

Abstract
We measured oxidized phospholipids (OxPL), lipoprotein (a) [Lp(a)], and lipoprotein-associated phospholipase A2 (Lp-PLA2) pre- and postapheresis in 18 patients with familial hypercholesterolemia (FH) and with low(∼10 mg/dl; range 10–11 mg/dl), intermediate (∼50 mg/dl; range 30–61 mg/dl), or high (>100 mg/dl; range 78–128 mg/dl) Lp(a) levels. By using enzymatic and immunoassays, the content of OxPL and Lp-PLA2 mass and activity were quantitated in lipoprotein density fractions plated in microtiter wells, as well as directly on apoB-100, Lp(a), and apoA-I immunocaptured within each fraction (i.e., OxPL/apoB and Lp-PLA2/apoB). In whole fractions, OxPL was primarily detected in the Lp(a)-containing fractions, whereas Lp-PLA2 was primarily detected in the small, dense LDL and light Lp(a) range. In lipoprotein capture assays, OxPL/apoB and OxPL/apo(a) increased proportionally with increasing Lp(a) levels. Lp-PLA2/apoB and Lp-PLA2/apoA-I levels were highest in the low Lp(a) group but decreased proportionally with increasing Lp(a) levels. Lp-PLA2/apo(a) was lowest in patients with low Lp(a) levels and increased proportionally with increasing Lp(a) levels. Apheresis significantly reduced levels of OxPL and Lp-PLA2 on apoB and Lp(a) (50–75%), particularly in patients with intermediate and high Lp(a) levels. In contrast, apheresis increased Lp-PLA2-specific activity (activity/mass ratio) in buoyant LDL fractions. The impact of apheresis on Lp(a), OxPL, and Lp-PLA2 provides insights into its therapeutic benefits beyond lowering apoB-containing lipoproteins.
Keywords: atherogenesis, inflammation, myocardial infarction
Lipoprotein (a) [Lp(a)] is now generally accepted as a causal, independent, genetic risk factor for cardiovascular disease (CVD). Consensus documents by the European Atherosclerosis Society (1) and the National Lipid Association (2) have recommended screening patients for elevated Lp(a) levels at moderate to high risk for CVD and also suggested an ideal Lp(a) level is <50 mg/dl. Lp(a) is enriched in proinflammatory oxidized phospholipids (OxPL) and lipoprotein-associated phospholipase A2 (Lp-PLA2) (3). Epidemiological outcome studies have shown that the OxPL, Lp(a), and Lp-PLA2 collectively mediate additive risk for CVD (4–6).
We have proposed that a physiological role of Lp[a] may be to bind and transport proinflammatory oxidized phospholipids (OxPL) (4, 7, 8). OxPL mediate direct proinflammatory effects on macrophages, endothelial, and smooth muscle cells, and upregulate inflammatory gene responses and expression of inflammatory cytokines. Plasma levels of OxPL on apoB-100 (OxPL/apoB), detected by monoclonal antibody E06, are elevated in subjects with coronary artery disease (CAD) and predict the presence and progression of carotid and femoral atherosclerosis and higher risk of future death, myocardial infarction, and stroke (reviewed in Ref. 3). Furthermore, they enhance the predictive value of the Framingham Risk Score and increase the area under the curve using c-index statistics, reflecting its clinical utility (3, 5, 6). Lp(a) is potentially more atherogenic than native LDL because it binds with increased affinity to arterial intimal proteoglycans, resulting in delivery of both atherogenic LDL and proinflammatory OxPL. OxPL, oxidized LDL, and Lp(a) trigger apoptosis in endoplasmic reticulum (ER)-stressed macrophages through CD36 and Toll-like receptor 2, suggesting a mechanism through which OxPL/Lp(a) may destabilize atherosclerotic plaques (9).
OxPL on Lp(a) may be modulated through Lp-PLA2, which cleaves OxPL into lysophosphatidylcholine (lysoPC) and a free oxidized fatty acid (10, 11). In normolipidemic individuals, Lp-PLA2 is mainly associated with LDL and is preferentially associated with atherogenic small dense LDL (12). In primary hypercholesterolemia, the LDL-associated Lp-PLA2 is increased in parallel with the severity of hypercholesterolemia, the highest levels being present in homozygous familial hypercholesterolemia (FH) (11). Interestingly, in individuals with elevated Lp(a) levels, Lp-PLA2 activity is higher on Lp(a) than on equimolar amounts of LDL (13, 14). Lp-PLA2 is present in vulnerable plaques, and increased Lp-PLA2 levels are associated with increased CVD risk in both primary and secondary populations (15).
Patients with FH generally have significantly higher Lp(a) levels than patients without FH (16), despite the fact that the LDL receptor is not thought to be involved in its clearance. Lp(a) and its associated proinflammatory OxPL and Lp-PLA2 may be particularly atherogenic and lead to increased CV events. In view of the pathophysiological relationship of OxPL, Lp(a), and Lp-PLA2, we assessed the acute changes and distribution of apheresis on these atherogenic moieties in patients with FH as a potential mechanism of clinical benefit.
METHODS
Study subjects with varying Lp(a) levels and blood sampling
Eighteen patients with Type IIa FH (10 men and 8 women) from the Haemobiotherapy Unit at Pitie-Salpetriere University Hospital in Paris undergoing LDL-apheresis every 2 or 3 weeks were selected for this study (17). For the purpose of this study, patients were a priori recruited into three groups according to their Lp(a) levels: (low 10 mg/dl; range, 10–11 mg/dl), intermediate (∼50 mg/dl; range 30–61 mg/dl), and high (>100 mg/dl; range, 78–128 mg/dl). The diagnosis of FH was validated by DNA analysis in all patients, except for two in which no mutation was found. DNA sequencing was extended to LDLR, APOB, and PCSK9 but not to ARH genes. However, all patients had phenotypic FH by family history. Four patients were homozygous for the LDL-R gene mutation, 6 were heterozygous, and 5 were compound heterozygotes with two mutations of the LDLR gene. One patient was a double heterozygote (mutations in LDLR gene and APOB3500). All patients were treated once daily by lipid-lowering drugs in combination therapy: atorvastatin/ezetimibe (80 mg/10 mg; n = 16), simvastatin/ezetimibe (80 mg/10 mg; n = 1), or rosuvastatin/ezetimibe (20 mg/10 mg; n = 1).
Blood samples were obtained pre-LDL apheresis before coupling the instrument to the cephalic vein and immediately post-LDL apheresis, and were collected into sterile EDTA-containing tubes (final concentration 1 mg/ml). Plasma was immediately separated from blood cells by low-speed centrifugation at 2,500 rpm for 20 min at 4°C and frozen at −80°C until analysis. The study was performed in accordance with the ethical principles set forth in the Declaration of Helsinki. Written informed consent was obtained from all subjects.
LDL-apheresis
Three types of columns were used for LDL-apheresis (see Table 1). To avoid blood coagulation in the column, 3,000–13,000 units of unfractionated heparin were added during preliminary rinsing of the column and prior to coupling the machine to the patient. Some patients (n = 4) received a heparin bolus (1,000–2,000 U) at the beginning of the procedure and a supplemental heparin infusion during the procedure (500 U/h for three of the four patients and 2000 U/h for one). The duration of each LDL-apheresis session was >2 h.
TABLE 1.
Baseline characteristics of the familial hypercholesterolemia study group
| Characteristic | Low Lp(a) Group (n = 6) | Intermediate Lp(a) Group (n = 6) | High Lp(a)Group (n = 6) | P |
| Lipoprotein(a), mg/dl | 10.7 ± 4.0 (range, 7–15) | 54.5 ± 22.5 (range, 29–81) | 102.5 ± 42.4 (range, 52–138) | <0.001 |
| Number of KIV repeats (major isoform) | 19.8 ± 2.9 | 22.8 ± 4.7 | 18.2 ± 3.3 | 0.15 |
| Sum of both KIV isoforms | 45.5 ± 9.5 | 46.6 ± 9.4 | 40.5 ± 7.2 | 0.51 |
| Age, yr | 37 ± 15 | 35 ± 16 | 49 ± 16 | 0.28 |
| Age at the beginning of LDL-apheresis, yr | 26 ± 16.2 | 20 ± 18.9 | 41 ± 23 | 0.001 |
| Male/female | 3/3 | 1/5 | 4/2 | 0.19 |
| Column type (DALI/KAN WB/KAN PL) | 3/2/1 | 3/2/1 | 3/2/1 | N/A |
| Body mass index | 24 ± 5 | 23 ± 4 | 24 ± 5 | 0.91 |
| Number of LDL-apheresis sessions | 182 ± 124 | 369 ± 137 | 180 ± 230 | 0.12 |
| Lipid parameters, mg/dl | ||||
| Total cholesterol | 494.0 ± 146.5 | 645.8 ± 214.8 | 331.8 ± 71.9 | <0.001 |
| Triglyceride | 88.0 ± 42.9 | 154.8 ± 47.8 | 169.6 ± 125.0 | 0.004 |
| LDL-cholesterol | 423.0 ± 156.7 | 598.8 ± 226.0 | 267.4 ± 87.1 | <0.001 |
| HDL-cholesterol | 47.8 ± 21.9 | 29.6 ± 6.5 | 49.8 ± 11.5 | <0.001 |
DALI, direct adsorption of lipoproteins (Fresenius HemoCare Adsorber Technology, St. Wendel, Germany); KAN PL, Kaneka plasma (Liposorber LA, Kaneka Corp., Osaka, Japan); KAN WB, Kaneka whole blood (Liposorber D, Kaneka Corp., Osaka, Japan.
Isolation of lipoprotein density fractions
Lipoproteins were isolated from plasma by a single-step, nondenaturing density gradient ultracentrifugation in a Beckman SW 41 Ti rotor at 40,000 rpm for 44 h in a Beckman L80 at 15°C by method of Chapman et al., with a slight modification as previously described (8, 17). Fractions 1–25 correspond to the following densities: <1.015, 1.015–1.016, 1.016–1.017, 1.017–1.019, 1.019–1.022, 1.022–1.025, 1.025–1.029, 1.029–1.033, 1.033–1.038, 1.038–1.044, 1.044–1.051, 1.051–1.059, 1.059–1.067, 1.067–1.075, 1.075–1.084, 1.084–1.094, 1.094–1.105, 1.105–1.116, 1.116–1.128, 1.128–1.139, 1.139–1.150, 1.150–1.163, 1.163–1.176, 1.176–1.189, >1.189 g/ml, respectively.
Lipid and protein analysis
Plasma, total cholesterol, triglycerides, LDL-cholesterol (LDL-C), HDL-cholesterol (HDL-C), apolipoprotein A-I (apoA-I), apolipoprotein B-100, high-sensitivity C-reactive protein (hsCRP), and Lp(a) were quantified by methods described earlier (18). Apo(a) isoform sizes were determined by immunoblot analysis with an apo(a)-specific antibody (19).
Determination of Lp-PLA2 mass and activity in plasma and density fractions and sPLA2 mass and activity in plasma
Plasma Lp-PLA2 mass in plasma and density gradients were measured in a blinded fashion as previously described (PLAC® Test, diaDexus, Inc.) (20, 21). The assay has a lower detection limit of 2 ng/ml and an interassay coefficient of variation (CV) of 6–7%. A threshold of >200 ng/ml is considered elevated (22). Lp-PLA2 activity was measured in a 96-well microplate with a colorimetric substrate that is converted on hydrolysis by the phospholipase enzyme (CAM™ assay, diaDexus, Inc.). The specific activity of Lp-PLA2 as the Lp-PLA2 activity/mass ratio was determined by dividing the Lp-PLA2 activity by the Lp-PLA2 mass. Serum sPLA2 type IIA mass and activity levels were measured only in plasma at the Paris Cardiovascular Research Center, as previously described (20, 21).
Determination of the distribution of apoB-100, apo(a), OxPL, and Lp-PLA2 mass in density gradient fractions by direct plating of fractions on microtiter well plates
To determine the distribution of apoB, apo(a), OxPL, and Lp-PLA2 mass in each density fraction directly on microtiter well plates, we incubated an aliquot containing a saturating amount of each fraction (1:200 dilution for apoB, 1:100 for apo(a) and OxPL, and 1:20 for Lp-PLA2) on microtiter wells overnight at 4°C. The content of apoB-100, apo(a), OxPL, and Lp-PLA2 were determined with the biotinylated secondary antibodies goat anti-human apoB-100 (Pierce), LPA4, E06, and 4B4, respectively, using ELISA (3), as illustrated in Fig. 1A. Determination of Lp-PLA2 mass by direct plating was used as a second complementary technique to the diaDexus method. Each sample was assayed in triplicate, and data are expressed as relative light units (RLU) per 100 milliseconds.
Fig. 1.
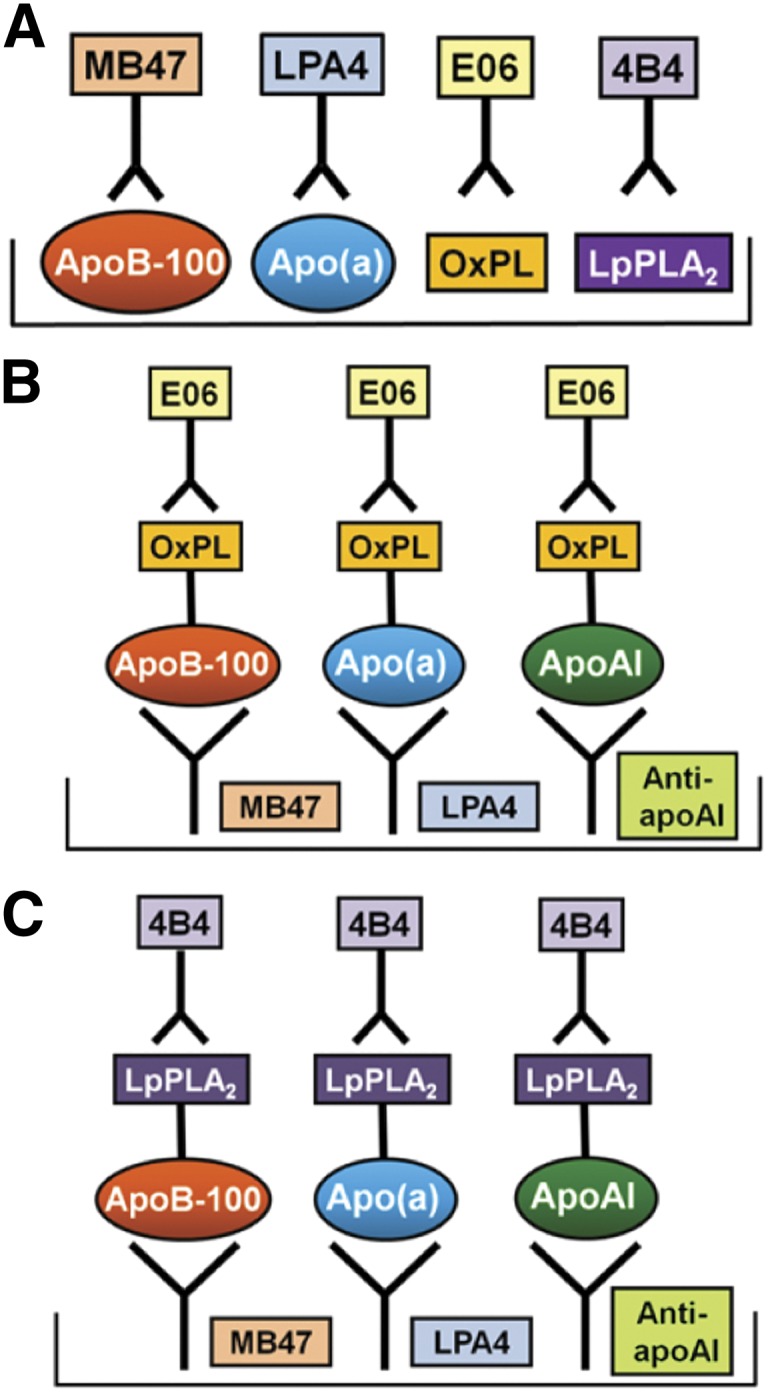
Methodology of various ELISA. (A) Detection of lipoproteins, OxPL, and Lp-PLA2 on density gradient fractions that were directly plated on microtiter well plates. (B) Detection of OxPL on apoB-100, apo(a), and apoA-I. (C) Detection of Lp-PLA2 on apoB-100, apo(a), and apoA-I.
Determination of the presence of OxPL and Lp-PLA2 mass directly on isolated apoB, apo(a), and apoA-I
The content of OxPL (Fig. 1B) or Lp-PLA2 mass (Fig. 1C) on isolated apoB-100, apo(a) and apoA-I captured on microtiter well plates with specific antibodies were determined by chemiluminescent sandwich ELISA in a similar format as above. For this set of assays, the antibodies MB47, LPA4, and guinea pig anti-human apoA-I were plated on microtiter well plates (5 mg/ml) to capture apoB-100, Lp(a), and apoA-I, respectively. A fraction (1:25 dilution) of each aliquot was added, the unbound plasma was washed off, and the amount of OxPL or Lp-PLA2 was determined with biotinylated E06 or 4B4, respectively. The assays for OxPL or Lp-PLA2 present on apoB-100 are reported as OxPL/apoB and Lp-PLA2/apoB, on Lp(a) as OxPL/apo(a) and Lp-PLA2/apo(a), and on apoA-I as OxPL/apoA-I and Lp-PLA2/apoA-I to denote the detection of OxPL or Lp-PLA2 on the captured lipoproteins. In this format, the amount of apoB-100, Lp(a), and apoA-I captured generally reflects their content in each aliquot (i.e., nonsaturating). The only exception to this is that because apoB is at a large excess compared with Lp(a) and apoA-I, it was generally saturating the plates (Fig. 2, top panels). The assays were set up in this manner to be able to consistently observe changes in all measurements pre- and postapheresis.
Fig. 2.

An aliquot of each density gradient fraction was directly plated onto microtiter well plates and assessed for the presence of apoB, apo(a), OxPL, or Lp-PLA2 pre- and postapheresis in the low Lp(a), intermediate Lp(a), and high Lp(a) groups. Due to lower values, the scale is different for Lp-PLA2. Each of the 25 individual data points pre- and postapheresis represents the mean of six patients.
RESULTS
Baseline demographic and biochemical characteristics of the study groups
Baseline characteristics of the three study groups on first presentation and segregated a priori into low (∼10 mg/dl, range 7–15 mg/dl), intermediate (∼50 mg/dl, range 29–81 mg/dl) or high (>100 mg/dl, range 52–138 mg/dl) Lp(a) levels are shown in Table 1. These subjects are relatively young-to-middle-aged adults, have very high baseline LDL-C levels, and have been on chronic apheresis for at least two years with a large number of sessions. The efficacy on the lipid profile achieved by these apheresis techniques is consistent with findings in the literature (17, 18).
Plasma lipids, lipoproteins, hsCRP, Lp-PLA2, sPLA2, and OxPL on lipoproteins pre- and postapheresis
Table 2 reports the biochemical variables on blood samples of patients on chronic apheresis that were recruited at the same time for this study. As expected, apheresis led to significant reduction in total cholesterol and LDL-C, apoB, triglycerides, and hsCRP in all Lp(a) groups (Table 2). HDL-C and ApoA-I were decreased by ∼15% in each Lp(a) group. Lp(a) levels were significantly reduced in the intermediate and high Lp(a) groups but not in the low Lp(a) group.
TABLE 2.
Changes in plasma lipids, lipoproteins, Lp-PLA2, and OxPL on lipoproteins and hsCRP pre- and postapheresis
| Low Lp(a) Group (n = 6) | Intermediate Lp(a) Group (n = 6) | High Lp(a) Group (n = 6) | |||||||
| Pre | Post | P | Pre | Post | P | Pre | Post | P | |
| Total cholesterol, mg/dl | 269 ± 64 | 88 ± 27 | <0.001 | 312 ± 110 | 100 ± 26 | <0.005 | 243 ± 28 | 94 ± 20 | <0.001 |
| Triglyceride, mg/dl | 77 ± 36 | 24 ± 14 | <0.005 | 101 ± 45 | 65 ± 75 | 0.12 | 159 ± 79 | 95 ± 44 | 0.07 |
| HDL-C, mg/dl | 39 ± 12 | 33 ± 12 | <0.05 | 37 ± 9 | 31 ± 8 | <0.001 | 40 ± 12 | 34 ± 11 | <0.001 |
| LDL-C, mg/dl | 215 ± 66 | 50 ± 32 | <0.001 | 255 ± 112 | 57 ± 27 | <0.005 | 171 ± 23 | 41 ± 27 | <0.001 |
| ApoA-I, mg/dl | 113 ± 32 | 97 ± 31 | <0.001 | 108 ± 16 | 90 ± 11 | <0.005 | 122 ± 30 | 101 ± 21 | <0.01 |
| ApoB, mg/dl | 135 ± 28 | 34 ± 15 | <0.001 | 167 ± 61 | 42 ± 14 | <0.005 | 133 ± 9 | 39 ± 15 | <0.0001 |
| Lp(a), mg/dl | 10 ± 0.4 (range, 10–11) | 10 ± 0.0 (range, 10–10 | 0.36 | 42 ± 11 (range, 30–61) | 12 ± 2.5 (range, 10–15 | <0.001 | 103 ± 20 (range, 78–158 | 28 ± 11 (range, 17–48) | <0.0005 |
| hsCRP, g/dl | 0.06 ± 0.08 | 0.02 ± 0.02 | 0.23 | 0.14 ± 0.26 | 0.04 ± 0.08 | 0.25 | 0.07 ± 0.08 | 0.05 ± 0.07 | <0.05 |
| Lp-PLA2 mass, ng/ml | 235 ± 37 | 114 ± 32 | <0.001 | 294 ± 41 | 160 ± 29 | <0.001 | 274 ± 71 | 137 ± 56 | 0.004 |
| Lp-PLA2 activity, mmol/ml/min | 189 ± 41 | 69 ± 32 | <0.001 | 215 ± 44 | 89 ± 24 | <0.001 | 179 ± 61 | 78 ± 56 | 0.014 |
| sPLA2 mass, ng/ml | 5.0 ± 2.5 | 4.2 ± 2.3 | 0.11 | 2.5 ± 1.1 | 1.7 ± 0.4 | 0.23 | 4.4 ± 1.9 | 3.0 ± 2.3 | 0.004 |
| sPLA2 activity, mmol/ml/min | 62.2 ± 23.0 | 65.0 ± 16.0 | 0.50 | 45.1 ± 7.1 | 60.4 ± 13.1 | 0.16 | 62.0 ± 6.8 | 62.4 ± 13.1 | 0.91 |
| OxPL/apoB, RLU | 1821 ± 871 | 2432 ± 1471 | 0.40 | 5247 ± 2807 | 2731 ± 1354 | 0.076 | 16723 ± 4718 | 6462 ± 1673 | <0.001 |
| OxPL/apo(a), RLU | 4314 ± 1455 | 1541 ± 930 | 0.003 | 23110 ± 11277 | 4791 ± 1887 | 0.003 | 41686 ± 11422 | 14944 ± 3830 | <0.001 |
| OxPL/apoA-I, RLU | 103 ± 92 | 87 ± 120 | 0.80 | 441 ± 192 | 313 ± 202 | 0.29 | 486 ± 90 | 386 ± 165 | 0.22 |
Lp-PLA2 mass and activity were significantly reduced in all three groups following apheresis (Table 2). As a comparison, sPLA2 mass, which is not known to circulate significantly with apoB-containing lipoproteins, was only sig nificantly reduced in the high Lp(a) group. There were no significant differences in sPLA2 activity pre- and postapheresis. When all three Lp(a) groups were combined, there was a reduction in sPLA2 mass (4.1 ± 7.7 versus 3.1 ± 8.4, P < 0.001) but not sPLA2 activity (57.6 ± 30.1 versus 62.7 ± 33.7, P = 0.12).
Plasma OxPL/apoB was not reduced in the low Lp(a) group, but the levels were very low and near the level of detection of this assay. There was a strong trend for reduction (48%) in OxPL/apoB in the intermediate Lp(a) group, and there was a significant decline (62%) in the high Lp(a) group (Table 2). When all three Lp(a) groups were combined, there was a reduction in OxPL/apoB (8,259 ± 7,299 versus 3,913 ± 2,424 RLU, P = 0.003). OxPL/apo(a) was significantly reduced in all groups, and OxPL/apoA-I levels were very low [approximately 40-fold lower than OxPL/apo(a)] and were not affected by apheresis.
ApoB, apo(a), OxPL, and Lp-PLA2 mass and activity on individual density gradient fractions measured by direct plating on microtiter well plates: effect of apheresis
In patients with low, intermediate, and high Lp(a) levels, apoB is mainly present in fractions 5–12 corresponding to density range 1.019–1.059 g/ml (Fig. 2). At this dilution, apoB was plated to be saturating to provide a comparison for other variables. Thus, the apoB values reflect the content on the plate rather than the plasma content. Apo(a) and OxPL, which are not saturating at the dilutions plated, are present in fractions 10–20 (density range 1.038–1.139 g/ml) and 11–17 (density range 1.044–1.105 g/ml), respectively. In the low Lp(a) group, OxPL are minimally present. In the intermediate and high Lp(a) groups, the level of apo(a) peaks at tubes 12–14, which represents density range of 1.051–1.075 g/ml. Note that all of the OxPL immunoreactivity coincides with the apo(a) peak and not with the main apoB peak. As the Lp(a) levels increase among groups, the OxPL peaks increase in size accordingly.
Distribution and changes in Lp-PLA2 mass pre- and postapheresis are shown in the bottom panels of Fig. 2. Lp-PLA2 mass was present primarily in fractions 9–16 corresponding to densities 1.033–1.094 g/ml, which represents the range of both small dense LDL, as previously shown for Lp-PLA2 activity and Lp(a) (12, 13, 23). There appeared to be a similar amount of Lp-PLA2 mass preapheresis in the low and intermediate Lp(a) groups, whereas the levels were lower in the high Lp(a) groups. Interestingly, Lp-PLA2 mass was also present in the very dense fractions 23–25, representing densities 1.163 → 1.189 g/ml. This is suggestive of dissociation of a fraction of Lp-PLA2 from apoB lipoproteins as previously described for separation of lipoproteins using ultracentrifugation compared with FPLC (24). Lp-PLA2 activity appeared qualitatively similarly distributed among the three groups and in a similar pattern to Lp-PLA2 mass, although only two patients in each group were available for measurements.
Using the diaDexus sandwich Lp-PLA2 mass and activity ELISA (Fig. 3), qualitatively similar information was noted compared with the direct-plating assays shown in Fig. 2. In this analysis, we were able to calculate the specific activity of Lp-PLA2 as the Lp-PLA2 activity/mass ratio. The specific activity was highest in fractions 8–10, which correspond to fractions between LDL-3 and LDL-4 [LDL-3 (d = 1.029–1.039 g/ml), LDL-4 (d = 1.039–1.050 g/ml)], which is in accordance with prior findings showing that despite the high Lp-PLA2 mass and activity associated with LDL-5 [LDL-5 (d = 1.050–1.063 g/ml)], the enzyme-specific activity (activity/mass) in LDL-5 is the lowest among all LDL subfractions (12, 23). This is also apparent in Fig. 2 using the direct-plating method where Lp-PLA2 mass in fractions 10 and higher almost disappear postapheresis.
Fig. 3.

The distribution of Lp-PLA2 mass, Lp-PLA2 activity, and Lp-PLA2 activity/mass ratio as a measure of the specific activity on individual density gradient aliquots was measured by the diaDexus ELISA pre- and postapheresis in the low Lp(a), intermediate Lp(a), and high Lp(a) groups. Each of the 25 individual data points pre- and postapheresis represents the mean of six patients, except for Lp-PLA2 activity, for which only two patients in each group were measured.
Following apheresis, reductions (∼50–75%) were noted in apoB, apo(a), OxPL, and Lp-PLA2 mass, and these changes were most pronounced in the intermediate and high Lp(a) groups (Figs. 2 and 3). Following apheresis, Lp-PLA2-specific activity actually increased in the intermediate and high Lp(a) groups. This may denote that, after apheresis, the remaining LDL particles are deficient in small dense LDL particles and enriched in large buoyant particles.
Distribution of OxPL levels on isolated apoB, apo(a), and apoA-I lipoproteins in FH patients: effect of apheresis
OxPL/apoB and OxPL/apo(a) levels progressively increased in parallel with increasing Lp(a) levels, being present primarily in fractions 11–15, representing densities of 1.044–1.084 g/ml (Fig. 4). In contrast, minor amounts of OxPL were detected on apoA-I particles. Following aphe resis, reductions were noted in both OxPL/apoB and OxPL/apo(a).
Fig. 4.

Capture assays were performed to detect the presence of OxPL on apoB, apo(a), or apoA-I lipoproteins captured on microtiter wells with specific murine monoclonal antibodies pre- and postapheresis in the low Lp(a), intermediate Lp(a), and high Lp(a) groups. Each of the 25 individual data points pre- and postapheresis represents the mean of six patients.
Distribution of levels of Lp-PLA2 on isolated lipoproteins apoB, apo(a), and apoA-I in FH patients: effect of apheresis
Preapheresis, Lp-PLA2/apoB and Lp-PLA2/apoA-I were highest in the low Lp(a) group, but they diminished with increasing Lp(a) levels (Fig. 5). In contrast, Lp-PLA2/apo(a) was lowest in the low Lp(a) group and increased with increasing Lp(a) levels. Interestingly, a significant amount of Lp-PLA2/apo(a) and Lp-PLA2/apoA-I were noted in the most dense fractions, suggesting the presence of Lp-PLA2 mass on poorly lipidated lipoproteins. Following apheresis, reductions were noted in Lp-PLA2/apoB and Lp-PLA2/apo(a), with more modest changes in Lp-PLA2/apoA-I.
Fig. 5.

Capture assays were performed to detect the presence of Lp-PLA2 on apoB, apo(a), or apoA-I lipoproteins captured on microtiter wells with specific murine monoclonal antibodies pre- and postapheresis in the low Lp(a), intermediate Lp(a), and high Lp(a) groups. Each of the 25 individual data points pre- and postapheresis represents the mean of six patients.
DISCUSSION
Patients with heterozygous FH have higher Lp(a) levels than non-FH patients (16) and represent a worldwide prevalence of about ten million individuals. Homozygous FH is thought to be present in ten to fifteen thousand subjects (25). In this study, apheresis significantly reduced levels of proatherogenic and proinflammatory OxPL and Lp-PLA2 on apoB and Lp(a), particularly in patients with high Lp(a) levels, while increasing Lp-PLA2-specific activity in buoyant LDL particles. These data provide a scientific rationale for understanding how apheresis may provide clinical benefits beyond lowering apoB-containing lipoproteins (26) and in understanding the pathophysiological relationships among Lp(a), OxPL, and Lp-PLA2 in collectively mediating CVD (5, 6, 9).
The clinical relevance of these findings is reflected by the fact that Lp(a) is now accepted as an independent genetic risk factor of CVD (1, 2), that Lp(a) is a carrier of OxPL and Lp-PLA2 (3, 4), and that apheresis is an accepted treatment for patients with FH and high Lp(a) levels (18). This study also demonstrates several novel insights into the potential pathophysiogical role of OxPL, Lp(a), and Lp-PLA2 in FH: i) OxPL were almost exclusively detected in the Lp(a)-containing fractions and not on HDL ApoA-I particles; ii) OxPL/apoB and OxPL/apo(a) increased proportionally with increasing Lp(a) levels; iii) Lp-PLA2/apoB and Lp-PLA2/apoA-I levels were highest in patients with low Lp(a) but decreased proportionally with increasing Lp(a) levels; and iv) Lp-PLA2/apo(a) was lowest in patients with low Lp(a) levels and increased proportionally with increasing Lp(a) levels.
Three retrospective, hypothesis-generating studies (26–28) demonstrated that maximally tolerated medications plus apheresis significantly reduced rate of major cardiac adverse events compared with maximally tolerated medications alone. Apheresis has been shown to significantly reduce Lp(a) concentrations (17, 18, 29, 30), and a small study suggested it reduces Lp-PLA2 mass (31). In this study, we demonstrate the powerful effect of apheresis on reduction of OxPL and Lp-PLA2 mass and activity physically present on associated apoB-containing lipoproteins. The basis of this decrease is likely due to the fact that the lipoprotein carriers of OxPL and Lp-PLA2, Lp(a) and apoB, respectively, also decreased in relative proportion following apheresis. Interestingly, the Lp-PLA2-specific activity increased in the intermediate and high Lp(a) groups, suggesting that the remaining LDL particles are deficient in small dense LDL particles with the lowest specific Lp-PLA2 activity and enriched in large buoyant particles exhibiting higher specific activity. These data are also consistent with previous data showing that apheresis enhances endothelium-dependent vasodilation in the brachial (32) and coronary arteries (33).
Treatments for elevated Lp(a) levels include niacin and the emerging cholesterol ester transport and PCSK9 inhibitors, although their underlying mechanisms of action are not well defined. Antisense oligonucleotides to apoB have shown promise in significantly reducing both LDL-C and Lp(a) levels (34, 35). In addition, specific antisense oligonucleotides to apo(a) have been shown to reduce apo(a) and their associated OxPL by ∼85% in apo(a)-transgenic mice (36). As they do not affect other lipoprotein levels to any great extent, it may be clinically feasible to ultimately test the hypothesis that lowering elevated Lp(a) a priori results in clinical benefits.
We also demonstrate that the majority of immunologically detected OxPL are physically present on Lp(a) particles and not significantly on non-Lp(a)-apoB particles or apoA-I particles. The fact that E06-detectable OxPL are associated with clinical events (3) suggests that they reflect a pathophysiologically important and clinically relevant biological activity of Lp(a). In corroborating evidence, we have also shown that OxPL/apoB levels primarily reflect OxPL associated with high Lp(a) levels and small apo(a) isoforms, i.e., the most atherogenic particles (37). Because apo(a) isoforms are not normally measured clinically, the OxPL/apoB measurement provides a unique biological assay that summates the cardiovascular risk of both LPA alleles (38). In preliminary data, we have documented that OxPL are present on Lp(a) in two compartments, one covalently bound to apo(a) and another noncovalently bound in the lipid phase of Lp(a). Further work needs to be performed to understand the relative proportion of OxPL in each component of Lp(a), particularly in different disease states, to define the sites on apo(a)/Lp(a) where OxPL are present, and to determine the specific species of OxPL responsible for clinical risk.
The distribution of Lp-PLA2 mass and activity was shown to be in the small dense LDL fraction, as previously documented by several studies (12, 23), along with a significant portion present on lipoproteins present in the poorly lipidated densities. Interestingly, as the Lp(a) levels change, the proportion of Lp-PLA2 on these particles changes significantly with the underlying lipid content. For example, in patients with low Lp(a) levels, the Lp-PLA2 is primarily present on apoB. However, as the Lp(a) levels increase, there is proportionately less Lp-PLA2 on apoB and more on Lp(a). This is consistent with the fact that on a molar basis, Lp(a) contains more Lp-PLA2 mass (1.5–2 times) and activity (7 times) than LDL (13, 14). However, as LDL is in excess of Lp(a) in most patients, there is globally more Lp-PLA2 on LDL than Lp(a).
In conclusion, these findings describe a pathophysiological relationship among Lp(a), OxPL, and Lp-PLA2 in patients with FH and suggest that additional benefits of apheresis, beyond lowering apoB-containing lipoproteins, may be to reduce their associated proinflammatory OxPL and Lp-PLA2.
Footnotes
Abbreviations:
- CAD
- coronary artery disease
- CVD
- cardiovascular disease
- FH
- familial hypercholesterolemia
- HDL-C
- HDL-cholesterol
- hsCRP
- high-sensitivity C-reactive protein
- LDL-C
- LDL-cholesterol
- Lp(a)
- lipoprotein (a)
- Lp-PLA2
- lipoprotein-associated phospholipase A2
- OxPL
- oxidized phospholipid
- RLU
- relative light unit
This work was supported by the Fondation Leducq, by National Institutes of Health Grants R01-HL-086559 (S.T. and J.L.W.) and HL-088093 (S.T. and J.L.W.), and by the General Clinical Research Center, University of California, San Diego, with funding provided by the National Center for Research Resources Grant M01-RR-00827. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. S.T. and J.L.W. are inventors of patents owned by the University of California for the clinical use of oxidation-specific antibodies and of biomarkers related to oxidized lipoproteins and phospholipase enzymes. Z.M. and A.T are co-inventors of patents related to phospholipase enzymes.
REFERENCES
- 1.Nordestgaard B. G., Chapman M. J., Ray K., Boren J., Andreotti F., Watts G. F., Ginsberg H., Amarenco P., Catapano A., Descamps O. S., et al. 2010. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur. Heart J. 31: 2844–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davidson M. H., Ballantyne C. M., Jacobson T. A., Bittner V. A., Braun L. T., Brown A. S., Brown W. V., Cromwell W. C., Goldberg R. B., McKenney J. M., et al. 2011. Clinical utility of inflammatory markers and advanced lipoprotein testing: advice from an expert panel of lipid specialists. J. Clin. Lipidol. 5: 338–367 [DOI] [PubMed] [Google Scholar]
- 3.Taleb A., Witztum J. L., Tsimikas S. 2011. Oxidized phospholipids on apolipoprotein B-100 (OxPL/apoB) containing lipo proteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark. Med. 5: 673–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsimikas S., Tsironis L. D., Tselepis A. D. 2007. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 27: 2094–2099 [DOI] [PubMed] [Google Scholar]
- 5.Kiechl S., Willeit J., Mayr M., Viehweider B., Oberhollenzer M., Kronenberg F., Wiedermann C. J., Oberthaler S., Xu Q., Witztum J. L., et al. 2007. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler. Thromb. Vasc. Biol. 27: 1788–1795 [DOI] [PubMed] [Google Scholar]
- 6.Tsimikas S., Mallat Z., Talmud P. J., Kastelein J. J., Wareham N. J., Sandhu M. S., Miller E. R., Benessiano J., Tedgui A., Witztum J. L., et al. 2010. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J. Am. Coll. Cardiol. 56: 946–955 [DOI] [PubMed] [Google Scholar]
- 7.Tsimikas S., Witztum J. L. 2008. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr. Opin. Lipidol. 19: 369–377 [DOI] [PubMed] [Google Scholar]
- 8.Bergmark C., Dewan A., Orsoni A., Merki E., Miller E. R., Shin M. J., Binder C. J., Horkko S., Krauss R. M., Chapman M. J., et al. 2008. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 49: 2230–2239 [DOI] [PubMed] [Google Scholar]
- 9.Seimon T. A., Nadolski M. J., Liao X., Magallon J., Nguyen M., Feric N. T., Koschinsky M. L., Harkewicz R., Witztum J. L., Tsimikas S., et al. 2010. Atherogenic lipids and lipoproteins trigger CD36–TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 12: 467–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stafforini D. M. 2009. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2). Cardiovasc. Drugs Ther. 23: 73–83 [DOI] [PubMed] [Google Scholar]
- 11.Tellis C. C., Tselepis A. D. 2009. The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochim. Biophys. Acta. 1791: 327–338 [DOI] [PubMed] [Google Scholar]
- 12.Tselepis A. D., Dentan C., Karabina S. A., Chapman M. J., Ninio E. 1995. PAF-degrading acetylhydrolase is preferentially associated with dense LDL and VHDL-1 in human plasma. Catalytic characteristics and relation to the monocyte-derived enzyme. Arterioscler. Thromb. Vasc. Biol. 15: 1764–1773 [DOI] [PubMed] [Google Scholar]
- 13.Karabina S. A., Elisaf M. C., Goudevenos J., Siamopoulos K. C., Sideris D., Tselepis A. D. 1996. PAF-acetylhydrolase activity of Lp(a) before and during Cu(2+)-induced oxidative modification in vitro. Atherosclerosis. 125: 121–134 [DOI] [PubMed] [Google Scholar]
- 14.Blencowe C., Hermetter A., Kostner G. M., Deigner H. P. 1995. Enhanced association of platelet-activating factor acetylhydrolase with lipoprotein (a) in comparison with low density lipoprotein. J. Biol. Chem. 270: 31151–31157 [DOI] [PubMed] [Google Scholar]
- 15.Thompson A., Gao P., Orfei L., Watson S., Di Angelantonio E., Kaptoge S., Ballantyne C., Cannon C. P., Criqui M., Cushman M., et al. 2010. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 375: 1536–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kraft H. G., Lingenhel A., Raal F. J., Hohenegger M., Utermann G. 2000. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 20: 522–528 [DOI] [PubMed] [Google Scholar]
- 17.Orsoni A., Saheb S., Levels J. H., Dallinga-Thie G., Atassi M., Bittar R., Robillard P., Bruckert E., Kontush A., Carrie A., et al. 2011. LDL-apheresis depletes apoE-HDL and pre-beta1-HDL in familial hypercholesterolemia: relevance to atheroprotection. J. Lipid Res. 52: 2304–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompsen J., Thompson P. D. 2006. A systematic review of LDL apheresis in the treatment of cardiovascular disease. Atherosclerosis. 189: 31–38 [DOI] [PubMed] [Google Scholar]
- 19.Marcovina S. M., Hobbs H. H., Albers J. J. 1996. Relation between number of apolipoprotein(a) kringle 4 repeats and mobility of isoforms in agarose gel: basis for a standardized isoform nomenclature. Clin. Chem. 42: 436–439 [PubMed] [Google Scholar]
- 20.Ravandi A., Boekholdt S. M., Mallat Z., Talmud P. J., Kastelein J. J., Wareham N. J., Miller E. R., Benessiano J., Tedgui A., Witztum J. L., et al. 2011. Relationship of IgG and IgM autoantibodies and immune complexes to oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J. Lipid Res. 52: 1829–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryu S. K., Mallat Z., Benessiano J., Tedgui A., Olsson A. G., Bao W., Schwartz G. G., Tsimikas S., Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering (MIRACL) Trial Investigators 2012. Phospholipase A2 enzymes, high-dose atorvastatin, and prediction of ischemic events after acute coronary syndromes. Circulation. 125: 757–766 [DOI] [PubMed] [Google Scholar]
- 22.Davidson M. H., Corson M. A., Alberts M. J., Anderson J. L., Gorelick P. B., Jones P. H., Lerman A., McConnell J. P., Weintraub H. S. 2008. Consensus panel recommendation for incorporating lipoprotein-associated phospholipase A2 testing into cardiovascular disease risk assessment guidelines. Am. J. Cardiol. 101: 51F–57F [DOI] [PubMed] [Google Scholar]
- 23.Gazi I., Lourida E. S., Filippatos T., Tsimihodimos V., Elisaf M., Tselepis A. D. 2005. Lipoprotein-associated phospholipase A2 activity is a marker of small, dense LDL particles in human plasma. Clin. Chem. 51: 2264–2273 [DOI] [PubMed] [Google Scholar]
- 24.McCall M. R., La Belle M., Forte T. M., Krauss R. M., Takanami Y., Tribble D. L. 1999. Dissociable and nondissociable forms of platelet-activating factor acetylhydrolase in human plasma LDL: implications for LDL oxidative susceptibility. Biochim. Biophys. Acta. 1437: 23–36 [DOI] [PubMed] [Google Scholar]
- 25.Goldberg A. C., Hopkins P. N., Toth P. P., Ballantyne C. M., Rader D. J., Robinson J. G., Daniels S. R., Gidding S. S., de Ferranti S. D., Ito M. K., et al. 2011. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 5: S1–S8 [DOI] [PubMed] [Google Scholar]
- 26.Jaeger B. R., Richter Y., Nagel D., Heigl F., Vogt A., Roeseler E., Parhofer K., Ramlow W., Koch M., Utermann G., et al. 2009. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat. Clin. Pract. Cardiovasc. Med. 6: 229–239 [DOI] [PubMed] [Google Scholar]
- 27.Gordon B. R., Kelsey S. F., Dau P. C., Gotto A. M., Jr, Graham K., Illingworth D. R., Isaacsohn J., Jones P. H., Leitman S. F., Saal S. D., et al. 1998. Long-term effects of low-density lipoprotein apheresis using an automated dextran sulfate cellulose adsorption system. Liposorber Study Group. Am. J. Cardiol. 81: 407–411 [DOI] [PubMed] [Google Scholar]
- 28.Mabuchi H., Koizumi J., Shimizu M., Kajinami K., Miyamoto S., Ueda K., Takegoshi T. 1998. Long-term efficacy of low-density lipo pro tein apheresis on coronary heart disease in familial hypercholesterolemia. Hokuriku-FH-LDL-Apheresis Study Group. Am. J. Cardiol. 82: 1489–1495 [DOI] [PubMed] [Google Scholar]
- 29.Otto C., Berster J., Otto B., Parhofer K. G. 2007. Effects of two whole blood systems (DALI and Liposorber D) for LDL apheresis on lipids and cardiovascular risk markers in severe hypercholesterolemia. J. Clin. Apher. 22: 301–305 [DOI] [PubMed] [Google Scholar]
- 30.Keller C. 2007. Apheresis in coronary heart disease with elevated Lp (a): a review of Lp (a) as a risk factor and its management. Ther. Apher. Dial. 11: 2–8 [DOI] [PubMed] [Google Scholar]
- 31.Moriarty P. M., Gibson C. A. 2005. Effect of low-density lipoprotein apheresis on lipoprotein-associated phospholipase A2. Am. J. Cardiol. 95: 1246–1247 [DOI] [PubMed] [Google Scholar]
- 32.Tamai O., Matsuoka H., Itabe H., Wada Y., Kohno K., Imaizumi T. 1997. Single LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humans. Circu lation. 95: 76–82 [DOI] [PubMed] [Google Scholar]
- 33.Mellwig K. P., Baller D., Gleichmann U., Moll D., Betker S., Weise R., Notohamiprodjo G. 1998. Improvement of coronary vasodilatation capacity through single LDL apheresis. Atherosclerosis. 139: 173–178 [DOI] [PubMed] [Google Scholar]
- 34.Raal F. J., Santos R. D., Blom D. J., Marais A. D., Charng M. J., Cromwell W. C., Lachmann R. H., Gaudet D., Tan J. L., Chasan-Taber S., et al. 2010. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 375: 998–1006 [DOI] [PubMed] [Google Scholar]
- 35.Merki E., Graham M. J., Mullick A. E., Miller E. R., Crooke R. M., Pitas R. E., Witztum J. L., Tsimikas S. 2008. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 118: 743–753 [DOI] [PubMed] [Google Scholar]
- 36.Merki E., Graham M., Taleb A., Leibundgut G., Yang X., Miller E. R., Fu W., Mullick A. E., Lee R., Willeit P., et al. 2011. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J. Am. Coll. Cardiol. 57: 1611–1621 [DOI] [PubMed] [Google Scholar]
- 37.Tsimikas S., Clopton P., Brilakis E. S., Marcovina S. M., Khera A., Miller E. R., de Lemos J. A., Witztum J. L. 2009. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: results from the Dallas Heart Study. Circulation. 119: 1711–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clarke R., Peden J. F., Hopewell J. C., Kyriakou T., Goel A., Heath S. C., Parish S., Barlera S., Franzosi M. G., Rust S., et al. 2009. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 361: 2518–2528 [DOI] [PubMed] [Google Scholar]