

Diazonamide A (1, Figure 1) is a marine-derived natural product with potent antimitotic activity and an unusual architecture.[1] Its mechanism of action has been studied, and though it displays differential cytotoxicity in the NCI COMPARE[2] screen consistent with a tubulin-active agent,[3] recent studies by Harran, Wang and McKnight suggest a unique mechanism of action involving the mitochondrial matrix enzyme, ornithinine δ-amino transferase (OAT).[4] Prior to these studies, OAT had no known mitotic function, and diazonamide A does not inhibit the amino transferase activity of this enzyme, yet it disrupts its interaction with mitotic spindle promoting proteins. These same workers showed that a close synthetic analogue of diazonamide A lacking the two chlorine atoms retains the cytotoxicity of the natural product, but does not display overt toxicity nor does it cause weight loss, change in overall physical appearance, or evidence of neutropenia in mice.[4a] The combination of limited supply, unique biological activity, and structural complexity, renders this molecule and analogues thereof important targets for chemical synthesis.
Figure 1.
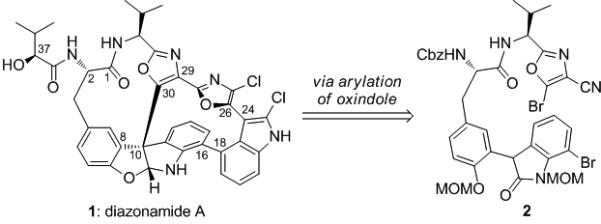
The structure and retrosynthesis of diazonamide A
The synthetic challenge posed by diazonamide A lies, in great part, in the stereoselective construction of the highly hindered C10 quaternary carbon, which has attracted the attention of numerous synthetic groups.[5] These efforts have resulted in three total syntheses by Nicolaou and by Harran, and a formal total synthesis by Magnus.[6] We have studied a new approach wherein we construct the C10 quaternary carbon via the arylation of a 3-aryloxindole.[7] We initially chose to study this construction via a Pd-catalyzed arylation,[8] and embarked on a model study to examine the order of bond formation in the synthesis of this subunit. We studied these reactions using 3-substituted oxindoles 3 and 4, which were subjected to arylation with bromobenzene or bromooxazole 5,[9] respectively, using modified Hartwig conditions[10] (Pd(OAc)2 or Pd(dba)2 and tBu3PHBF4[11] in toluene at reflux, Scheme 1). We found that while the combination of substrate 3 and bromobenzene did not provide the desired product, substrate 4 reacted cleanly with bromooxazole 5 producing compound 6 in 79% yield.
Scheme 1.
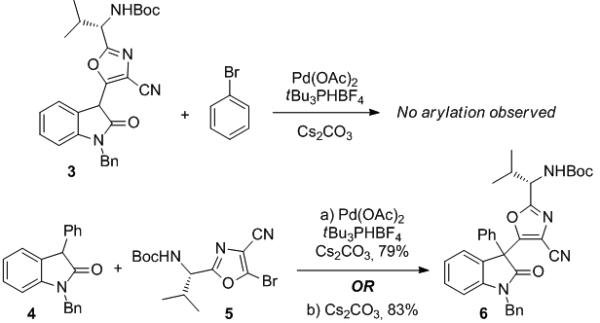
Arylation of 3-aryloxindoles 3 & 4.
This method can readily form a very hindered quaternary carbon, and in an effort to optimize the reaction, we reduced the catalyst loading from 5% to incrementally lower levels. Surprisingly, we were able to eliminate the Pd entirely with no decrease in yield, indicating that the reaction can proceed via an SNAr mechanism in the absence of Pd (Scheme 1).[12] We are currently examining further applications of this reaction to the formation of quaternary carbon centers.
In order to apply this reaction to the synthesis of diazonamide A, we prepared the bis-MOM protected cyclization precursor 2 (Scheme 2). N-Cbz-L-tyrosine methyl ester (7)[13] was treated with iPrMgCl, and the resulting phenoxide was coupled with N-MOM-7-bromoisatin (8) to provide alcohol 9.[14, 15] The phenolic hydroxyl group of compound 9 was then protected (MOMCl) and the tertiary alcohol was reduced (SOCl2 then Zn/HOAc) to provide oxindole 12.[15] Saponification of the methyl ester of 12 followed by amidation of the resulting carboxylic acid with aminooxazole 13 (EDC/HOBt) provided cyclization precursor 2.[15]
Scheme 2.
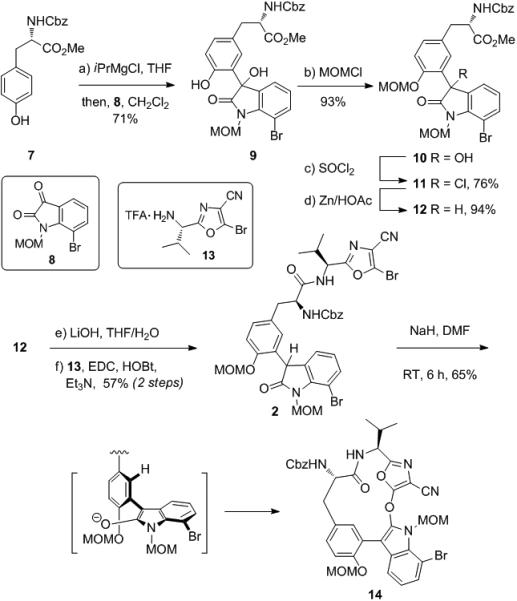
Preparation and cyclization of bis-MOM protected oxindole 2.
The cyclization of 2 was studied under a variety of conditions with variations in the base, solvent and temperature. The formation of a cyclic product was observed under many conditions, e.g., NaH in DMF, RT, 65%; however, we were disappointed to find that in all cases only the O-arylation product (14) was formed (Scheme 2).
We hypothesized that the desired C-arylation was hindered by the ortho-substituent of the tyrosine moiety, which prevents the formation of a coplanar enolate as shown in Scheme 2. The orthogonal aryl group would thereby render the carbon center too hindered to approach the bromooxazole. We wished to test this hypothesis and therefore synthesized cyclization precursor 22 which lacks an ortho-substituent and should be capable of adopting a planar conformation with the enolate. The synthesis of 22 was similar to that of 2, but required the reduction of the tyrosine phenol. Typical hydrogenolysis conditions are not compatible with the Cbz protecting group on the C2 nitrogen or the bromine at C16, so the Cbz was replaced with a Boc group, and the bromine was not installed. Thus, compound 17[15] was prepared by addition of N-Boc-L-tyrosine methyl ester (15)[16] to N-MOM-isatin (16), and the phenolic hydroxyl group of the product converted to the triflate with Comins’ reagent (18)[17] to provide 19[15] (Scheme 3). This was then subjected to hydrogenolysis (Pd/C, H2) to provide the doubly reduced product 20[15] in 69% yield along with the partially reduced byproduct 21 in 21% yield. These compounds were separated, and 20 was saponified with LiOH and coupled with aminooxazole 13 to provide cyclization precursor 22.[15] Subjection of compound 22 to Cs2CO3 in DMF at 65 °C provided the desired C-arylated cyclization product 23 in 70% yield. The stereochemistry of this product was assigned by analogy to that of 34 (vide infra) and no other stereoisomers were observed by NMR.
Scheme 3.
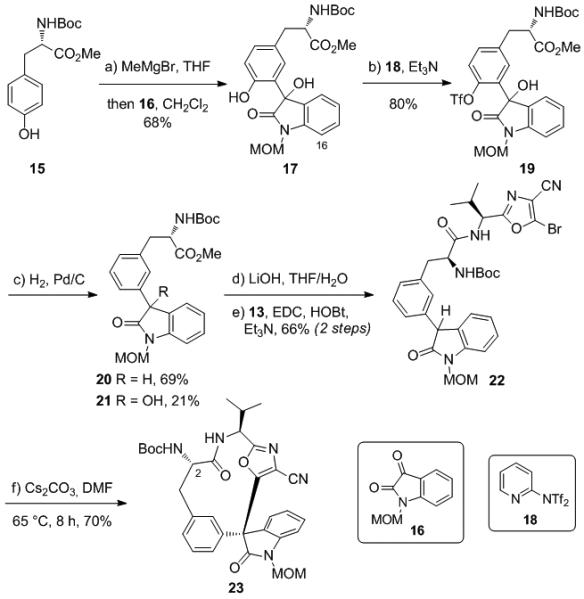
Cyclization of substrate 22 lacking an ortho-substituent on the tyrosine moiety.
This result suggests that an arene lacking ortho-functionalization can achieve coplanarity with the oxindole enolate and undergo cyclization on carbon. However, we still required a method for the synthesis of the C-arylated product bearing the tyrosine hydroxyl group. We reasoned that we could force co-planarity of the tyrosine arene and the oxindole enolate by taking advantage of hydrogen bonding between the phenol of the tyrosine and the enolate alkoxide. This is expected to be a strong interaction due to the acidity of the phenol and the basicity of the enolate. Cyclization precursor 25[15] was therefore synthesized as shown in Scheme 4 from 17 by reduction of the tertiary hydroxyl group (H2/Pd(OH)2/C) followed by saponification (LiOH) and coupling with 13 (EDC/HOBt). We were pleased to find that compound 25 underwent cyclization in 46% yield upon treatment with Na2CO3 in DMF at 65 °C to provide the desired C-arylation product 26 as a single diastereomer (Scheme 4; stereochemistry assigned by analogy to that of 34). More reactive bases, such as Cs2CO3 or LiHMDS, were unsatisfactory as they provided complex mixtures.
Scheme 4.
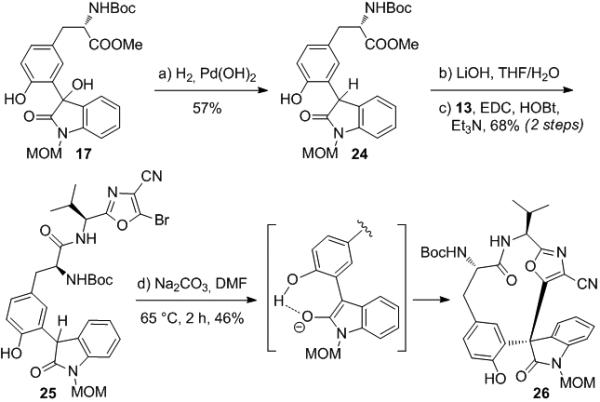
Cyclization of free-phenol substrate 25.
In order for this cyclization to be viable for a synthesis of the natural product, we required a handle at C16 to allow for the formation of the C16-C18 bond. We therefore prepared cyclization precursors 28[15] and 29,[15] bearing a bromine at C16 starting from compounds 10 and 27, respectively, by a route analogous to that shown in Scheme 2. Unfortunately, we did not obtain the desired cyclization products under a variety of conditions; in all cases either recovered starting material or decomposition was observed (Scheme 5). We hypothesize that these cyclizations are inhibited by the conformation of the MOM group, which is likely altered due to the sterics of the bromine at C16. We therefore studied cyclization precursor 33 which lacks the N-MOM protecting group.
Scheme 5.
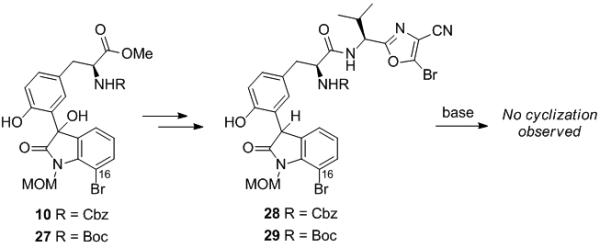
Attempted cyclization of substrates 28 & 29.
Synthesis of 33[15] proceeded by the addition of N-Cbz-L-tyrosine methyl ester (7) to 7-bromoisatin (30) to provide 31[15] in 74% yield (Scheme 6). This was followed by reduction of the resulting tertiary alcohol by Nicolaou's procedure (SOCl2 then NaCNBH3)[6c,d] to provide 32[15] in 82% yield over two steps. Saponification followed by coupling of the resulting acid with aminooxazole 13 provided cyclization precursor 33 in 72% yield over two steps. We were delighted to find that subjection of 33 to Na2CO3 in DMF at 65 °C for 20 hours provided the desired C- cyclized product in 56% yield. The stereochemistry of this material was determined by X-ray crystallography and found to be consistent with the natural product.[18] We observed no other isomers in this reaction; the remainder of the material was a mixture of starting material and an unidentified non-isomeric side product bearing two bromine atoms by mass spectrometry that co-elutes with the starting material. Under the same conditions, other carbonate bases either provided comparable yields (K2CO3, 40 – 50%), no reaction (Li2CO3), or a complex mixture (Cs2CO3). Acetonitrile provides comparable yields to DMF while DMA and DMSO provide slightly diminished yields (~40%).
Scheme 6.
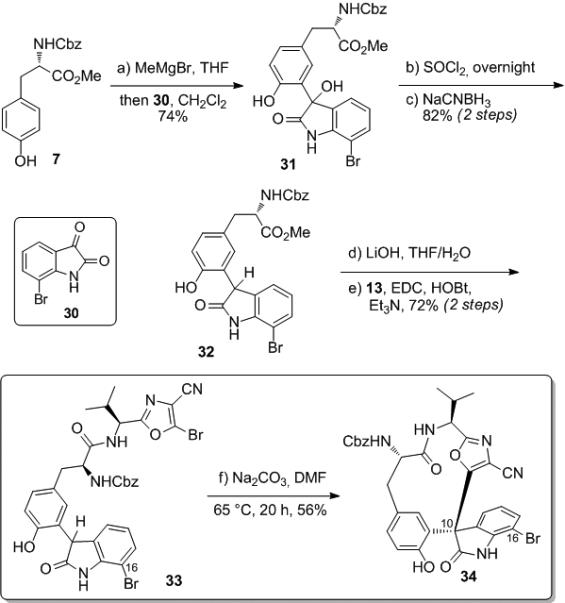
Cyclization of unprotected phenol-oxindole 33.
Compound 34 was then converted to 36[6a,b] by a two step sequence involving hydrolysis of the nitrile to carboxamide 35 using Parkins’ catalyst (37)[19] in 95% ethanol in a sealed tube at 120 °C (92%). This was followed by reduction to the primary alcohol using SmI2 and H2O (51%, Scheme 7).[20] Nicolaou has converted this intermediate to diazonamide A by an 11-step sequence,[6a,b] and as such, this constitutes a formal total synthesis.
Scheme 7.
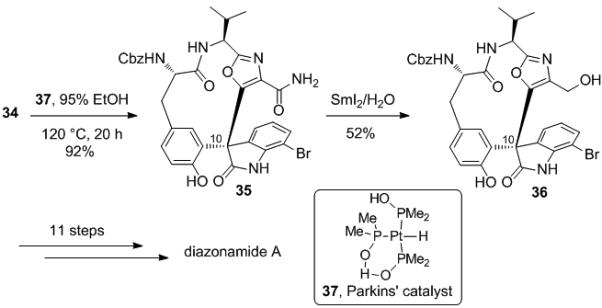
Correlation with Nicolaou's First Synthesis of Diazonamide A.
In conclusion, a formal total synthesis of diazonamide A has been described. The key step in this synthesis is the diastereoselective intramolecular arylation of a 3-aryloxindole via an SNAr reaction, thereby forming the hindered C10 quaternary stereocenter in an efficient and stereoselective fashion. Interestingly, the cyclization occurs under very mild conditions using sodium carbonate as the base such that no protecting groups are required on the phenol or oxindole nitrogen.
Footnotes
This work was supported by a generous grant from the National Institutes of Health (GM 48498). We wish to thank Dr. Joseph H. Reibenspies (Texas A&M University) for acquiring the X-ray crystal structure of 34 and Dr. Richard Shoemaker (University of Colorado) for expert assistance with acquiring NMR data.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For isolation and structure revision of diazonamide A, see: Lindquist N, Fenical W, Van Duyne GD, Clardy J. J. Am. Chem. Soc. 1991;113:2303–2304.Li J, Jeong S, Esser L, Harran PG. Angew. Chem. 2001;113:4901–4906. doi: 10.1002/1521-3773(20011217)40:24<4765::aid-anie4765>3.0.co;2-1.Angew. Chem. Int. Ed. 2001;40:4765–4769. doi: 10.1002/1521-3773(20011217)40:24<4765::aid-anie4765>3.0.co;2-1.Li J, Burgett AWG, Esser L, Amezcua C, Harran PG. Angew. Chem. 2001;113:4906–4909.Angew. Chem. Int. Ed. 2001;40:4770–4773. doi: 10.1002/1521-3773(20011217)40:24<4770::aid-anie4770>3.0.co;2-t.
- 2.Shoemaker RH. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 3.Cruz-Monserrate Z, Vervoort HC, Bai R, Newman DJ, Howell SB, Los G, Mullaney JT, Williams MD, Pettit GR, Fenical W, Hamel E. Mol. Pharm. 2003;63:1273–1280. doi: 10.1124/mol.63.6.1273. [DOI] [PubMed] [Google Scholar]
- 4.For biological studies of diazonamide A and analogues, see: Williams NS, Burgett AWG, Atkins AS, Wang X, Harran PG, McKnight SL. Proc. Natl. Acad. Sci. USA. 2007;107:2074–2079. doi: 10.1073/pnas.0611340104.Wang G, Shang L, Burgett AWG, Harran PG, Wang X. Proc. Natl. Acad. Sci. USA. 2007;104:2068–2073. doi: 10.1073/pnas.0610832104.Harran P, Wang X, Wang G. University of Texas System; 2007. US 2007/0287679 A1.
- 5.For a recent review of the synthesis of diazonamide A, see: Lachia M, Moody CJ. Nat. Prod. Rep. 2008;25:227–253. doi: 10.1039/b705663j.
- 6.For the synthesis of diazonamide A, see: Nicolaou KC, Bella M, Chen DY-K, Huang X, Ling T, Snyder SA. Angew. Chem. 2002;114:3645–3649. doi: 10.1002/1521-3773(20020916)41:18<3495::AID-ANIE3495>3.0.CO;2-7.Angew. Chem. Int. Ed. 2002;41:3495–3499. doi: 10.1002/1521-3773(20020916)41:18<3495::AID-ANIE3495>3.0.CO;2-7.Nicolaou KC, Chen DY-K, Huang X, Ling T, Bella M, Snyder SA. J. Am. Chem. Soc. 2004;126:12888–12896. doi: 10.1021/ja040092i.Nicolaou KC, Rao PB, Hao J, Reddy MV, Rassias G, Huang X, Chen DY-K, Snyder SA. Angew. Chem. 2003;115:1795–1800. doi: 10.1002/anie.200351112.Angew. Chem. Int. Ed. 2003;42:1753–1758. doi: 10.1002/anie.200351112.Nicolaou KC, Hao J, Reddy MV, Rao PB, Rassias G, Snyder SA, Huang X, Chen DY-K, Brenzovich WE, Giuseppone N, Giannakakou P, O'Brate A. J. Am. Chem. Soc. 2004;126:12897–12906. doi: 10.1021/ja040093a.Burgett AWG, Li Q, Wei Q, Harran PG. Angew. Chem. 2003;115:5111–5116.Angew. Chem. Int. Ed. 2003;42:4961–4966. doi: 10.1002/anie.200352577.Cheung C-M, Goldberg FW, Magnus P, Russell CJ, Turnbull R, Lynch V. J. Am. Chem. Soc. 2007;129:12320–12327. doi: 10.1021/ja0744448.
- 7.For the arylation of oxindoles, see: Durbin MJ, Willis MC. Org. Lett. 2008;10:1413–1415. doi: 10.1021/ol800141t.Altman RA, Hyde AM, Huang X, Buchwald SL. J. Am. Chem. Soc. 2008;130:9613–9620. doi: 10.1021/ja803179s. For the asymmetric arylation of 3-alkyloxindoles, see: Taylor AM, Altman RA, Buchwald SL. J. Am. Chem. Soc. 2009;131:9900–9901. doi: 10.1021/ja903880q. For a related approach, see: Lin J, Gerstenberger BS, Stessman NYT, Konopelski JP. Org. Lett. 2008;10:3969–3972. doi: 10.1021/ol8014336.
- 8.Bellina F, Rossi R. Chem. Rev. 2009 doi: 10.1021/cr9000836. ASAP. DOI: 10.1021/cr9000836; Johansson CCC, Colacot TJ. Angew. Chem. 2010;122:686–718.Angew. Chem. Int. Ed. 2010;49:676–707. doi: 10.1002/anie.200903424.
- 9.Jeong S, Chen X, Harran PG. J. Org. Chem. 1998;63:8640–8641. [Google Scholar]
- 10.Jørgensen M, Lee S, Liu X, Wolkowski JP, Hartwig JF. J. Am. Chem. Soc. 2002;124:12557–12565. doi: 10.1021/ja027643u. [DOI] [PubMed] [Google Scholar]
- 11.Netherton MR, Fu GC. Org. Lett. 2001;3:4295–4298. doi: 10.1021/ol016971g. [DOI] [PubMed] [Google Scholar]
- 12.Palmer DC, Venkatraman S. In: The Chemistry of Heterocyclic compounds, Oxazoles: Synthesis, Reactions, and Spectroscopy: Part A. Palmer DC, editor. Vol. 60. John Wiley & Sons, Inc.; Hoboken, New Jersey: 2003. pp. 138–149. [Google Scholar]
- 13.Lin S, Yang Z-Q, Kwok BHB, Koldobskiy M, Crews CM, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:6347–6355. doi: 10.1021/ja049821k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hewawasam P, Erway M. Tetrahedron Lett. 1998;39:3981–3984. [Google Scholar]
- 15.Prepared as a 1:1 mixture of diastereomers.
- 16.Richter JM, Whitefield BW, Maimone TJ, Lin DW, Castroviejo MP, Baran PS. J. Am. Chem. Soc. 2007;129:12857–12869. doi: 10.1021/ja074392m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299–6302. [Google Scholar]
- 18.CCDC 753609 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
- 19.a Ghaffar T, Parkins AW. Tetrahedron Lett. 1995;56:8657–8660. [Google Scholar]; b Ghaffar T, Parkins AW. J. Mol. Catal. A: Chem. 2000;160:249–261. [Google Scholar]
- 20.Kamochi Y, Kudo T. Chem. Lett. 1993;22:1495–1498. [Google Scholar]