of the remnant kidney mediated by metabolic acidosis and endothelin receptors. Kidney Int. 2008;73:192–199. doi: 10.1038/sj.ki.5002647. [DOI] [PubMed] [Google Scholar]
- 10.Mahajan A, Simoni J, Sheather SJ, Broglio KR, et al. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int. 2010;78:303–309. doi: 10.1038/ki.2010.129. [DOI] [PubMed] [Google Scholar]
- 11.Frassetto LA, Lanham-New SA, Macdonald HM, Remer T, et al. Standardizing Terminology for Estimating the Diet-Dependent Net Acid Load to the Metabolic System. J Nutr. 2007;137:1491–1492. doi: 10.1093/jn/137.6.1491. [DOI] [PubMed] [Google Scholar]
- 12.Frassetto LA, Todd KM, Morris RC, Jr, Sebastian A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am J Clin Nutr. 1998;68:576–583. doi: 10.1093/ajcn/68.3.576. [DOI] [PubMed] [Google Scholar]
- 13.Scialla JJ, Appel LJ, Astor BC, Miller ER, et al. Estimated Net Endogenous Acid Production and Serum Bicarbonate in African Americans with Chronic Kidney Disease. Clin J Am Soc Nephrol. 2011;6:1526–1532. doi: 10.2215/CJN.00150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Curhan GC, Forman JP. Diet-Dependent Net Acid Load and Risk of Incident Hypertension in United States Women. Hypertension. 2009;54:751–755. doi: 10.1161/HYPERTENSIONAHA.109.135582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eknoyan G, Levin A, Levin NW. Bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42:1–201. [PubMed] [Google Scholar]
- 16.Wesson DE, Simoni J. Increased tissue acid mediates a progressive decline in the glomerular filtration rate of animals with reduced nephron mass. Kidney Int. 2009;75:929–935. doi: 10.1038/ki.2009.6. [DOI] [PubMed] [Google Scholar]
- 17.Wesson DE, Simoni J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010;78:1128–1135. doi: 10.1038/ki.2010.348. [DOI] [PubMed] [Google Scholar]
- 18.Wesson DE, Simoni J, Broglio K, Sheather S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Physiol - Renal Physiol. 2011;300:F830–F837. doi: 10.1152/ajprenal.00587.2010. [DOI] [PubMed] [Google Scholar]
- 19.Phisitkul S, Khanna A, Simoni J, Broglio K, et al. Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int. 2010;77:617–623. doi: 10.1038/ki.2009.519. [DOI] [PubMed] [Google Scholar]
- 20.Astor BC, Hallan SI, Miller ER, III, Yeung E, et al. Glomerular Filtration Rate, Albuminuria, and Risk of Cardiovascular and All-Cause Mortality in the US Population. Am J Epidemiol. 2008;167:1226–1234. doi: 10.1093/aje/kwn033. [DOI] [PubMed] [Google Scholar]
- 21.KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis. 2007;49:S12–S154. doi: 10.1053/j.ajkd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Agodoa LY, Appel L, Bakris GL, Beck G, et al. Effect of Ramipril vs Amlodipine on Renal Outcomes in Hypertensive Nephrosclerosis: A Randomized Controlled Trial. JAMA. 2001;285:2719–2728. doi: 10.1001/jama.285.21.2719. [DOI] [PubMed] [Google Scholar]
- 23.Appel LJ, Middleton J, Miller ER, III, Lipkowitz M, et al. The Rationale and Design of the AASK Cohort Study. J Am Soc Nephrol. 2003;14:S166–172. doi: 10.1097/01.asn.0000070081.15137.c0. [DOI] [PubMed] [Google Scholar]
- 24.Sika M, Lewis J, Douglas J, Erlinger T, et al. Baseline Characteristics of Participants in the African American Study of Kidney Disease and Hypertension (AASK) Clinical Trial and Cohort Study. Am J Kidney Dis. 2007;50:78–e71. doi: 10.1053/j.ajkd.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Pak CYC, Odvina CV, Pearle MS, Sakhaee K, et al. Effect of dietary modification on urinary stone risk factors. Kidney Int. 2005;68:2264–2273. doi: 10.1111/j.1523-1755.2005.00685.x. [DOI] [PubMed] [Google Scholar]
- 26.Robinson J, Lupkiewicz S, Palenik L, Lopez L, et al. Determination of ideal body weight for drug dosage calculations. Am J Hosp Pharm. 1983;40:1016–1019. [PubMed] [Google Scholar]
- 27.Maroni BJ, Steinman TI, Mitch WE. A method for estimating nitrogen intake of patients with chronic renal failure. Kidney Int. 1985;27:58–65. doi: 10.1038/ki.1985.10. [DOI] [PubMed] [Google Scholar]
- 28.Bingham SA, Gill C, Welch A, Cassidy A, et al. Validation of dietary assessment methods in the UK arm of EPIC using weighed records, and 24-hour urinary nitrogen and potassium and serum vitamin C and carotenoids as biomarkers. Int J Epidemiol. 1997;26:S137. doi: 10.1093/ije/26.suppl_1.s137. [DOI] [PubMed] [Google Scholar]
- 29.Lewis J, Agodoa L, Cheek D, Greene T, et al. Comparison of cross-sectional renal function measurements in African Americans with hypertensive nephrosclerosis and of primary formulas to estimate glomerular filtration rate. Am J Kidney Dis. 2001;38:744–753. doi: 10.1053/ajkd.2001.27691. [DOI] [PubMed] [Google Scholar]
- 30.Fine JP, Gray RJ. A Proportional Hazards Model for the Subdistribution of a Competing Risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 31.Appel LJ, Wright JT, Greene T, Agodoa LY, et al. Intensive Blood-Pressure Control in Hypertensive Chronic Kidney Disease. N Engl J Med. 2010;363:918–929. doi: 10.1056/NEJMoa0910975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Appel LJ, Wright JT, Jr, Greene T, Kusek JW, et al. Long-term Effects of Renin-Angiotensin System-Blocking Therapy and a Low Blood Pressure Goal on Progression of Hypertensive Chronic Kidney Disease in African Americans. Arch Intern Med. 2008;168:832–839. doi: 10.1001/archinte.168.8.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garabed E, Adeera L, Nathan WL. Bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42:1–201. [PubMed] [Google Scholar]