

Abstract
Dendritic cells (DCs) and macrophages are critical early initiators of innate immunity in the kidney; they also orchestrate inflammation subsequent to ischemia-reperfusion injury. Hence, these cells hold great promise as targets for pharmacological interventions. Macrophages and DCs are the most abundant leukocytes present in the kidney, and they represent a heterogeneous population of cells that are capable of inducing sterile inflammation after reperfusion, directly through the production of proinflammatory cytokines, chemokines, and other soluble inflammatory mediators, or indirectly through activation of effector T lymphocytes and natural killer T cells. In addition, recent studies have indicated that macrophages participate in tissue repair and DCs possess tolerogenic functions in normal and disease states. Understanding the function of DCs and macrophages as well as the microenvironment that governs their phenotype will shed light on the pathogenesis of kidney disease and offer novel drug targets.
INTRODUCTION
Thank you Dr. Falk for the kind introduction. Throughout my career I have always had many supportive individuals and I am grateful that Dr. W. Kline Bolton, you and many others have supported my career. Today I would like to discuss a topic that has been most enigmatic to many investigators since 1973 when dendritic cells (DCs) were discovered. These peculiar cells have recently piqued the interest of many investigators in the field of kidney disease pathogenesis, and today I would like to highlight our interest in this field from the context of acute kidney injury. Before I do this, however, it would be most appropriate to recognize the individual who discovered DCs, Dr. Ralph Steinman. In 1973, Dr. Ralph Steinman, along with Dr. Zanvil Cohn, discovered a new cell type, known as dendritic cells, that contribute importantly to activating the adaptive immune system (1, 2). This discovery has opened the field of immunology and has led to our understanding of immunity, autoimmunity, and novel approaches to the treatment of cancer, infectious diseases, organ transplantation, and other disorders of the immune system. Ultimately, his seminal discovery led to a world-wide effort among researchers and clinicians in developing a pioneering immunotherapy approach to treat pancreatic cancer, a disease that ultimately took his life. Dr. Steinman's battle against pancreatic cancer lasted 4.5 years, much longer than most patients with pancreatic cancer. For his own personal treatment, he used a combination of conventional chemotherapy and autologous immunotherapy with DCs. Dr. Ralph Steinman was awarded the Nobel Prize in Medicine or Physiology on October 3, 2011, 3 days after he died. What cells did Dr. Steinman discover? Two publications described the morphological and functional properties of DCs (1, 2). Single cell suspensions were prepared and cultured on plastic or petri dishes, and non-adherent cells were removed. The remaining 10% to 50% of nucleated cells from mouse spleen were examined by phase contrast microscopy. Four types of nucleated adherent cells were described; three of them have characteristics of lymphocytes, mononuclear phagocytes, and granulocytes. A fourth cell type was large and had a variety of shapes: elongate, stellate, or dendritic; the cytoplasm contained large, phase-dense granules, but there was no evidence of active endocytosis. These DCs had high levels of major histocompatibility (MCH) complex products, and they were potent in lymphocyte reactions, as a ratio of one DC to 100 lymphocytes produced a strong proliferative response.
In the kidney, DCs are major constituents of the heterogeneous mononuclear phagocytic system. Identification of these cells is enabled by using a transgenic mouse in which the coding exon of one allele for the chemokine receptor, CX3CR1, has been replaced with the gene encoding green fluorescent protein GFP (3); heterozygotes had normal phenotypic and functional properties. Examination of the kidneys of these mice using laser-scanning confocal microscopy identified a contiguous network of CX3CR1+ DCs throughout the kidney interstitium and mesangium; cells that are poised to respond and activate innate and adaptive immunity (4, 5).
KIDNEY DENDRITIC CELLS
Kidney DCs reside in the interstitial extracellular compartment and are strategically positioned to interact with substances transported from the tubule lumen into the peritubular capillaries (6), endogenous molecules released from cells or exogenous invading organisms, or resident or infiltrating immune cells including lymphocytes, natural killer T cells (NKT cells), epithelial cells, and fibroblasts. DCs are activated by danger-associated molecular patterns (DAMPS) or pathogen-associated molecular patterns (PAMPS) (7). In the presence of PAMPS or DAMPS, DCs are key initiators, potentiators, and effectors of the innate immune system (which is comprised of neutrophils, monocytes/macrophages, DCs, natural killer [NK] cells and NKT cells). In kidney ischemia-reperfusion injury (IRI) DCs induce injury either directly or through inflammatory signals. DCs play an important role in the early antigen-independent inflammatory response after reperfusion, including the production of cytokines/chemokines that drive neutrophil infiltration via activation of NKT cells and the IL-17/IL23 signal pathway (4, 8). However, because of their functional and phenotypic plasticity, DCs can also induce tolerance. Tolerogenic DCs are functionally immature, express inadequate positive or enhanced negative co-stimulatory signals and reduced pro-inflammatory cytokines, and can generate immune tolerance by inducing T cell anergy or deletion or regulatory T (Treg) cell induction or expansion (9, 10) (Figure 1). However, mature DCs can also promote tolerance (9, 11). Also, it is evident that both immature and mature DCs can prime Treg cells that prevent autoimmunity.
Fig. 1.
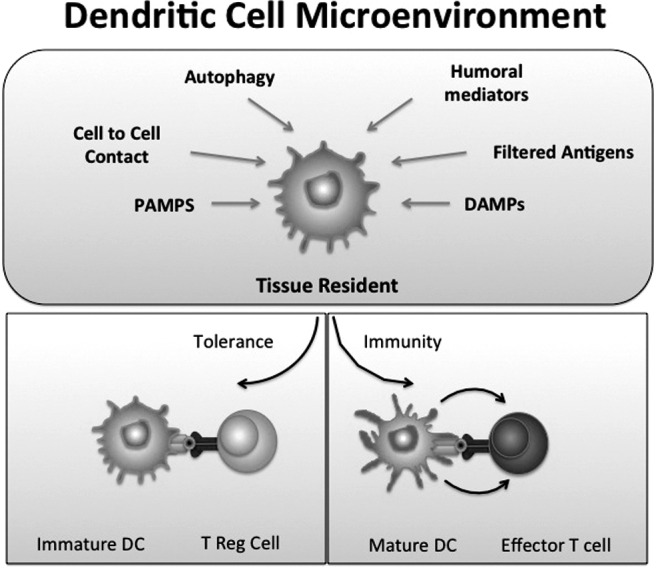
Dendritic cells (DCs) have a dual role and can induce tolerance or immunity. In the steady state, immature DCs situated in the renal interstitial microenvironment are affected by autophagy of proteins from dying cells, cell-to-cell contact, PAMPS, DAMPS, humoral mediators, or filtered antigens. In the absence of inflammatory signals these immature DCs express low amounts of costimulatory molecules and may induce tolerance. During an infection or in the presence of maturation-inducing inflammatory signals, mature DCs induce the development of effector T-cell responses.
The role of DCs in IRI can be assessed specifically by using transgenic mice expressing the human diphtheria toxin receptor (DTR; human heparin binding epidermal growth factor–like growth factor) in CD11c+ cells (CD11c-DTR mouse) (12). Transgenic expression of human DTR renders normally diphtheria toxin–resistant mouse cells diphtheria toxin–sensitive. Using this inducible lineage ablation method, exposure of CD11c-DTR mice to low-dose diphtheria toxin (DT) will kill primarily CD11c+ DCs. Kidney CD11c+GFP+ DCs were efficiently depleted in CD11c-DTR/GFP mice by DT but not by catalytically inactive mutant DT before IRI; and kidney injury, as indicated by an increase in plasma creatinine (Figure 2) or H&E staining, was significantly less in DT-treated CD11c-DTR/GFP mice compared to mutant DT-treated CD11c-DTR/GFP mice (13). The protective effect of DC depletion was not found in DT- or mutant DT-treated WT control mice. Thus, DCs contribute to the early innate response in IRI. However, not all studies show that DCs contribute to immune injury in acute kidney injury (AKI). In cisplatin-induced nephrotoxicity, DC ablation in DTR mice increased injury, indicating the importance of dendritic DCs for tissue protection in this model (14). Fundamental differences in these models — cisplatin injury primarily induces proximal tubule injury whereas the pathogenesis of IRI is much more heterogeneous — have serendipitously revealed disparate roles of tissue resident DCs in AKI. In addition, kidney IRI is recognized as inducing a systemic inflammatory response and different subsets of myeloid and lymphoid CD11c+ DCs in the local kidney and other lymphoid organs (lymphoid node, spleen, et al.) may play a different role in IRI compared with cisplatin organ-targeted injury. We hypothesize that the interstitial environment created by different pathogenic circumstances, such as cisplatin toxicity or IRI, governs DC function.
Fig. 2.

Dendritic cells (DC) are important in the pathogenesis of the kidney IRI. (A) CD11c+GFP+ cells (arrows) in mouse kidney sections from CD11c-DTR/GFP mice (which express the human diphtheria toxin receptor [DTR] as a GFP fusion protein) were revealed immunohistochemically with GFP antibody (FITC fluorescence, green). Mutant diphtheria toxin (DT) had no effect on the number of GFP-positive CD11c cells in the CD11c-DTR/GFP sham and IRI mouse kidneys; however, there were significantly less GFP-labeled cells in DT-treated CD11c-DTR/GFP mouse kidneys after sham or IRI. (Scale bar = 50 μmol/L.) (B) The plasma creatinine level was measured in wild-type (WT) and CD11c-DTR/GFP mice that received mutant (control) or WT DT (4 μg/g) 48 hours before surgery. Values are mean ± SE; N = 2 to 11; *** P < 0.001. (C) H&E staining of kidneys from WT and CD11c-DTR/GFP mice reveals marked tubule injury in WT kidneys after IRI. Pretreatment with WT but not mutant DT before surgery protected CD11c-DTR mouse kidneys from injury (arrows, necrotic tubules; Scale bar = 100 μm). Data in A and C are representative of more than three experiments. From Li and Okusa (13).
RENAL INTERSTITIAL MICROENVIRONMENT
The kidney has regenerative capacity and should possess a unique immunological microenvironment. Kaissling recently provided a detailed histological description of the renal interstitial compartment (15). This compartment is situated between basement membranes of epithelia and vessels and contains interstitial fibroblasts and DCs. The fibroblasts express ecto-5'-nucleotidase (CD73) in their plasma membrane. The resident DCs belong to the mononuclear phagocyte system and express MHC class II and CD11c. Interestingly, CD73 is highly expressed on epithelial cells and fibroblasts and is a cell surface enzyme that is involved in the final step in the sequential dephosphorylation of extracellular ATP and ADP to AMP (CD39) and AMP to adenosine (CD73) (16). Adenosine is a purine nucleoside that is produced by tissue under metabolic stress, such as ischemia, hypoxia, and inflammation. In the kidney, adenosine is an important regular of metabolic cellular activity (17). Concentrations of adenosine, which are normally less than 1 μmol/L, increase to more than 6-fold after 2 minutes of ischemia. Once released into the interstitial microenvironment, adenosine can bind to different types of adenosine receptors (i.e., adenosine A1, A2A, A2B, A3Rs) expressed on various innate immune cells such as neutrophils, macrophages, DCs, natural killer cells (18), and modulate cellular responses (13, 19, 20). Koshiba et al. used a monoclonal antibody to show that both T and B cells express A2ARs (21).
ADENOSINE 2A RECEPTORS AND TOLERANCE INDUCTION IN ACUTE KIDNEY INJURY
The hypothesis that DC A2ARs dampen the immune response supports our in vivo studies in which we show that A2AR activation attenuates acute kidney injury. A2AR agonists ameliorate renal IRI in rat kidneys (22, 23) by ∼70% when infusion is induced before, or at the time of, ischemia and is continued for 24 hours during the reperfusion period (22, 24).
To determine whether the DC-mediated activation of the innate response to IRI is attenuated by A2ARs expressed in DCs, we are currently using a strategy that combines the method of site-specific recombination with gene targeting to achieve lineage/cell type-specific knockouts in mice. To investigate the functional consequences of A2AR inactivation and the role of A2AR signaling in DCs, we crossed Adora2afl/fl mice and Cd11c-Cre mice, in which Cre recombinase is expressed in DCs. Mice homologous for the floxed A2AR that also harbor a cell type–specific Cre recombinase transgene are entirely normal except in the cell type expressing Cre recombinase, in which the A2AR gene will bear a deletion between the two loxP sites flanking the target gene, thereby yielding a mouse in which the DCs are deficient in A2ARs. We anticipate that mice deficient of DC A2ARs have worse injury and will fail to respond to A2ARR agonists. These results would then suggest that expression of A2ARs on DCs is necessary for the protective effects of A2AR agonists, that endogenous adenosine may modulate and tolerize DC activity, and that the microenvironment may determine the final DC phenotype. Although DCs contribute to immune activation, they also induce tolerance. Tolerogenic DCs have been used as potential therapeutic tools and represent a new and promising immunotherapeutic approach for ameliorating or preventing graft rejection or to treat autoimmune disorders, cancers and other serious conditions (9). Immature myeloid DCs, which express low surface levels of MHC class II and co-stimulatory molecules, can induce T cell tolerance, whereas mature myeloid DCs, which express much higher levels of these molecules, induce T cell immunity. Bone marrow (BM)-derived DCs have been rendered tolerogenic by exposure to cytokines, growth factors, or pharmacological mediators, or by genetic engineering (9,25–28). Tolerogenic DCs can produce T cell death, T cell anergy or regulatory T cell expansion, and they can regulate autoreactive or alloreactive T cell responses and promote or restore antigen-specific tolerance in experimental animal models. Tolerogenic DCs of either donor or host origin can promote transplant tolerance induction. We have shown that DC-mediated NKT cell activation plays a key role in initiating the immune response to kidney IRI (29), and that A2AR agonists protect mouse kidneys from IRI through their actions on BM-derived leukocytes (30), including suppressing NKT cell migration and Th1 type cytokine (IFN-γ production (19). At present, there is no information about the tolerogenic DC effect on NKT cell activation in the inflammation disease model. We believe that A2AR agonists may tolerize DCs and attenuate kidney injury by suppressing NKT cell activation and affecting resident immune cells. If so, we believe that cell-based therapy, in which DCs are tolerized by drugs such as A2AR agonists ex vivo and then administered to subjects, may be a novel form of targeted cell-based therapy of AKI.
ACKNOWLEDGMENTS
We gratefully acknowledge support from the National Institutes of Health (NIH DK 5 R01 DK062324) (to MDO) and the American Heart Association (AHA) National Scientist Development Grant (to LL).
Footnotes
Potential Conflicts of Interest: None disclosed.
DISCUSSION
May, Gainesville: Oftentimes, pharmacologic agents like ATL wind up mimicking endogenous molecules. During stress, it may cause ischemic renal injury. Are there molecules that act like they try to down-regulate dendritic cells (for example, an adenosine antagonist)?
Okusa, Charlottesville: The adenosine 2A (A2A) agonist mimics the breakdown product of ATP, adenosine. Adenosine is a nonspecific agonist to four G-protein coupled receptors, A1, A2A, A2B, and A3 receptors. An A2A agonist, such as the ATL compound, is a modification of adenosine that gives it specificity for the A2A receptor. So, when there is stress, such as that caused by ischemia or inflammation, there is actually an up-regulation of the A2A receptor in various immune cells; this up-regulation might be an adaptive response to try to suppress the inflammatory effect. So, there is an adaptive response, but in a positive way.
May, Gainesville: Sure, but is there a regulatory response that would be on the negative side of that that maybe is not recognized but if there was then this would be a way of trying try to augment that in patients at risk for renal injury during surgery?
Okusa, Charlottesville: If we go back to your original question regarding the endogenous molecule mimicked by ATL, adenosine may have negative effects depending on the specific receptor that it activates. For example, if it binds to A3 receptors, adenosine may have a negative effect. This could occur depending on the relative expression of the four different adenosine receptor subtypes. If there is a more dominant expression of A3 receptors, there could be a negative effect of high levels of the endogenously produced adenosine. Now, if you are asking are there ways of augmenting endogenous molecules to reduce injury, the answer is yes. Mesenchymal stem cells have been studied in acute kidney injury; it has been shown that these cells do not necessarily repopulate these injured tubules and replace dead epithelial cells or fuse with injured cells but more likely, they have a paracrine effect to allow for the release of growth factors that allow for the repair of the kidney. There are human studies conducted at the University of Utah in US veterans who are post-surgical CABG. So what they've shown in very early reports is over a 6 month period of time, based upon the subjects that have received the mesenchymal stem cells versus a historical control, that there actually is a difference in terms of the plasma creatinine after several months. They do not report any significant adverse events that I am aware of. With regard to dendritic cells, there might be some differences in that whereas mesenchymal stem cells are probably importantly involved in releasing growth factors, dendritic cells might be more important in activating the innate system. In our prior studies, we have shown that natural killer T cells, which are presented with a glycolipid antigen from the dendritic cell that is an important activator of that natural killer T cell, are important in AKI. This leads to a subsequent downstream inflammatory response such as activation of IL-17 and IL-23. When you tolerize these dendritic cells, they cannot activate these natural killer T cells, and there is a reduction of interferon-gamma, IL-17, and IL-23, and a reduction of kidney injury. So, the mechanisms might be a little bit different, but I wouldn't exclude the possibility that there might be some overlapping areas.
Quesenberry, Providence: Camuzzi's group from Toronto demonstrated that mesenchymal stem cells may repair kidney by microvesicle delivery.
Okusa, Charlottesville: That is correct — they showed that microvesicle delivery of nucleic acid content may participate in reprogramming cells and begin the repair process. I think that's very interesting.
Abboud, Iowa City: Very nice results on adenosine 2A receptors. There are other receptors, though, on dendritic cells that could modulate significantly the response to the receptors; angiotensin receptors, for example, and recently nicotinic acetylcholine receptors. Any thoughts on the relative contributions of all these receptors on dendritic cells?
Okusa, Charlottesville: That is a very good question. I certainly don't want to exclude any other humoral mediators, but certainly the summation of response from humoral mediators likely will determine the ultimate phenotype of dendritic cells. In my presentation I did want to highlight the one that we are interested in. However, I think that there is much that we don't know about the renal interstitial microenvironment in which these dendritic cells reside because it is very difficult to study. Brigitte Kaissling recently published beautiful micrographs that highlight the important morphological and cell-to-cell relationships that occur in that microenvironment. In particular, the epithelial cell plays probably a very important role in communicating to the dendritic cell, and I don't know if there are any direct cell communications or not, but certainly the fibroblasts and macrophages as well as a number of different molecules that can alter the dendritic cell phenotype are likely candidates.
Luke, Cincinnati: I was impressed with the number of dendritic cells in the kidney and I have two questions. (1) How does it compare with other organs, like the heart, in terms of the percent dendritic cells in some objective relationship?; and (2) Are there chronic models? You know we spend forever trying to reduce proteinuria and the inflammation it produces. Are those more chronic situations?
Okusa, Charlottesville: Yes. So, I don't know the relative abundance of dendritic cells within the kidney compared to other organs. I suspect that organs such as the lung and the gut are going to be organs that have a high density of dendritic cells because they are likely to be exposed to more pathogens. There are additional complications in quantifying dendritic cells because of the classification of dendritic cells. The dendritic cell is a component of a heterogeneous mononuclear phagocytic system where on the one end you have classic macrophages and on the other end you have classic dendritic cells. There are also cells that are in between; therefore, the system is heterogeneous and reference to dendritic cells sometimes is unclear. Now, in terms of chronic models, you raise a very important point. In lupus nephritis, it is likely that dendritic cells contribute to the early initiation of disease, leading to down stream inflammatory processes culminating in chronic disease.
REFERENCES
- 1.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–62. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp Med. 1974;139:380–97. doi: 10.1084/jem.139.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L, Huang L, Sung SS, et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74:1509–11. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soos TJ, Sims TN, Barisoni L, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70:591–6. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 6.Kaissling B, Le Hir M. Characterization and distribution of interstitial cell types in the renal cortex of rats. Kidney Int. 1994;45:709–20. doi: 10.1038/ki.1994.95. [DOI] [PubMed] [Google Scholar]
- 7.Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol JASN. 2011;22:416–25. doi: 10.1681/ASN.2010040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Huang L, Vergis AL, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120:331–42. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7:610–21. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 10.Hu J, Wan Y. Tolerogenic dendritic cells and their potential applications. Immunology. 2011;132:307–14. doi: 10.1111/j.1365-2567.2010.03396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buch T, Heppner FL, Tertilt C, et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2:419–26. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010;30:268–77. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tadagavadi RK, Reeves WB. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J Am Soc Nephrol. 2010;21:53–63. doi: 10.1681/ASN.2009040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130:247–62. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar V, Sharma A. Adenosine: an endogenous modulator of innate immune system with therapeutic potential. Eur J Pharmacol. 2009;616:7–15. doi: 10.1016/j.ejphar.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Hansen PB, Schnermann J. Vasoconstrictor and vasodilator effects of adenosine in the kidney. Am J Physiol Renal Physiol. 2003;285:F590–9. doi: 10.1152/ajprenal.00051.2003. [DOI] [PubMed] [Google Scholar]
- 18.Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–52. [PMC free article] [PubMed] [Google Scholar]
- 19.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203:2639–48. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gessi S, Varani K, Merighi S, Ongini E, Borea PA. A2A-adenosine receptors in human peripheral blood cells. Br J Pharmacol. 2000;129:2–11. doi: 10.1038/sj.bjp.0703045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koshiba M, Rosin DL, Hayashi N, Linden J, Sitkovsky M. Patterns of A2A-extracelluar adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometric studies with anti-A2A-receptor monoclonal antibodies. Mol Pharmacol. 1999;55:614–24. [PubMed] [Google Scholar]
- 22.Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A-adenosine receptor activation during reperfusion reduces ischemia-reperfusion injury in rat kidney. Am.J.Physiol. 1999;277:F404–F12. doi: 10.1152/ajprenal.1999.277.3.F404. [DOI] [PubMed] [Google Scholar]
- 23.Lee HT, Emala CW. Systemic adenosine given after ischemia protects renal function via A(2a) adenosine receptor activation. Am J Kidney Dis. 2001;38:610–8. doi: 10.1053/ajkd.2001.26888. [DOI] [PubMed] [Google Scholar]
- 24.Okusa MD, et al. Enhanced protection from renal ischemia-reperfusion injury with A2A-adenosine receptor activation and PDE 4 inhibition. Kidney Int. 2001;59:2114–25. doi: 10.1046/j.1523-1755.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 25.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 26.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010;11:647–55. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 27.Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol. 2002;80:477–83. doi: 10.1046/j.1440-1711.2002.01115.x. [DOI] [PubMed] [Google Scholar]
- 28.Hawiger D, Inaba K, Dorsett Y, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Huang L, Sung SJ, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 30.Day Y-J, Huang L, McDuffie MJ, et al. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–91. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]