

Abstract
Despite vaccination, varicella-zoster virus (VZV) remains an important pathogen. We investigated VZV latency in autopsy specimens from vaccinees, in gastrointestinal tissue removed surgically, and in a guinea pig model. We propose that retrograde transport from infected skin and viremia deliver VZV to neurons in which it becomes latent. Wild type (WT) VZV was found to be latent in many ganglia of vaccinated children with no history of varicella, suggesting that subclinical infection with WT-VZV occurs with subsequent viremic dissemination. The 30% to 40% rate of WT-VZV zoster reported in vaccinees and occasional trigeminal zoster due to vaccine type VZV (vOka) are consistent with viremic delivery of VZV to multiple ganglia. Most human intestinal specimens contained latent VZV within neurons of the enteric nervous system (ENS). Induction of viremia in guinea pigs led to VZV latency throughout the ENS. The possibility VZV reactivation in the ENS is an unsuspected cause of gastrointestinal disease requires future investigation.
INTRODUCTION
VZV establishes latent infection in neurons of sensory ganglia during primary infection. Subsequently, in an estimated 30% of individuals who have had the natural infection, the latent virus reactivates, causing zoster, characteristically a unilateral skin infection in a dermatomal distribution. Zoster occurs in healthy individuals, but its incidence is much higher in those who are immunocompromised or immunosenescent (1). Zoster is most often found in patients who have acquired wild-type (WT) VZV as a result of clinical varicella but it also occurs, albeit at a lower rate, in persons who have been vaccinated (2).
How VZV reaches neurons to establish latency has not previously been determined. One possibility is that latency is established when the highly infectious virions that are released within the epidermis during varicella infect adjacent intra-epidermal sensory nerve endings (3). Virions may then travel by retrograde axonal transport to the cell bodies of neurons in dorsal root ganglia (DRG) and cranial nerve ganglia (CNG) (3,4). A second potential viral route to ganglia is to be carried there in the T cells that transport VZV during the viremia associated with varicella (5). Infected lymphoctyes could infect neurons in sensory ganglia as well as keratinocytes of the epidermis (6).
The current study was undertaken to determine whether VZV establishes latency in DRG and CNG via retrograde transport from the skin or viremia or both. We studied ganglia for the presence of VZV latency from vaccinated children who came to autopsy after sudden death. We reasoned that the ganglionic location and viral genotype would provide clues as to how the virus established latent infection.
Because evidence of VZV had been reported in patients with gastrointestinal disease (7,8), it seemed plausible that VZV might establish latency in enteric neurons as well as in DRG and CNG. Therefore, we asked whether latent VZV could be detected in surgically removed specimens of human intestine. We also developed an animal model of latent infection of the guinea pig intestine. These experiments, performed with vOka expressing green fluorescent protein (GFP) under the control of the VZV ORF66 gene (VZVOKA66.GFP), permitted us to test the hypothesis that an experimental viremia is able to establish VZV latency in the bowel. These studies led to a newly recognized site of VZV latency, the enteric nervous system (ENS).
MATERIALS AND METHODS
Two groups of samples of human ganglia were studied. One set of ganglia was obtained from 10 children who died suddenly and were autopsied in Albuquerque, NM, between November 2008 and May 2009. Although the autopsies were legally required, informed consent of parents for examining DRG and CNG was obtained for this research. A brief medical history was obtained from the parents, specifically including the varicella vaccination status and prior experience, if any, of varicella. The cause of death was noted, as well as the interval between death and autopsy. Ganglia were submitted to the laboratory in a coded fashion to protect confidentiality. These children ranged in age from less than 1 year old to 10 years old.
The second set of human ganglia was present in full-thickness surgical specimens of gut removed from 20 surviving children. Surgery was performed for medically indicated reasons; after removal of the bowel, tissue was coded and sent to the laboratory accompanied only with information about each child's age, varicella vaccination status, and history of varicella. The Institutional Review Board of Columbia University approved these studies. The children ranged in age from 2 months old to 17 years old.
Ganglia removed at autopsy or during surgery were divided in half. One half was placed in RNAlater™ (Ambion, Applied Biosystems, Austin, TX), whereas the other half was fixed with 4% formaldehyde (from paraformaldehyde) at pH 7.4. The specimens from New Mexico were then placed on wet ice and shipped to New York by rapid courier and immediately stored at 4° C until use. Surgical specimens of gut obtained at New York Presbyterian Hospital were transmitted immediately to the laboratory for processing.
Nested PCR and RT-PCR
DNA was extracted (DNeasy Blood and Tissue Kit; Qiagen, Valencia, CA) from tissue or peripheral blood mononuclear cells (PBMCs). DNA (100 ng) was amplified (Eppendorf SmartCycler) in 20 μL of reaction mixture for 35 cycles. The PCR product (1.0 μL) was subjected to further amplification with sets of nested primers for each gene for an additional 32 cycles. Extraordinary precautions were taken to avoid contamination because nested amplification was used. Negative control samples of tissue DNA (gut tissue removed from human neonates or uninfected guinea pigs) were analyzed simultaneously with experimental samples. Cellular DNA was also examined; primers for human β-globin and guinea pig glyceraldehyde 3-phosphate dehydrogenase primers were used to verify the presence of human and guinea pig DNA, respectively. Primers for PCR and nested PCR were designed to amplify VZV open reading frames (ORFs 4, 29, 31, 61, 62, 63, 66, 67, and 68) (9, 10). PCR products were separated on 2% agarose gels, and digital images were obtained for documentation with a Kodak Image Station 440.
RNA was extracted with Trizol (Invitrogen, Carlsbad, CA), following the manufacturer's instructions, from the same tissue from which DNA was extracted as described above. RNA was treated with DNase I (4U/100 μL) to remove DNA contamination. Total RNA (3.0 μg) was subjected to reverse transcription with Maloney Murine Leukemia Virus reverse transcriptase (Promega, Madison, WI) in 25 μL of reaction mixture to obtain cDNA. The cDNA (1.0 μL) was amplified in 20 μL of reaction mixture for each VZV gene. PCR amplication with β-actin universal primers was used as a control for RNA extraction and cDNA preparation.
Identification of Wild-type (WT) and Vaccine (vOka) Viruses
A screening procedure was used to discriminate WT-VZV from vOka. DNA from open reading frames (ORF) 38 and ORF54 were PCR-amplified and characterized by restriction fragment length polymorphisms (RFLPs) (11). A PstI restriction site is present in ORF38 from WT-VZV that is not present in vOka. ORF54 from vOka has a BglI restriction site that is lacking in 80% of US WT strains (11). Because it is possible (on rare occasions in the US) to misidentify vOka with this screening procedure (12), further analysis of single nucleotide polymorphisms (SNPs) was performed to confirm the identification of all specimens that were initially identified as vOka. The PCR-amplified ORF62 fragment (7 μL) (13) was digested with SmaI (0.5 Ll; 10 U) (New England Biolabs, Ipswich, MA) and 12 μL of water. The enzymatic reaction was performed overnight at room temperature and the digested products were separated by gel electrophoresis on 4% agarose. The presence of a SmaI restriction site at nucleotide position 106262 identifies vOka (13); the region of ORF62 between bases 106035-106303 was sequenced to further confirm the identification of vOka, which has C at 106262, whereas WT-VZV has T at that location.
Infection of Guinea Pig Peripheral Blood Mononuclear Cells (PBMC) and In Vivo Infection of Guinea Pigs
VZV-infected PBMC were prepared by co-culture with VZVORF66.GFP-infected human embryonic lung fibroblasts (HELF) (9, 14). VZVORF66.GFP was a gift from Professor Paul Kinchington of the University of Pittsburgh Medical Center. Four-week-old Hartley guinea pigs (Charles River, MA) were injected into the posterior ocular sinus with 1 × 106 VZVORF66.GFP –infected PBMCs in 50μL. The animals were euthanized 2 days to 6 weeks later and the intestinal wall was dissected into layers to permit the ENS to be examined in whole mounts as flat laminar preparations. The layers examined consisted of the submucosa containing the submucosal plexus and the longitudinal muscle with adherent myenteric plexus.
Immunocytochemistry.
Fixed ganglia from autopsy specimens were cryoprotected overnight in 30% sucrose, embedded in Neg50™ medium (Richard Allen Scientific Co, Kalamazoo, MI) and frozen with liquid nitrogen. Sections were cut at widths of 8 μm to 10 μm with a cryostat-microtome. Preparation and characterization of polyclonal antibodies to ORF29p, 63p and 68p have previously been described (15). Monoclonal antibodies to ORF62 were purchased from Biodesign International (Saco, ME). Secondary antibodies conjugated with Alexa 350, 488, or 594 were obtained from Invitrogen, Inc (Eugene, OR). Sections were blocked with 5% horse serum in 0.1% Triton-PBS and primary antibodies were applied overnight at 4° C. Sections were examined with a Leica CTR6000 microscope. Images were captured with a cooled CCD camera; brightness and contrast were adjusted with a Macintosh computer running Velocity 4™ software (Improvision; PerkinElmer, Waltham, MA).
For immunocytochemistry on whole mounts of bowel, specimens were blocked with 10% normal horse serum for 30 minutes at room temperature and then incubated for 48 hours or 72 hours at 4°C with primary antibodies. Bound primary antibodies were visualized with streptavidin-Alexa 594 for mouse monoclonal antibodies, and donkey antibodies to goat, rabbit, or sheep IgG coupled to Alexafluor 350, 488, 594, or 680 [infrared] (diluted 1:200; Invitrogen). The use of species-specific secondary antibodies coupled to contrasting fluorophores enabled as many as four primary antibodies to be located simultaneously. Preparations were washed with PBS, mounted in buffered glycerol, and examined with a Leica CTR 6000 microscope. Images were obtained with a cooled CCD camera and analyzed with computer assistance (Velocity 5.4 imaging software; Improvision). DNA was visualized with bisbenzimide (1 μm/mL; Sigma Aldrich, St. Louis, MO). Biotinylated mouse monoclonal antibodies to the neuronal marker Hu C/D were obtained from Invitrogen, and diluted 1:50.
In situ hybridization.
Frozen sections of ganglia were permeabilized with proteinase K (100 μg/mL) for 20 minutes, washed with PBS, and post-fixed with 4% formaldehyde (from paraformaldehyde) for 10 minutes. The sections were then treated with 0.3M NaOH for 5 minutes and neutralized with 0.4M Tris-HCl pH7.4 for 15 minutes. The sections were washed with PBS and dehydrated with 50% and 100% ethanol, respectively. Prehybridization was performed in buffer (50% formamide, 1 XSSC, 1X Denhardt's, 500 μg/mL salmon sperm DNA) at room temperature for 2 hours to 3 hours. Specimens were then covered with a hybridization buffer (prehybridization buffer supplemented with a digoxigenin-conjugated oligonucleotide probe targeted to VZV ORFs 54 and 68, and 10% dextran sulfate) and incubated for 10 minutes at 85°C and overnight at 37°C. After hybridization, slides were washed with 2x and 0.1x SSC. Sites of bound probe were detected with alkaline phosphatase conjugated antibodies to digoxingenin (Roche Diagnostics, Mannheim, Germany), which were applied for 2 hours and visualized after washing and histochemical detection of alkaline phosphatase activity (15). Levamizol (0.5 mmol/L) was added to the buffer solution to inhibit the endogenous alkaline phosphatase activity of neurons.
RESULTS
Autopsies were conducted on four male and six female children (Table 1). Autopsies were performed soon after death (mean, 17.0 ± 1.5 hours). None of the children had a history of varicella. Six had been vaccinated against varicella (1 dose). Vaccination status was uncertain for one subject. Three children had not been vaccinated and were less than 1 year of age; they served as controls.
TABLE 1.
Information on Sensory Ganglia Obtained From 10 Children at Autopsy, Soon After Demise (Mean, 17 Hours + 1.5 Hours)
| Age (y) | Vaccination | Cause of Death |
|---|---|---|
| 1 (control) | No | Seizure |
| 1 (control) | No | ? |
| 1 (control) | No | Automobile accident |
| 1.75 | Yes | ? |
| 2 | Yes | ? |
| 2 | Yes | Automobile accident |
| 2 | ? | ? |
| 4 | Yes | Automobile accident |
| 8 | Yes | Gunshot |
| 10 | Yes | Surgical complications |
No VZV DNA or RNA was detected in any of the ganglia examined in the three children who were less than 1 year of age and had no known exposure to Oka or WT VZV (Table 2). In contrast, despite the lack of any record of clinical varicella, VZV was found in at least some ganglia of all of the other subjects.
TABLE 2.
No VZV DNA or RNA Was Found by PCR in Patients <1 Year Old With No History of Varicella or Varicella Vaccine
| Patient | Trigeminal |
Cervical |
Thoracic |
Lumbar |
||||
|---|---|---|---|---|---|---|---|---|
| Age (Y) | Right | Left | Right | Left | Right | Left | Right | Left |
| 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
DNA examined: ORFs 4, 31, 63, 66, 67; RNA examined: ORFs 4, 40, 63, 66
Bilateral trigeminal, cervical, thoracic, lumbar, and sacral ganglia were analyzed from the seven older children, who ranged from almost 2 years to 10 years of age (Table 3). Six of the seven were known to have been vaccinated. VZV DNA and RNA were extensively and often bilaterally present in trigeminal, cervical, thoracic, and lumbar ganglia from these seven patients.
TABLE 3.
Latent VZV (DNA/RNA) Was Found by PCR in Ganglia at Multiple Levels, Often Bilaterally: Distribution of VZV DNA/RNA in Ganglia of Vaccinated Patients
| Patient Age | Vaccinated | Trigeminal |
Cervical |
Thoracic |
Lumbar |
Type | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Right | Left | Right | Left | Right | Left | Right | Left | |||
| 1.75 | Yes | 0 | 0 | 0 | 0 | 0 | + | + | 0 | Oka |
| 2 | Yes | 0 | 0 | 0 | 0 | + | 0 | + | 0 | Wild |
| 2 | ? | + | + | 0 | 0 | + | 0 | 0 | + | Wild |
| 2 | Yes | + | + | + | + | + | + | + | + | Wild |
| 4 | Yes | 0 | 0 | + | + | + | + | + | 0 | Wild |
| 8 | Yes | 0 | 0 | 0 | 0 | + | 0 | 0 | 0 | Wild |
| 10 | Yes | + | + | 0 | 0 | + | 0 | + | 0 | Wild |
DNA examined: ORFs 4, 31, 63, 66, 67; RNA examined: ORFs 4, 40, 63, 66
Nucleic acid analysis from these ganglia showed the following: in one 1.75-year-old patient, VZV DNA and RNA were identified in two ganglia (Table 3). The DNA was PstI negative (ORF 38), BglI positive (ORF 54), and had a Sma1 restriction site at position 106262 in ORF 62 (data not shown), indicating that it was the Oka strain.
In the six other children, VZV DNA and RNA were identified in multiple ganglia. The DNAs were all PstI positive, and there was no restriction site at position 106262 in ORF 62, indicating that these children had latent infection with WT VZV (data not shown). DNA fragments in ORF 62 at positions 106035-106303 were amplified by PCR and sequenced. A number of nucleotide variations were identified at positions 106209, 106229, 106347, and 106262. DNA from five patients exhibited differences in A, T, or C, indicating that the identification of WT-VZV could not have been due to laboratory contamination during the analysis (data not shown). No latent Oka strain VZV was identified in these six children.
In Situ Hybridization and Immunocytochemistry
In situ hybridization and immunocytochemistical analyses on ganglia from autopsied children indicated both that the virus was harbored in neurons and that VZV was latent and not lytic. The latency signature of VZV is as follows: lytic genes (for example, ORFS 31, 61, and 68) are not expressed, and proteins of latency-associated genes (ORFS 4, 21, 29, 62, 63, and 66) are expressed but restricted to the cell cytoplasm (16).
In situ hybridization indicated that VZV DNA was present in neurons. Ganglia from two patients (aged 10 years and 4 years) are shown in Figure 1. These show that VZV DNA encoding ORF 54 and 68 are found in neurons.
Fig. 1.
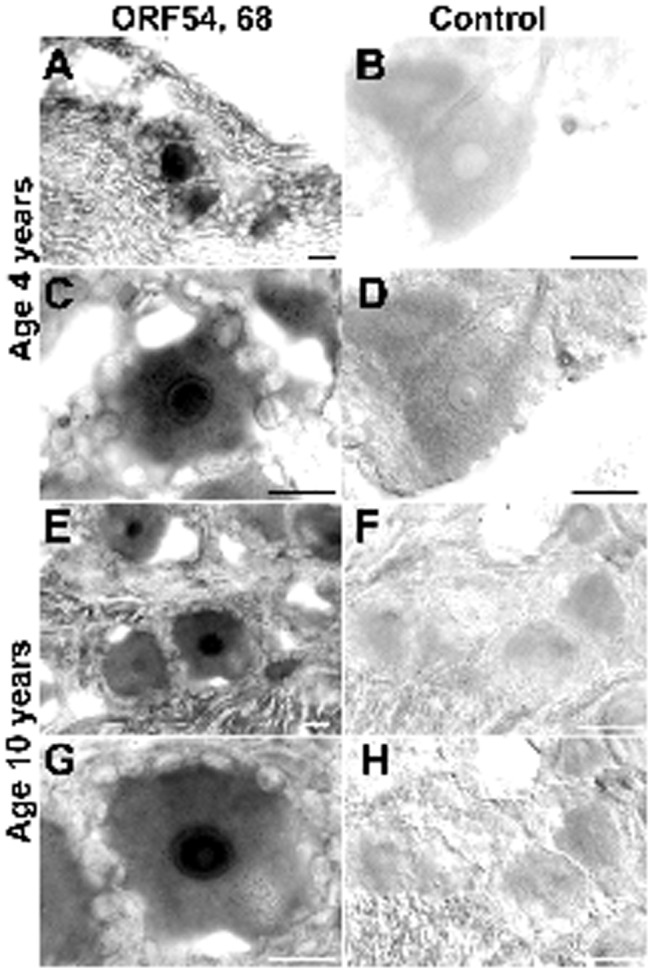
In situ hybridization confirms that VZV DNA is present in and restricted to neurons in human DRG. A-D, Sections cut through a DRG of a 4-year-old child. E-F, Sections cut through a DRG of a 10-year-old child. The column of panels on the left were hybridized with probes against VZV ORFs 54 and 68. The column of panels on the right were hybridized with a control probe. A, At low magnification, hybridizing neurons stand out. Notice that little or no hybridizing DNA can be detected in connective tissue cells or nerve fibers. B, Bright field. Neurons do not hybridize with the control probe. C, At higher magnification, the nuclear concentration of hybridizing DNA can be seen. The edges of the hybridizing neuron are scalloped by unstained satellite cells. D, The same field as shown in B, but illuminated with interference contrast to help visualize the non-hybridizing structures of the DRG. E, At low magnification, several hybridizing neurons can be seen. F, No neurons hybridize with a control probe. G, The nuclear concentration of hybridizing VZV DNA is evident. H, Section serial to that shown in F. Interference contrast. No hybridizing neurons are evident. The markers = 20 μm.
Ganglia from autopsied children were immunocytochemically analyzed to determine whether VZV latency-associated proteins could be detected. Ganglia from two patients (aged 10 years and 4 years) are shown (Figure 2). Immunoreactivity of latency-associated VZV ORF 29p, ORF 62p, and ORF 63p was observed; the immunoreactivity was entirely confined to cytoplasm. No ORF 68 immunoreactivity characteristic of lytic infection was found, nor was there immunoreactivity in ganglia from control patients (Figure 2). The immunocytochemistry indicates that only latent VZV infection was present in ganglia (Figure 2).
Fig. 2.

The immunoreactivity of latency associated VZV proteins can be detected in the cytoplasm of human DRG neurons. A, Immunoreactivity of ORF29p. Note that no immunoreactivity is present in the neuronal nucleus. B, ORF 63p. A cluster of DRG neurons is immunoreactive. Immunoreactivity is cytoplasmic and extends into proximal axons. C, Control. In the absence of antibodies to VZV proteins, there is no immunoreactivity. D, ORF29p. The restriction of immunoreactivity to the cytoplasm is evident. E, ORF63p. The restriction of immunoreactivity to the cytoplasm is evident. F, ORF62p. Immunoreactivity is restricted to cytoplasm and extends into proximal axons. G, ORF68. There is no immunoreactivity of this glycoprotein (gE), which is produced only during lytic infection. The markers = 20 μm.
VZV RNA Surgical Specimens of Gut From Children Undergoing Gastrointestinal Surgery
Surgical specimens from the intestines of 13 children were analyzed (17) (Table 4). Expression of transcripts encoding latency-associated genes (ORFs 4, 29, 62, 63, and 66) was identified in six of six patients who had a history of varicella. The most commonly detected transcript was ORF63, followed by ORFs4 and 66. No transcripts characteristic of lytic infection (ORFs 31, 61, and 68) were found. Similarly, in six of seven vaccinated children, transcripts encoding latency-associated genes were found in intestinal specimens; however, no transcripts associated with lytic infection were found. No VZV transcripts were identified in the control specimens.
TABLE 4.
VZV RNA Was Often Present in Surgical Specimens of Gut From Patients With a History of Varicella or Vaccination
| Transcript | 4 | 29 | 31 | 61 | 62 | 63 | 66 | 68 | Total |
|---|---|---|---|---|---|---|---|---|---|
| History | β | ||||||||
| Varicella | 4/6 | 0/6 | 0/6 | 0/6 | 0/6 | 5/6 | 1/6 | 0/6 | 6/6 |
| % | 67 | 0 | 0 | 0 | 0 | 83 | 17 | 0 | 100 |
| Vaccine | 4/7 | 0/7 | 0/7 | 0/7 | 2/7 | 6/7 | 1/7 | 0/7 | 6/7 |
| % | 57 | 0 | 0 | 0 | 29 | 86 | 14 | 0 | 86 |
| No VZV | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 |
| % | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Transcripts that were detected were latency-associated. Transcripts encoding proteins that were characteristic of lytic infection (31, 61, 68) were not detected.
Establishment of VZV Latency in Enteric Neurons by Viremia in Experimental Animals
Guinea pig PBMC were infected by co-culture with VZVORF66.GFP-infected HELF. VZVORF66.GFP –infected PBMC (1 × 106) in 50μL was injected into the posterior ocular sinus of 4-week-old guinea pigs. Infection of enteric neurons was detected within 2 days. The fluorescence of GFP was coincident with the immunofluorescence of the neuronal marker, HuC/D, which identified the VZV-infected cells as enteric neurons (Figures 3, 4). GFP fluorescence was confined to neurons, with fluorescence confined to the cytoplasm of cells. No GFP fluorescence was observed in the ENS of control uninfected guinea pigs (Figure 4).
Fig. 3.

Intravenous injection of guinea pig lymphocytes infected with VZVOKA66.GFP infects and establishes latency in guinea pig enteric neurons. The longitudinal muscle was dissected with adherent myenteric plexus from the wall of the guinea pig small intestine and processed as a flat whole mount of the laminar preparation. The neurons and all of their neuritic processes are immunostained with antibodies to β3-tubulin (red). The cell bodies of neurons, which also exhibit the green fluorescence of GFP and thus express ORF66p, are doubly stained in this merged image and appear yellow. Axons within interganglionic connectives and the tertiary plexus do not express ORF66-GFP and thus display only the red immunofluorescence of β3-tubulin. DNA has been stained with bisbenzimide (blue). The widespread nuclei throughout the preparation reveal the underlying muscle and connective tissue has not been infected with VZVOKA66.GFP. The marker = 50 μm.
Fig. 4.

VZVOKA66.GFP establishes latency in guinea pig enteric neurons. The longitudinal muscle with adherent myenteric plexus was dissected from the wall of the guinea pig colon and processed as a laminar whole mount. A-C, The guinea pig had been injected intravenously 4 days before euthanasia with guinea pig lymphocytes that had been infected with VZVOKA66.GFP. DNA is stained with bisbenzimide, which is visible in all panels. A, Immunofluorescence of the ELAV proteins, HuC/D identifies neuronal cell bodies. B, GFP immunofluorescence identifies cells infected with VZVOKA66.GFP. C, Merged image. Yellow fluorescence identifies cells that display both GFP and HuC/D immunoreactivities. Notice that virtually all of the neurons express GFP and thus contain VZVOKA66.GFP. The fluorescence of ORF66-GFP is restricted to neuronal cytoplasm; therefore VZVOKA66.GFP is latent. D-F, Uninfected guinea pig control, processed identically to the guinea pig illustrated above. D, Immunofluorescence of HuC/D identifies neuronal cell bodies. E, GFP immunofluorescence is absent. F, Merged image. Only the red immunofluorescence of HuC/D immunofluorescence is seen. The marker = 50 μm.
DISCUSSION
The overarching aim of these studies was to determine the route or routes by which VZV achieves latent neuronal infection; the first experiments involved examination of ganglia from children who underwent autopsy for medico-legal reasons. Because an autopsy was required, we were able to study their ganglia to determine whether they harbored latent VZV. Because the children had been vaccinated before they died, we wished to determine the frequency with which vOka causes latent infection. We also wished to test the hypothesis that infection of DRG and CNG neurons occurs as a result of retrograde transport from the VZV-infected epidermis. If that hypothesis were to be true, the establishment of VZV latency in DRG and CNG would require skin infection. That would, in turn, restrict to latency to those DRG that innervate sites of vaccination. If there had been no skin infection, there would be no latent VZV.
The idea that VZV is transported in the retrograde direction within nerves from the skin to DRG and CNG was suggested over a century ago by the observation that the distribution of skin lesions in cases of zoster reflects the distribution of the preceding varicella rash (18). Thus, it thus became accepted wisdom that VZV reaches neurons mainly or even exclusively by retrograde axonal transport from skin lesions (4, 18, 19). In line with this concept, the incidence of zoster was shown to be significantly higher in leukemic vaccinees who experienced a skin rash due to VZV after immunization, than in vaccinees who had no VZV rash (19). Studies in guinea pigs in which VZV was injected into the skin and later latent infection was found in corresponding DRG supported the mechanism of axonal transport (20).
In the study of ganglia from autopsied children, we therefore anticipated that evidence of latent vOka would be found in DRG that innervate areas of skin where the vaccine had been administered. Because the vaccine is usually injected into an arm, that would mean that transcripts encoding VZV genes might be restricted to DRG at thoracic and cervical axial levels. Soon after our study was underway, however, reports appeared indicating that vOka can occasionally cause zoster on the face, and that 30% to 40% of reported zoster cases in vaccinees are due to WT VZV despite the absence of a history of varicella (21). These reports were inconsistent with the idea that latency resulted only from retrograde axonal transport of VZV from skin infection to neurons.
Results of the autopsy study showed VZV DNA in DRG and CNG in each of seven vaccinated children, but not in the three control children. VZV latency was widespread in vaccinees, frequently bilateral, and overwhelmingly due to WT VZV. Because each region of skin is unilaterally innervated and infection was bilateral and not restricted to a limited number of dermatomes, it is apparent that retrograde transport from the injection site is not the only means by which VZV gains access to DNG and CNG to establish latency. VZV was latent only in neurons within ganglia. Only latency-associated genes (ORFs 4, 29, 62, 63, 66) were expressed and none of those associated with lytic infection. We confirmed the PCR data by showing latent VZV by immunocytochemistry and in situ hybridization, as shown in Figures 1 and 2.
The results of our study of autopsied children were surprising, not only in that many ganglia on both sides of the body were found to contain latent VZV, but also because the virus was overwhelmingly WT, despite vaccination and the absence of a history of varicella. These observations suggest that although varicella vaccine may successfully prevent clinical varicella, it does not necessarily prevent subclinical infection or superinfection of individuals with WT VZV or the ability of WT VZV to establish latency. Presumably, infection with WT VZV was suppressed sufficiently by vaccine-derived immunity for the vaccinee to remain asymptomatic, but nevertheless allowed WT VZV to disseminate, possibly via viremia, and replace vOka. Alternatively, it is conceivable that after a single dose of vaccine, vOka provides some level of immune protection without establishing latency in a subset of individuals. In that case, subclinical infection with WT VZV would not replace latent vOka, but find naïve ganglia to infect and establish latency in component neurons. It is possible that the two-dose vaccination schedule that has now been adopted for children will boost immunity sufficiently to prevent superinfection with WT VZV and replacement of vOka or infection of a host in which vOka failed to establish latency. If there were to be less frequent superinfection or subclinical infection with WT VZV, the incidence of zoster may decrease. That would assume that the tendency of vOka to reactivate is less than that of WT VZV (22). In any case, our results suggest that skin rash is not necessary for WT VZV to reach sensory ganglia. These observations are compatible with the idea that viremia carries VZV to the ganglia in which it establishes latency.
That viremia can lead to latency was also strongly suggested by the widespread latent infection with WT VZV in DRG and particularly in CNG. Whereas establishment of latent infection of neurons by the hematogenous route has not previously been proven for VZV, hematogenous spread of simian varicella virus (SVV) to DRG has been reported within days after experimental inoculation, before rash has had time to develop, in monkeys (6).
We developed an animal model to explore the possibility that viremic transport of VZV causes latent infection. We infected guinea pig PBMC with vOka expressing green fluorescent protein (GFP) under the control of the VZV ORF66 gene (VZVOKA66.GFP). The VZVOKA66.GFP-infected PBMC were then given intravenously to guinea pigs. Within 2 days, we were able microscopically to identify latent VZV in neurons of the ENS of all animals, as shown in Figures 3 and 4. Latent VZV was also detected in the DRG of these guinea pigs (20). These experiments show that, at least in the guinea pig model, lymphocytes that carry VZV can deliver the virus to sensory and enteric ganglia to enable latency to be established.
Even before these studies, we suspected that VZV might become latent in the ENS. We had found that we could induce latent VZV infection in isolated guinea pig enteric neurons in vitro (23). In addition, gastrointestinal diseases due to VZV such as achalasia and gastritis have been described in humans (24–27). Another intestinal condition that has clinically been linked to VZV is Ogilvie's syndrome, in which a severe and sometimes lethal pseudoobstruction occurs in the bowel (7, 8, 28, 29).
Examination of surgical specimens of the human gut showed that latent VZV infection commonly occurs in the ENS after varicella or administration of the varicella vaccine (Table 4) (20). Latent VZV that reactivates has the potential to cause gastrointestinal disease, and in fact may be the cause of the intestinal conditions discussed above. On the other hand, it is also possible that VZV in the gut also serves a useful purpose by participating in long-term maintenance of immunity. Cryptic reactivations that do not cause disease may function as autologous boosters of immunity. The gastrointestinal tract is the largest lymphoid organ in the body and thus may, in most individuals, be able to keep VZV in tight enough control to remain beneath a clinical threshold.
CONCLUSION
Our observations suggest that viremia as well as retrograde transport from the skin enable VZV to establish latent infection in sensory and enteric neurons (20). The only explanation for the widespread and bilateral presence of latent WT VZV in vaccinated children with no history of VZV rash is that they experienced viremia after a subclinical infection or viral superinfection. Because vOka was also widespread, it is likely that viremia can disseminate VZV after vaccination. Viremia leading to latency in many ganglia is also consistent with the reported 30% to 40% rate of WT VZV causing zoster in vaccinees and the cases of trigeminal zoster caused by vOka. In support of the viremic mechanism of establishment of latency, we were able to show that PBMC infected with VZVOKA66.GFP lead to latent infection of neurons in the ENS and DRG after their intravenous injection into guinea pigs. Finally, we showed that VZV is commonly latent in the human gut in the neurons of intestinal ganglia. This last finding is particularly novel and interesting because subclinical reactivations of VZV in the highly immunocompetent gut might explain the long-lasting immunity to varicella. Reactivation of latent VZV within the ENS might also be a cause of gastrointestinal disorders the pathophysiology of which is currently unexplained.
ACKNOWLEDGMENT
The authors thank Paul Kinchington, PhD, for his generous gift of VZV ORF66.GFP.
Footnotes
Potential Conflicts of Interest: None disclosed.
DISCUSSION
Oates, Vanderbilt: We tend to look on the reactivation syndrome in adults as being a disease of the skin. Do you think that there is a possibility that any intestinal consequences might occur with reactivation in the enteric neurons?
Gershon, New York: There is an illness called Oglvies's syndrome, which was described long before anyone thought that VZV might be involved in its causation. These patients present with gastrointestinal pseudo-obstruction; there is no anatomical lesion causing the obstruction. A number of patients have gone to surgery, but no masses have been identified. Some patients have developed zoster of the skin, before or after the pseudo-obstruction, which implicates VZV. In at least one case, VZV was demonstrated in the muscularis mucosa. One way that the virus could get to the intestine is by reactivation in the gut leading to lytic infection there. Obviously, just finding DNA in the gut in a patient would not necessarily mean reactivation but in certain cases VZV has been identified. I think it's interesting that this would suggest that reactivation might be occurring in more than one place at once. I also think that for years we've felt that if you didn't have VZV on the skin that you didn't have active VZV infection. We are beginning to see patients with encephalitis and no skin lesions identified by a positive VZV DNA PCR in cerebrospinal fluid. Many but not all are immune-compromised. So, we are now in an era where we are finding VZV or other infection without skin lesions. So, I think VZV can reactivate in neurons and if the immune response cannot control the reactivated virus, skin manifestations result. Clearly, VZV causes more complicated infections than we realized previously.
Oates, Vanderbilt: Thank you.
Wolf, Boston: Just a comment, John. People develop muscle palsies with herpes zoster, so it's not just an infection of the skin. They actually have palsies, so we know that muscles are involved as well. The question I have is in your animal model, if you immunize the guinea pigs with the VOka strain and then expose them to wild-type virus, do they get low-grade wild-type infections, which would explain what we are seeing in the patients, and if you give them two exposures to VOka, does that not happen, which is your hypothesis?
Gershon, New York: Yes. You propose a great experiment. Unfortunately, this animal model is too new for us to have done anything like that but I think someone should do this in the future.
Wolf, Boston: I will accept co-authorship.
Gershon, New York: The major thing we are trying to do right now is to reactivate the virus from latency in the guinea pig.
Vercellotti, Minnesota: I have one naïve question. Why the neuron? What's the tropism for these lymphocytes to go to the neuron, and is that an area where you might consider an immunological blockade. The second is, I got my ACIP recommendations for vaccines, a very nice book that the ACIP put out, and I wanted to know in individuals over 65 who are vaccinated, what is the incidence of viremia leading to either chicken pox or zoster?
Gershon, New York: I'm not sure we can answer either of your questions, but I can give you some thoughts about them. One of the reasons that Mike (Gershon) and I were very doubtful that this virus could cause latency by viremia was that we couldn't figure out how it would know to go to the neuron, and I guess one possibility is that the neuron has the ability to survive when infected with this virus whereas other cells may not. We do know that the cellular receptor for VZV is the mannose 6-phosphate receptor, which is present on axons, so that is the route that the virus could take; the ligand of that particular receptor is present on the viral envelope. People who are studying latency are looking at apoptosis in neurons because VZV is capable of preventing apoptosis from occurring and therefore it may be a survival tactic for the virus for it to become latent in the nerve.
Regarding viremia in 65-year-olds, we don't know much about it because VZV is a very difficult virus on which to work. Many investigators have looked elsewhere for projects because VZV is difficult to propagate. VZV is highly cell-associated and very labile; we only really began to learn about this virus in any detail after the development of molecular techniques, which don't require one to grow the virus. Therefore, we don't know much about viremia in the natural situation. It is rare, however, to develop either clinical varicella or zoster as a direct result of vaccination.
On the other hand, in addition to varicella and zoster infections without skin lesions, we do know that there are forms of zoster in which the virus reactivates but doesn't reach the outer layers of the skin but does produce dermatomal pain, which may be severe. You may be able to demonstrate VZV DNA in saliva in these patients, and it may be that that this phenomenon is some kind of a reflection of viremia. We don't know how VZV gets to saliva or what it's doing there, but it's a wonderful non-invasive means to demonstrate whether somebody is experiencing reactivation of VZV. I want to say another thing about this subject. We think that with reactivation without symptoms, in other words, low-level reactivation, that we may be boosting immunity internally, which has important implications for VZV vaccines. Many people who are critical of varicella vaccine are fond of saying that in the vaccine era we will have less circulation of the wild-type virus, which will lead to less opportunity for people to have exogenous boosting of their VZV immunity. Based on computer modeling, they fear that we are going to have a resultant epidemic of zoster. Fortunately, investigators at the CDC have been studying this and, although the incidence of zoster in our population has been increasing, it's been doing so since the 1950s, not just since the vaccine was introduced. Most experts in the field don't think that the increase in zoster in our population is related to vaccine or if it is, it plays only a minor part. It is very important, however, to establish whether we have internal boosting of immunity to VZV. I'd like to remind everybody that the largest immune organ in the body is the gut, so latent VZV may be doing something good there as well as something bad.
Vercelloti, Minnesota: Thank you.
Billings, Baton Rouge: I have a couple of thoughts. The first is: when someone develops shingles, when they are over the shingles, should they be vaccinated?
Gershon, New York: The recommendation is yes and the reasoning is that maybe they'll get more boost from the Zostavax than you do from the natural infection. I don't know whether this is true or not, but the Zostavax is 14 times as potent as the varicella vaccine, so possibly, it's true. What we can say about giving Zostavax to somebody who has already had shingles is that it appears to be safe. The Zostavax is, I think, safer than the varicella vaccine and I think that's probably because even if you are going to get shingles, you have some residual immunity and that serves to protect you from bad side effects. The problem with the zoster vaccine is that it doesn't work as well as the chicken pox vaccine and so even though it provides 50% to 60% protection against zoster and post-herpetic neuralgia, PHN, which is what we really worry about, we'd like protection to be better than 50% to 60%. The varicella vaccine after two doses is 98% protective.
Billings, Baton Rouge: If you have a patient who is in their 50s or 60s who is about to begin chemotherapy, do you vaccinate those patients and, if so, are there data that show that the vaccination works? For instance, if you have a patient with non-Hodgkin's lymphoma or Hodgkin's disease and the patient is undergoing vigorous treatment, does the vaccination work in the midst of all that? Should you wait until chemotherapy is completed? I have had a number of patients who have developed shingles in the midst of chemotherapy, suggesting whatever immunity they had before I got hold of them, or their disease got hold of them, was no longer there. So do they get vaccinated while you are treating them or after you're treating them? Should you treat everybody at the end of chemotherapy realizing that you may have knocked out their immunity for varicella zoster?
Gershon, New York: This is a difficult question and I think that there are no really specific recommendations, particularly about cancer patients. One place where we have made some progress, or are making some progress, is in HIV-infected patients who are well-controlled immunologically. They are getting now varicella vaccine, and there are studies in the ACTG in which they are getting the Zostavax. I believe that the studies have shown that Zostavax appears to be safe so far in these patients. The studies are still underway, and I think that they have almost completed enrollment, but we don't have any information about whether it works or not. I am particularly concerned about patients with lymphoma who get VZV vaccine and also transplant patients. There have been a couple of reported problems with it, so I am circumspect about immunizing them and basically would not do it. One area, however, in which people are thinking about giving the vaccine is prior to renal transplantation. That might work and after a long period following chemotherapy or a reasonable period before somebody is going to get chemotherapy would seem to be an optimal time. However, I don't think there are any data on this approach, so I think one has to proceed very carefully with these patients. We are fortunate that we have good antiviral therapy for VZV infections, which we can use very quickly, as soon as the diagnosis is suspected. That may be the best approach until we have more data. One thing that may help is that there are subunit-adjuvanted vaccines aimed to prevent zoster that are in clinical trials. Those, of course, won't establish latent infection and won't induce a viremic phase, so that may be something that it is important on the horizon. There are, however, no data yet.
Billings, Baton Rouge: Thank you.
REFERENCES
- 1.Gershon A, Silverstein S. Varicella-zoster virus. In: Richman D, Whitley R, Hayden F, editors. Clinical Virology. 3rd Ed. Washington, DC: ASM Press; 2009. pp. 451–73. [Google Scholar]
- 2.Hardy I, Gershon AA, Steinberg SP, et al. The incidence of zoster after immunization with live attenuated varicella vaccine. A study in children with leukemia. Varicella Vaccine Collaborative Study Group. N Engl J Med. 1991;325:1545–50. doi: 10.1056/NEJM199111283252204. [DOI] [PubMed] [Google Scholar]
- 3.Chen JJ, Zhu Z, Gershon AA, et al. Mannose 6-phosphate receptor dependence of varicella zoster virus infection in vitro and in the epidermis during varicella and zoster. Cell. 2004;119:915–26. doi: 10.1016/j.cell.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Oxman MN. Zoster vaccine: current status and future prospects. Clin Infect Dis. 2010;51:197–213. doi: 10.1086/653605. [DOI] [PubMed] [Google Scholar]
- 5.Koropchak C, Solem S, Diaz P, Arvin A. Investigation of varicella-zoster virus infection of lymphocytes by in situ hybridization. J Virol. 1989;63:2392–5. doi: 10.1128/jvi.63.5.2392-2395.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahalingham R, Wellish M, Soike K, White T, Kleinschmidt-DeMasters GD. Simian varicella virus infects ganglia before rash in experimentally infected monkeys. Virology. 2001;279:339–42. doi: 10.1006/viro.2000.0700. [DOI] [PubMed] [Google Scholar]
- 7.Edelman DA, Antaki F, Basson MD, Salwen WA, Gruber SA, Losanoff JE. Ogilvie syndrome and herpes zoster: case report and review of the literature. J Emerg Med. 2009;39:696–700. doi: 10.1016/j.jemermed.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Pui JC, Furth EE, Minda J, Montone KT. Demonstration of varicella-zoster virus infection in the muscularis propria and myenteric plexi of the colon in an HIV-positive patient with herpes zoster and small bowel pseudo-obstruction (Ogilvie's syndrome) Am J Gastroenterol. 2001;96:1627–30. doi: 10.1111/j.1572-0241.2001.03808.x. [DOI] [PubMed] [Google Scholar]
- 9.Gan L, Wang M, Chen J, Gershon M, Gershon AA. New persectives on varicella-zoster infection in a guinea pig model. In preparation. 2011 [Google Scholar]
- 10.Gershon A, Chen J, LaRussa P, Steinberg S. Varicella-zoster virus. In: Murray PR, Baron E, Jorgensen J, et al., editors. Manual of Clinical Microbiology. 9th Ed. Washington, DC: ASM Press; 2007. pp. 1537–48. [Google Scholar]
- 11.LaRussa P, Lungu O, Hardy I, Gershon A, Steinberg S, Silverstein S. Restriction fragment length polymorphism of polymerase chain reaction products from vaccine and wild-type varicella-zoster virus isolates. J Virol. 1992;66:1016–20. doi: 10.1128/jvi.66.2.1016-1020.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pahud BA, Glaser CA, Dekker CL, Arvin AM, Schmid DS. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316–23. doi: 10.1093/infdis/jiq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loparev VN, Argaw T, Krause PR, Takayama M, Schmid DS. Improved identification and differentiation of varicella-zoster virus (VZV) wild-type strains and an attenuated varicella vaccine strain using a VZV open reading frame 62-based PCR. J Clin Microbiol. 2000;38:3156–60. doi: 10.1128/jcm.38.9.3156-3160.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soong W, Schultz JC, Patera AC, Sommer MH, Cohen JI. Infection of human T lymphocytes with varicella-zoster virus: an analysis with viral mutants and clinical isolates. J Virol. 2000;74:1864–70. doi: 10.1128/jvi.74.4.1864-1870.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lungu O, Sun XW, Wright TC, Ferenczy A, Richart RM, Silverstein S. A polymerase chain reaction-enzyme-linked immunosorbent assay method for detecting human papillomavirus in cervical carcinomas and high-grade cervical cancer precursors. Obstet Gynecol. 1995;85:337–42. doi: 10.1016/0029-7844(94)00399-x. [DOI] [PubMed] [Google Scholar]
- 16.Cohen JI. Rodent models of varicella-zoster virus neurotropism. Curr Top Microbiol Immunol. 2010;342:277–89. doi: 10.1007/82_2010_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Gershon M, Wan S, et al. Latent infection of the human enteric nervous system by varicella zoster virus (VZV) Gastroenterology. 2009;136:W1689. [Google Scholar]
- 18.Head H, A.W. C. The pathology of herpes zoster and its bearing on sensory localization. Brain. 1900;23:353523. [Google Scholar]
- 19.Gershon A. Varicella-zoster virus: prospects for control. Adv Pediatr Infect Dis. 1995;10:93–124. [PubMed] [Google Scholar]
- 20.Chen J, Gershon AA, Li Z, Cowles RA, Gershon MD. Varicella zoster virus (VZV) infects and establishes latency in enteric neurons. J Neurovirol. 2011;17:578–89. doi: 10.1007/s13365-011-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galea SA, Sweet A, Beninger P, et al. The safety profile of varicella vaccine: a 10-year review. J Infect Dis. 2008;197(Suppl 2):S165–9. doi: 10.1086/522125. [DOI] [PubMed] [Google Scholar]
- 22.Shapiro ED, Vazquez M, Esposito D, et al. Effectiveness of 2 doses of varicella vaccine in children. J Infect Dis. 2011;203:312–5. doi: 10.1093/infdis/jiq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Gershon A, Silverstein SJ, Li ZS, Lungo O, Gershon MD. Latent and lytic infection of isolated guinea pig enteric and dorsal root ganglia by varicella zoster virus. J Med Virol. 2003;70:S71–8. doi: 10.1002/jmv.10325. [DOI] [PubMed] [Google Scholar]
- 24.McCluggage WG, Fox JD, Baillie KE, Coyle PV, Jones FG, O'Hara MD. Varicella zoster gastritis in a bone marrow transplant recipient. J Clin Pathol. 1994;47:1054–6. doi: 10.1136/jcp.47.11.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivera-Vaquerizo PA, Gomez-Garrido J, Vicente-Gutierrez M, Blasco-Colmenarejo M, Mayor-Lopez J, Perez-Flores R. Varicella zoster gastritis 3 years after bone marrow transplantation for treatment of acute leukemia. Gastrointest Endosc. 2001;53:809–10. doi: 10.1067/mge.2001.114421. [DOI] [PubMed] [Google Scholar]
- 26.Robertson CS, Martin BA, Atkinson M. Varicella-zoster virus DNA in the oesophageal myenteric plexus in achalasia. Gut. 1993;34:299–302. doi: 10.1136/gut.34.3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratman E. Visceral zoster as the presenting feature of disseminated herpes zoster. J Am Acad Dermatol. 2002;46:771–4. doi: 10.1067/mjd.2002.119091. [DOI] [PubMed] [Google Scholar]
- 28.Caccese WJ, Bronzo RL, Wadler G, McKinley MJ. Ogilvie's syndrome associated with herpes zoster infection. J Clin Gastroenterol. 1985;7:309–13. doi: 10.1097/00004836-198508000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Nomdedeu JF, Nomdedeu J, Martino R, et al. Ogilvie's syndrome from disseminated varicella-zoster infection and infarcted celiac ganglia. J Clin Gastroenterol. 1995;20:157–9. doi: 10.1097/00004836-199503000-00020. [DOI] [PubMed] [Google Scholar]