

Abstract
Successful development of drugs against novel targets crucially depends on reliable identification of the activity of the target gene product in vivo and a clear demonstration of its specific functional role for disease development. Here, we describe an immunological knockdown (IKD) method, a novel approach for the in vivo validation and functional study of endogenous gene products. This method relies on the ability to elicit a transient humoral response against the selected endogenous target protein. Anti-target antibodies specifically bind to the target protein and a fraction of them effectively neutralize its activity. We applied the IKD method to the in vivo validation of plasma PCSK9 as a potential target for the treatment of elevated levels of plasma LDL-cholesterol. We show that immunization with human-PCSK9 in mice is able to raise antibodies that cross-react and neutralize circulating mouse-PCSK9 protein thus resulting in increased liver LDL receptor levels and plasma cholesterol uptake. These findings closely resemble those described in PCSK9 knockout mice or in mice treated with antibodies that inhibit PCSK9 by preventing the PCSK9/LDLR interaction. Our data support the IKD approach as an effective method to the rapid validation of new target proteins.
Keywords: in vivo functional validation, animal model systems, cholesterol lowering treatments, active immunization
The development of drugs against novel target proteins requires a significant commitment in terms of time and cost (1). In many cases, new drugs fail in the clinic due to unacceptable side effects or lack of efficacy (2, 3). It is likely that the frequency of such failures could be reduced by more reliable target validation strategies in the drug development pathway. High-throughput technologies have been used extensively to characterize a wide number of genes and to gain insight into their functions. In particular, large-scale profiling of gene transcription, protein expression, protein-protein interactions and in silico data mining have been exploited to select potential targets for the treatment of human diseases (4–8). Although such data can be very informative, they are often insufficient to successfully drive a new drug discovery program and to set up strategies for therapeutic intervention. Therefore, it remains of key relevance to define the activity of the target gene product in vivo and clearly demonstrate its specific functional role in disease development.
Cardiovascular disease is one of the leading causes of death worldwide (9). Pro-protein convertase subtilisin-like/kexin type 9 (PCSK9) has recently emerged as a key determinant of liver low density lipoprotein receptor (LDLR) and LDL-cholesterol (LDL-c) plasma levels, and consequently of cardiovascular health in humans (10–13). PCSK9 belongs to the mammalian pro-protein convertase family of serine proteases (14, 15) and is expressed predominantly in the liver and small intestine (16). Following auto-cleavage in the endoplasmic reticulum (ER), the pro-protein is secreted into the plasma in an auto-inhibited form lacking enzymatic activity (17–21). The addition of PCSK9 to cultured cell medium has been shown to result in LDLR degradation, both overall and at the cell surface (22, 23), and in decreased LDL-c uptake (24). Consistently, several groups have reported that PCSK9 binds the LDLR ectodomain (17–19, 24–27) and that the complex is then internalized by a clathrin dependent mechanism (22). Together, these data are consistent with a mechanism whereby PCSK9 acts as a chaperon binding LDLR and shuttling the receptor to lysosomes for degradation (11).
Genetic knockout (KO) of PCSK9 in mice has been shown to result in reduced circulating cholesterol levels, increase in liver LDLR levels, and accelerated clearance of LDL-c (16, 28). Conversely, the introduction into mouse circulation of PCSK9 through parabiosis or by intravenous injection of the recombinant protein caused a strong reduction in hepatic LDLR levels (22, 29, 30) suggesting that plasma PCSK9 can act by binding to the receptor on the cell surface in vivo. Consistently, injection of neutralizing monoclonal antibodies (mAb) that binds PCSK9 and interfere with the PCSK9/LDLR interaction has been shown to decrease LDL-c by 70 to 80% in the nonhuman primate cynomologus monkey (31–33) and 65% in a human trial (34).
Here, we describe an immunological knockdown (IKD) method, a novel approach for the in vivo validation and functional study of endogenous gene products. This method relies on the ability to elicit a transient humoral response against the selected endogenous target protein. We show that this polyclonal response does include antibodies, which specifically bind to the target protein and effectively neutralize its activity. We applied the IKD method to the in vivo validation of plasma PCSK9 as a potential target in the treatment of elevated levels of plasma LDL-c.
EXPERIMENTAL PROCEDURES
Immunogens
Full-length human and mouse PCSK9 (indicated as hPCKS9 and mPCKS9, respectively) cDNAs were amplified from human or mouse fetal liver, respectively, and cloned under the transcriptional control of human cytomegalovirus (CMV) promoter in pVIJ expression plasmid (35). A pVIJ_hPCSK9 and pVIJ_mPCSK9 derivatives with C-terminal V5 and 6-His epitope tags were expressed in stably transfected HEK293 cell lines and purified as previously described (24).
Animal studies
Female BALB/c and C57BL6 mice were bred under specific pathogen-free conditions by Charles River Breeding Laboratories (Calco, Como, Italy). In all operations, mice were treated in accordance with European guidelines. Animals were maintained in standard conditions under a 12 h light-dark cycle, provided irradiated food (Mucedola, Settimo Milanese, Italy) and water ad libitum. Six-week-old mice were fully anesthetized with ketamine (Merial Italia, Milano, Italy) at 100 mg/kg of body weight and xylazine (BIO 98, Bologna, Italy) at 5.2 mg/kg. The immunization experiments were all performed at the I.R.B.M. Laboratory Animal Research, which has full AAALAC accreditation.
Immunization protocol
In xeno-DNA protocol mice were electro-injected intramuscularly with pVIJ_hPCSK9 (50 µg/mouse/injection: 3 times at 2 weeks intervals; days 0, 14, and 28) and CpG adjuvant (50 µg/mouse/injection; 3 times at 2 weeks intervals; day 7, 21, and 35: Fig. 1). A 20-mer (5′-TCCATGACGTTCCTGACGTT-3′) with a nuclease-resistant phosphorothioate backbone, which contains two CpG motifs with known immune-stimulatory effects on the murine immune response, was used as CpG in the studies we describe (36). Control mice were injected only with CpG (50 µg/mouse/injection; 3 times at 2 week intervals; days 7, 21, and 35). For the xeno-protein protocol the hPCSK9 protein formulated with CpG (100 µg protein and 50 µg adjuvant) was injected subcutaneously at the base of the tail on days 0, 3, 6, and 21. Control mice were injected only with CpG with the same schedule (Fig. 1). Both protocols showed a low and comparable reactivity in this as well as in other similar experiments for the control groups. This prompted us to use in the experiments reported in this work a unique “negative-ctrl” group populated by the mice from the two groups.
Fig. 1.

Schematic description of the immunization protocols tested. The leftmost column indicates the name of the protocol. Black-filled squares, gray-filled triangles, and white-filled circles refer to injection of hPCSK9 protein, plasmid DNA expressing hPCSK9, or CpG oligonucleotide adjuvant, respectively. ES, electrical stimulation. Time of analysis is indicated by an asterisk.
Clinical chemical parameters
Peripheral blood was collected and direct HDL-c, direct LDL-c, and total cholesterol were measured using the ADVIA 1200 IMS (Bayer Healthcare, Terrytown, NY).
ELISA detection of plasma anti-hPCSK9 and anti-mPCSK9 serum antibodies
Multi-well Maxisorp ELISA plates (Nunc, Roskild, Denmark) were coated overnight with PCSK9 protein at a concentration of 5 µg/ml in 50 mmol/L NaHCO3 (pH 9.6). Plates were then briefly rinsed with washing buffer (PBST, 0.05% Tween-20 in PBS) and incubated for 1 h at 37°C with blocking buffer (3% nonfat dry milk in PBST). Serial dilutions of preimmune or immune sera in PBST (ranging from 1:100 to 1/8,100) were added to the wells and incubated for 2 h at room temperature. Plates were washed and incubated with mAb anti-mouse IgG Fc-specific, alkaline phospatase (AP)-conjugated (Sigma-Aldrich Inc., St. Louis, MO) for 60 min at RT and AP activity detected by incubation with AP substrate solution (Sigma-Aldrich Inc., St. Louis, MO) in 10% diethanolamine/0.5 mmol/L MgCl2.
Titers of anti-hPCSK9 antibodies in the serum of immunized mice were computed as follows. Experimental data were acquired as (A405nm-A460nm) for each dilution of each sample. For each animal, prebleeds were included in duplicates at a 1:100-fold dilution together with the serial dilutions. We observed some variability in the prebleeds (A405nm-A460nm) between plates, but not within each plate. The baseline was therefore determined as the mean in addition to 3 standard deviations of the prebleed (A405nm-A460nm) for each plate, individually. Data referring to the same sample were fitted by a monotone Hermite spline. Titers were defined as the dilution at which the spline intersects the base line. When this intersection fell outside the dilution interval, titers were reported as the minimal or maximal dilutions, as appropriate.
Determination of plasma mPCSK9 concentration
Plasma mPCSK9 concentration was measured by ELISA as previously described (37). High binding 4HBX plates (ThermoLabsystems, Helsinki, Finland) were coated overnight at 4°C with 50 µl of 10 µg/ml of anti-mPCSK9 A1 antibody. Next day, the wells were first blocked for 1 h at room temperature with 250 µl of blocking solution (1% BSA and 0.05% Tween-20 in TBS) and then washed in a plate-washer. Purified mPCSK9 protein diluted in 1% BSA in PBS (as standard) or mouse plasma was added to the wells and incubated at 37°C for 2 h followed by a washing step. Then, 100 μl of 1 µg/ml biotinylated anti-mPCSK9 Fab A2, was added to the plate and after an additional washing step, 75 µl of 1:1,000 Streptavidin/Europium (Perkin Elmer, Waltham, MA) was added. Plates were incubated at room temperature for 20 min, followed by a last washing step and the addition of 100 µl of DELFIA enhancer (Perkin Elmer, Waltham, MA). After 1 h, the plates were read with a Europium reader.
Western blot analysis of mLDLR
Liver expression of mouse LDLR was evaluated by Western blot analysis and immunohistochemical staining. Liver crude protein extracts were prepared using RIPA Lysis buffer 500 μl per mg tissue (Santa Cruz Biotechnology Inc., Santa Cruz, CA) with phenylmethylsulfonyl fluoride, sodium orthovanadate, and proteinase inhibitors. NuPAGE 4-12% Bis-Tris gels (Life Technologies, Carlsband, CA) were loaded with 50 μg protein per lane. Blotted proteins were developed using a FITC-labeled anti mlDLR goat IgG (R and D Systems, Minneapolis, MI) diluted in primary Ab diluent buffer. FITC-labeled anti-Tubulin (Sigma, St Louis, MO) mouse mAb 3 was used to normalize data. Quantification of bands on X-Ray was made with ImageReader LAS 3000 (Fujifilm, Tokio, Japan).
Immunohistochemical staining for mouse LDLR
Tissues were fixed in 10% buffered formalin and embedded in paraffin. Sections of 10 µl were obtained using a microtome, cleared in xylol, and rehydrated. The unmasking procedure was carried out by incubating the samples in 10 × antigen retrieve solution (DAKO, Glostrup, Denmark) at 99°C for 40 min. After rinsing in PBS, the sections were incubated with blocking solution (15 μl goat serum in 1 ml PBS) for 30 min at RT and then without additional rinse, immediately incubated with rabbit polyclonal anti-LDLR (Abcam Inc., Cambridge, UK) for 1 h at room temperature. Samples were then rinsed in PBS and incubated with goat anti-rabbit IgG Peroxidase-conjugated antibody (Sigma-Aldrich Inc., St. Louis, MO) at the dilution of 1:200 for additional 60 min. After rinsing in PBS, the sections were stained with the diaminobenzidine staining kit (Vector Laboratories Inc., Burlingame, CA) and nuclei were stained with hematoxylin. The sections were dehydrated and mounted with Entellan (Merck KGaA, Darmstadt, Germany).
Statistical analyses
Differences in antibody titers were assessed using Wilcoxon's rank sum test due to lack of normality. Differences in LDL-c, HDL-c, cholesterol, and triglycerides levels were assessed using Welch's t-test. Normality assumptions were confirmed using the Shapiro-Wilk test. Correlations were assessed using Spearman's rank test. All reported p-values are two-sided.
RESULTS
Breaking tolerance against endogenous mouse PCSK9 protein
We explored different immunization protocols for their ability to raise antibodies against endogenous PCSK9 protein in mice. In a first set of experiments, BALB/c or C57BL6 mice were immunized by electro-injecting into the quadriceps muscles a DNA plasmid expressing mPCSK9 under the control of the human CMV promoter (pVIJ-mPCSK9), in conjunction with a CpG oligonucleotide adjuvant (35, 36). As expected, this syngeneic immunization failed to elicit a detectable antibody response against the target mPCSK9 protein in both mouse strains as assessed by ELISA (data not shown).
In a second set of experiments, a xenogeneic approach was evaluated by inducing expression of the hPCSK9 protein, which is 78% identical to mPCSK9. Six-week-old female BALB/c or C57BL6 mice were immunized by electro-injecting into the quadriceps muscles a DNA plasmid expressing hPCSK9 under the control of the human CMV promoter (pVIJ-hPCSK9). The DNA plasmid injections were performed at days 0, 14 and 28 and the CpG adjuvant injections were performed at days 7, 21 and 35 (xeno-DNA protocol; Fig. 1). As a negative control, a second group of mice was injected only with CpG adjuvant at days 7, 21 and 35 (DNA-ctrl protocol; Fig. 1). High titers of anti-hPCSK9 antibodies were detected by ELISA as early as 14 days after immunization (data not shown) and a fraction of these antibodies was also shown to cross-react with the mPCSK9 protein (Fig. 2). Similar effects were observed in BALB/c and C57BL6 mice. However, because anti-mPCSK9 titers were higher in BALB/c than in C57BL6 mice (data not shown), the former mouse strain was used for further immunizations.
Fig. 2.

Immunization with hPCSK9 elicits humoral response against the mouse endogenous gene product. Anti-mPCSK9 antibody titers were measured by ELISA in the serum of mice immunized according to the xeno-DNA (gray triangles) or xeno-protein (black squares) protocol. The corresponding negative controls (DNA-ctrl and protein-ctrl) showed a low and comparable reactivity in this as well as in the other reported experiments. We therefore referred to them as a unique group indicated a “negative-ctrl” (empty circles). Dashed lines indicate the dilution interval used as described in Materials and Methods. Data falling outside this interval were reported as the minimal or maximal dilution as appropriate.
To compare the efficacy of protein versus DNA immunization protocols, we performed a third set of experiments in which BALB/c mice were immunized by injection of highly purified hPCSK9 (xeno-protein protocol; Fig. 1). Recombinant hPCSK9 protein formulated with CpG adjuvant was subcutaneously injected at the base of the mice tail at days 0, 3, and 6, followed by a fourth injection at day 21. In a control experiment, mice were similarly injected only with CpG at days 0, 3, 6, and 21 (protein-ctrl). Fourteen days after the beginning of the immunization protocol, a very strong anti-hPCSK9 response was detected by ELISA in the serum of mice immunized with the hPCSK9 protein but not in the control animals (data not shown). As previously observed for the xeno-DNA vaccination protocol, a fraction of these antibodies cross-reacted with the endogenous mouse protein (Fig. 2). Of note, anti-mPCSK9 titers in the xeno-protein group were consistently higher than titers in the xeno-DNA group on days 14, 28, and 42 (P < 0.002, P < 0.001, and P = <0.017, respectively). At later times, this difference tended to decrease but it was still statistically significant at day 42. In addition the antibody titers in both groups differed strongly from those in the control animals at all time points (P < 0.001) demonstrating that there was no relevant effect of the CpG adjuvant alone (Fig. 2).
PCSK9 immunization decreases circulating cholesterol levels in mice
Mice immunized by electro-injection of pVIJ-hPCSK9 plasmid-DNA and those immunized by the injection of the recombinant hPCSK9 protein formulated with CpG adjuvant (same protocol described in Fig. 1) were monitored every 14 days for total cholesterol, HDL-c, LDL-c, and triglyceride levels (days 0, 14, 28, 42, and 56). Two weeks after the first injection, mice immunized with hPCSK9 protein showed total cholesterol levels reduced by 40% (Fig. 3A). Subsequently, the total cholesterol concentration gradually increased but after 42 days was still reduced by 28% compared with day 0. The level of HDL-c, the most abundant cholesterol fraction in mice, paralleled that of total cholesterol with a reduction of 42% and 22% of the initial HDL-c value at days 14 and 42, respectively (Fig. 3B). Strikingly, compared with the starting levels, at day 14, circulating LDL-c was reduced by 60% and remained consistently lower than in control mice for at least 8 weeks and approximately 35% lower than the initial value even at day 42 (Fig. 3C). The above changes were highly statistically significant (P < 0.0001 for all).
Fig.3.

Lipoprotein levels upon immunization. Total cholesterol (A), HDL-c (B), LDL-c (C) and triglycerides (D) were monitored every 14 days after the immunization start (days 0, 14, 28, 42, 56). Display items report data derived from the analysis of sera of mice immunized according to xeno-protein (black squares), to xeno-DNA (gray triangles), and negative-ctrl (empty circles). Bars indicate SEM.
A similar though milder phenotype was obtained in mice immunized with plasmid-DNA expressing the human protein where reduced levels of total cholesterol, HDL-c, and LDL-c (P = 0.005, P = 0.01, and P = 0.0001, respectively) were observed. These levels returned to their basal values by day 28 for LDL-c, HDL-c and total cholesterol (Fig. 3A–C). In contrast to the reduced cholesterol levels, in both protein- or DNA-immunized mice the triglyceride levels, did not significantly vary throughout the same period (Fig. 3D). Together these data demonstrate that immunization of mice with hPCSK9 protein results in an acute decrease of serum cholesterol.
Cholesterol levels inversely correlate with anti-mouse PCSK9 antibody titers in immunized mice
Analysis of the anti-mPCSK9 antibody levels reported above reveals that at day 14, the average antibody titers from mice immunized with the protein protocol were higher than the average titers measured on the DNA-immunized group (Fig. 2). Consistently, the average level of circulating LDL-c in mice immunized with the protein protocol was 49% lower than that measured in the DNA-immunized group which, in turn, was 23% lower of the average LDL-c level in the control-DNA group mice (Fig. 3C). Furthermore, in immunized mice, an inverse correlation between LDL-c levels and anti-mPCSK9 antibodies titers was observed with anti-mPCSK9 antibodies and LDL-c levels being respectively at the highest and lowest levels in the protein-immunized mice (Fig. 4). Thus, the immunization procedure using the hPCSK9 protein and, to a lesser extent, the hPCSK9 DNA elicited high anti-mPCSK9 titers that directly correlate with the reduction in plasma LDL-c levels.
Fig.4.

Correlation between LDL-c levels, anti-mPCSK9 antibody titers, and plasma mPCSK9 concentration in immunized mice. Correlation plot of circulating LDL-c levels with anti-mPCSK9 Ab titers measured at day 14 after immunization start according to xeno-protein (black squares), xeno-DNA (gray triangles; n = 10), and negative-ctrl (empty circles). Dashed lines indicate the dilution interval. Data falling outside this dilution interval were reported as the minimal or maximal dilution as appropriate. A significant anticorrelation was observed among LDL-c and the anti-mPCSK9 antibodies in immunized and in control mice (ρ=−0.79; P = 1.4e-9).
Circulating mPCSK9 protein levels in immunized mice
To monitor the variation on immunization in the levels of circulating mPCSK9, we used a highly sensitive ELISA (37). This assay uses a biotinylated mPCSK9-specific fragment binding antigen (Fab) and a neutralizing mAb that competes with LDLR/PCSK9 binding and allows an accurate detection of plasma mPCSK9. Two groups of mice each were immunized with hPCSK9 protein formulated with CpG or with CpG only and plasma mPCSK9 levels in the two groups were measured at day 14. In the control mice, the average level of detected circulating mPCSK9 was 238 ng/ml (SD = 50 ng/ml) whereas in the protein-immunized mice, the average level was significantly decreased (84 ng/ml; SD = 32 ng/ml). In line with previous experiments, circulating LDL-c was strongly reduced in immunized versus control mice and sustained antibody reactivity was observed against mPCSK9 in immunized but not in control mice. Thus, the vaccination protocol with hPCSK9 protein results in a marked decrease of circulating mPCSK9.
Hepatic LDLR levels increase in mice with high anti-mPCSK9 antibodies titers
Introduction of mPCSK9 into mice by parabiosis or by injection has been shown to strongly decrease hepatic LDLR levels (22, 29). Therefore, we sought to determine the effect of the immunization against PCSK9 on the levels and cellular distribution of the LDLR protein in the liver of mice from the protein and control immunization groups. In a first experiment, liver protein extracts from both groups were fractionated and blotted on a nitrocellulose filter. The LDLR was visualized by Western blotting with an anti-LDLR antibody and was quantified by densitometry scanning of the auto-radiogram (Fig. 5A, B). The level of hepatic LDLR protein was 2.1-fold higher in the immunized mice compared with the control group. Immuno-histochemistry analyses of the same liver samples were also performed. Using an anti-LDLR antibody, hepatic cell samples from the control group of mice showed a comparable staining of the cytoplasm and cell membrane, whereas samples from hPCSK9-immunized mice showed an enhanced staining at the membrane (Fig. 5C, D). Thus, our analysis reveals an increase in the cell surface levels of LDLR in immunized versus control mice, in line with the predicted functional activity of PCSK9 on the receptor and in agreement with the previous observations in PCSK9 KO mice (16).
Fig.5.
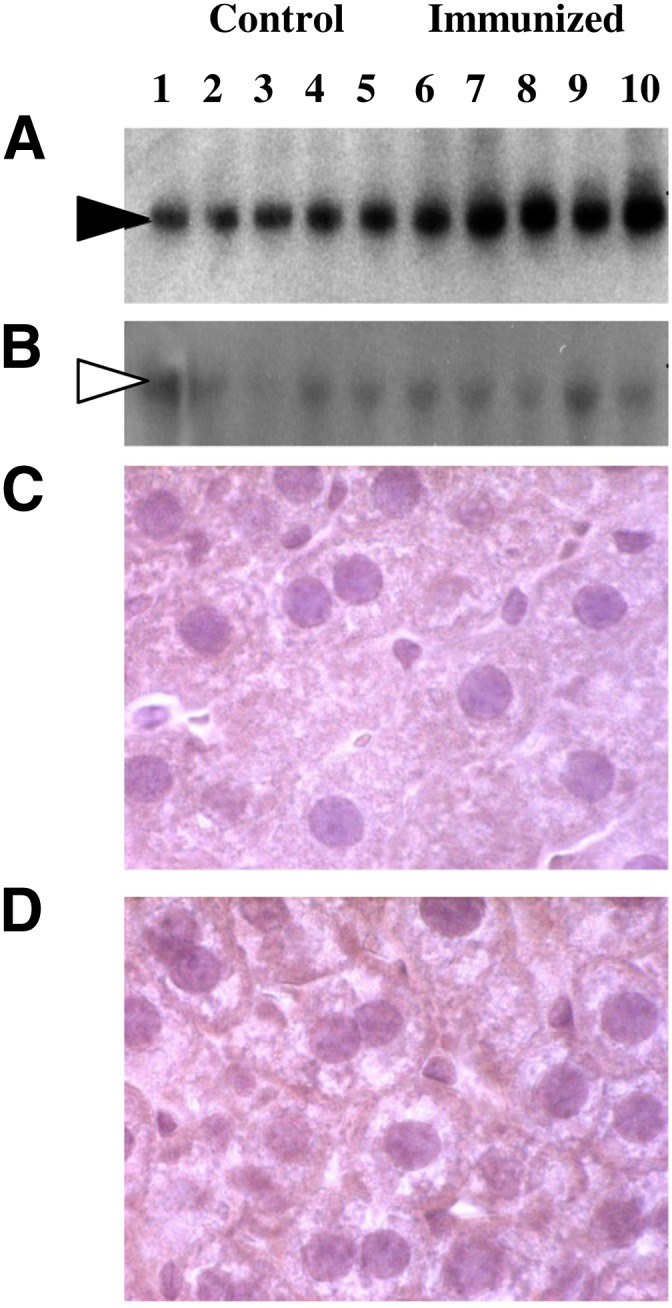
Levels and cellular distribution of LDLR in livers of immu nized mice. A, B: Liver extracts from mice immunized performed according to control-protein (samples 1–5) or xeno-protein (samples 6–10) protocol were separated by SDS-PAGE and blotted onto a nitro-cellulose filter. Following incubation with FITC-labeled anti-LDLR (A) or anti-tubulin (B) antibody, the bands corresponding to LDLR or α-tubulin proteins were visualized by autoradiography. The LDLR signal was measured by densitometric scanning and normalized on the corresponding α-tubulin signal. C, D: Cellular distribution of LDLR in immunized mice. Immuno-histochemistry analysis was performed on liver samples obtained from the same mice immunized with control (C) or xeno-protein (D) protocol using rabbit anti-LDLR antibodies detected by anti-rabbit IgG peroxidase-conjugated antibodies.
DISCUSSION
An essential step in the validation of a gene product as a target for therapeutic intervention is to assess the effects of its functional inhibition in vivo. A popular route to obtain this information is to create genetic KO mice where expression of a target gene product is abrogated by the deletion of the gene itself. Generating KO mice, however, requires time that cannot be easily reduced. A number of additional experimental approaches have been developed to target selected genes, including antisense oligonucleotides and small interfering RNA (siRNA). These are efficient, specific, and relatively nontoxic reagents. However, the delivery of these compounds into specific cells/organs is still a challenge and represents an obstacle in many cases (38).
An additional limitation of all these approaches is that they are based on the deletion of the gene or its transcribed product, which results in a reduction in the levels of expression of the target protein. Moreover, the obtained phenotype reflects their genetic origin and could differ from that induced by the pharmacological inhibition of the corresponding gene product in vivo. In contrast, it can be argued that the validation of a target protein is best done by modulating its activity, not its expression level, because this more closely mimics the effects of pharmacological intervention.
Here, we described an IKD method as a novel nongenetic approach for functional characterization and target validation in vivo. This method is based on raising an antibody-based immune response against a specific target, thus resulting in its inhibition. As such, this strategy is suitable for the in vivo functional validation of circulating proteins or proteins exposed on the extracellular surface. We applied IKD to the study of PCSK9, a potential target for the development of new LDL-c lowering agents (12, 39–42), and reported the effects of functional inhibition of endogenous circulating plasma PCSK9 on hepatic LDLR and plasma cholesterol levels.
We explored two immunization protocols in mice to assess the effects of both DNA and protein-based vaccination approaches in the inhibition of endogenous circulating PCSK9 and for the treatment of hyperlipidimia. Preliminary experiments revealed, as expected, that immunization with syn-DNA was ineffective in raising a detectable antibody response against the target mPCSK9 protein in mice. Moreover, analysis of the antibody titers raised by the xeno-protein and xeno-DNA immunization protocols revealed that although both approaches were effective in raising anti-mPCSK9 antibodies in mice, the titers and effects on LDL-c levels were higher when the recombinant hPCSK9 protein was used.
In the xeno-protein immunization protocol, the strong antibody response against hPCSK9 was accompanied by a 2-fold increase in the hepatic LDLR levels as revealed by Western blot comparisons of liver samples from immunized and control mice. Furthermore, a 40% decrease in total and plasma HDL-c and an approximately 60% decrease in LDL-c at day 14 were observed. The normal plasma LDL levels for BALB/c mice are generally very low but nevertheless the decrease in LDL-c concentration on hPCSK9 immunization is statistically significant and consistent with what was reported for PCSK9 KO mice (28) and in both nonhuman primates and humans injected with mAbs that interfere with the PCSK9/LDLR interaction (28, 31, 32, 34, 37). In addition, both the decrease in plasma lipoprotein levels and the increase in hepatic LDLR levels are similar to what was observed in PCSK9 KO mice and in mice treated with a neutralizing mAb against PCSK9 (16, 28, 31–33, 37). Of note, a similar decrease in HDL-c was previously reported for PCSK9 KO mice (28) and in mice injected with a neutralizing antibody against PCSK9 (33). Mouse HDL-c contains apo-E, which allows it to bind to LDLR possibly resulting in decreased plasma HDL-c levels upon increase in liver LDLR levels caused by antibody mediated PCSK9-inhibition (28, 33)
The decrease in LDL-c, increase in hepatic LDLR, and high anti-mPCSK9 titers were paralleled by a decrease in functionally active plasma mPCSK9. Using an ELISA that detects mPCSK9, we observed that 14 days after immunization, the concentration of detected circulating protein was decreased by 66% from 238 ng/ml, a value in the range of hPCSK9 observed in human plasma (50–600 ng/ml) (22), to 83 ng/ml. The ELISA uses an immobilized mAb that competes with the PCSK9/LDLR interaction, suggesting that a portion of the circulating mPCSK9 protein being detected might be still functional in LDLR binding and downregulation. One caveat of the ELISA is that plasma mPCSK9 molcules bound to antibodies that interfere with binding to the biotinylated anti-mPCSK9 Fab used in the development step are not detected and, as a result, the concentration of circulating and possibly functionally active plasma mPCSK9 protein in the immunized mice might be slightly underestimated. Taken together, these results suggest that it might not be necessary to completely inhibit circulating PCSK9 to have a robust KO-like effect on hepatic LDLR and plasma LDL-c levels.
In our experiment, we broke tolerance in mice using the human antigen. Similarly, antigens from different species might be effectively used. Of note, we and others have reported that just one amino-acid difference between the immunizing antigen and the endogenous mouse protein can suffice to elicit antibodies that cross-react with the mouse protein and remove it from the circulation (43, 44). Although we did not directly assess the anti-PCSK9 T-cell response in these mice, intact liver histology and lack of lymphocyte infiltration suggest that the main mechanism of action is not destruction of PCSK9 expressing cells but rather is antibody-mediated protein neutralization and possibly clearance. This mechanism is supported by the higher potency of protein versus DNA immunization as the first approach is known to induce mainly a B-cell response.
Several therapeutic strategies are being evaluated to inhibit PCSK9 in vivo (12, 31, 37, 40–42). The results presented here suggest that the IKD approach provides an effective method to transiently inhibit the activity of a desired gene product in vivo, thus mimicking the phenotype that might be achieved pharmacologically and enabling the rapid validation of a new target protein. Finally, we want to highlight the potential offered by the IKD strategy in establishing an active immunotherapy. This objective clearly requires a more detailed characterization of the system (including safety, specificity, efficiency, etc.), which is beyond the scope of this work. However, the data we report here suggest that the anti-self immune-response could be tamed and might be used as therapeutic tool. Such an approach would likely require fewer injections and would be less costly than a passive immunotherapy potentially allowing for a broader use.
Footnotes
Abbreviations:
- AP
- alkaline phosphatase
- CMV
- human cytomegalovirus
- ES
- electrical stimulation
- KO
- knockout
- IHC
- immune-histochemistry
- IKD
- immunological knockdown
- LDL-c
- low density lipoprotein cholesterol
- LDLR
- LDL receptor
- mAb
- monoclonal antibody
- PBST
- 0.05% Tween-20 in PBS
- RT
- room temperature
- siRNA
- small interfering RNA
All contributions to the submitted manuscript were generated while the authors were Merck Research Laboratories’ employees. Merck Research Laboratories provided complete research support.
REFERENCES
- 1.DiMasi J. A., Hansen R. W., Grabowski H. G. 2003. The price of innovation: new estimates of drug development costs. J. Health Econ. 22: 151–185 [DOI] [PubMed] [Google Scholar]
- 2.Schäfer S., Kolkhof P. 2008. Failure is an option: learning from unsuccessful proof-of-concept trials. Drug Discov. Today. 13: 913–916 [DOI] [PubMed] [Google Scholar]
- 3.Giacomini K. M., Krauss R. M., Roden D. M., Eichelbaum M., Hayden M. R., Nakamura Y. 2007. When good drugs go bad. Nature. 446: 975–977 [DOI] [PubMed] [Google Scholar]
- 4.Persidis A. 2000. Data mining in biotechnology. Nat. Biotechnol. 18: 237–238 [DOI] [PubMed] [Google Scholar]
- 5.Butcher E. C., Berg E. L., Kunkel E. J. 2004. Systems biology in drug discovery. Nat. Biotechnol. 22: 1253–1259 [DOI] [PubMed] [Google Scholar]
- 6.Xia X. G., Zhou H., Xu Z. 2006. Transgenic RNAi: accelerating and expanding reverse genetics in mammals. Transgenic Res. 15: 271–275 [DOI] [PubMed] [Google Scholar]
- 7.Lindsay M. A. 2003. Target discovery. Nat. Rev. Drug Discov. 2: 831–838 [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee-Kishore M., Miller C. P. 2005. Exploring the sounds of silence: RNAi-mediated gene silencing for target identification and validation. Drug Discov. Today. 10: 1559–1565 [DOI] [PubMed] [Google Scholar]
- 9.Lloyd-Jones D., Adams R., Carnethon M., De Simone G., Ferguson T. B., Flegal K., Ford E., Furie K., Go A., Greenlund K., et al. 2009. Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 119: 480–486 [DOI] [PubMed] [Google Scholar]
- 10.Horton J. D., Cohen J. C., Hobbs H. H. 2009. PCSK9: a convertase that coordinates LDL catabolism. J. Lipid Res. 50: S172–S177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton J. D., Cohen J. C., Hobbs H. H. 2007. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem. Sci. 32: 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seidah N. G. 2009. PCSK9 as a therapeutic target of dyslipidemia. Expert Opin. Ther. Targets. 13: 19–28 [DOI] [PubMed] [Google Scholar]
- 13.Costet P., Krempf M., Cariou B. 2008. PCSK9 and LDL cholesterol: unravelling the target to design the bullet. Trends Biochem. Sci. 33: 426–434 [DOI] [PubMed] [Google Scholar]
- 14.Henrich S., Lindberg I., Bode W., Than M. E. 2005. Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J. Mol. Biol. 345: 211–227 [DOI] [PubMed] [Google Scholar]
- 15.Seidah N. G., Prat A. 2007. The proprotein convertases are potential targets in the treatment of dyslipidemia. J. Mol. Med. 85: 685–696 [DOI] [PubMed] [Google Scholar]
- 16.Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., et al. 2008. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 48: 646–654 [DOI] [PubMed] [Google Scholar]
- 17.Piper D. E., Jackson S., Liu Q., Romanow W. G., Shetterly S., Thibault S. T., Shan B., Walker N. P. 2007. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. 15: 545–552 [DOI] [PubMed] [Google Scholar]
- 18.Hampton E. N., Knuth M. W., Li J., Harris J. L., Lesley S. A., Spraggon G. 2007. The self-inhibited structure of full-length PCSK9 at 1.9 Å reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. USA. 104: 14604–14609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., et al. 2007. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14: 413–419 [DOI] [PubMed] [Google Scholar]
- 20.Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., et al. 2004. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279: 48865–48875 [DOI] [PubMed] [Google Scholar]
- 21.Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. 2003. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA. 100: 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagace T. A., Curtis D. E., Garuti R., McNutt M. C., Park S. W., Prather H. B., Anderson N. N., Ho Y. K., Hammer R. E., Horton J. D. 2006. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Invest. 116: 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cameron J., Holla O. L., Ranheim T., Kulseth M. A., Berge K. E., Leren T. P. 2006. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum. Mol. Genet. 15: 1551–1558 [DOI] [PubMed] [Google Scholar]
- 24.Fisher T. S., Lo Surdo P., Pandit S., Mattu M., Santoro J. C., Wisniewski D., Cummings R. T., Calzetta A., Cubbon R. M., Fischer P. A., et al. 2007. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J. Biol. Chem. 282: 20502–20512 [DOI] [PubMed] [Google Scholar]
- 25.Zhang D. W., Garuti R., Tang W. J., Cohen J. C., Hobbs H. H. 2008. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. USA. 105: 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. 2008. Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. USA. 105: 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bottomley M. J., Cirillo A., Orsatti L., Ruggeri L., Fisher T. S., Santoro J. C., Cummings R. T., Cubbon R. M., Lo Surdo P., Calzetta A., et al. 2009. Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 284: 1313–1323 [DOI] [PubMed] [Google Scholar]
- 28.Rashid S., Curtis D. E., Garuti R., Anderson N. N., Bashmakov Y., Ho Y. K., Hammer R. E., Moon Y. A., Horton J. D. 2005. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking PCSK9. Proc. Natl. Acad. Sci. USA. 102: 5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grefhorst A., McNutt M. C., Lagace T. A., Horton J. D. 2008. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J. Lipid Res. 49: 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt R. J., Beyer T. P., Bensch W. R., Qian Y. W., Lin A., Kowala M., Alborn W. E., Konrad R. J., Cao G. 2008. Secreted proprotein convertase subtilisin/kexin type 9 reduces both hepatic and extrahepatic low-density lipoprotein receptors in vivo. Biochem. Biophys. Res. Commun. 370: 634–640 [DOI] [PubMed] [Google Scholar]
- 31.Chan J. C., Piper D. E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z., et al. 2009. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA. 106: 9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang H., Chaparro-Riggers J., Strop P., Geng T., Sutton J. E., Tsai D., Bai L., Abdiche Y., Dilley J., Yu J., et al. 2012. Proprotein convertase substilisin/kexin type 9 antagonism reduces low density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J. Pharmacol. Exp. Ther. 340: 228–236 [DOI] [PubMed] [Google Scholar]
- 33.Chaparro-Riggers J., Liang H., Devay R. M., Bai L., Sutton J. E., Chen W., Geng T., Lindquist K., Galindo Casas M., Boustany L. M., et al. 2012. Increasing serum half-life and extending cholesteroil lowering in vivo by engineering an antibody with pH sensitive binding to PCSK9. J. Biol. Chem. 287: 11090–11097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein E. A., Mellis S., Yancopoulos G. D., Stahl N., Logan D., Smith W. B., Lisbon E., Gutierrez M., Webb C., Wu R., et al. 2012. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 366: 1108–1118 [DOI] [PubMed] [Google Scholar]
- 35.Montgomery D. L., Shiver J. W., Leander K. R., Perry H. C., Friedman A., Martinez D., Ulmer J. B., Donnelly J. J., Liu M. A. 1993. Heterologous and homologous protection against influenza A by DNA vaccination: optimization of DNA vectors. DNA Cell Biol. 12: 777–783 [DOI] [PubMed] [Google Scholar]
- 36.Krieg A. M., Yi A. K., Schorr J., Davis H. L. 1998. The role of CpG dinucleotides in DNA vaccines. Trends Microbiol. 6: 23–27 [DOI] [PubMed] [Google Scholar]
- 37.Ni Y. G., Di Marco S., Condra J. H., Peterson L. B., Wang W., Wang F., Pandit S., Hammond H. A., Rosa R., Wood D. D., et al. 2011. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9)-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces free circulating PCSK9 and LDL-cholesterol in mice and rhesus monkeys. J. Lipid Res. 52: 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitehead K. A., Langer R., Anderson D. G. 2009. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 8: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lilly S. M., Rader D. J. 2007. New targets and emerging therapies for reducing LDL cholesterol. Curr. Opin. Lipidol. 18: 650–655 [DOI] [PubMed] [Google Scholar]
- 40.Lopez D. 2008. Inhibition of PCSK9 as a novel strategy for the treatment of hypercholesterolemia. Drug News Perspect. 21: 323–330 [DOI] [PubMed] [Google Scholar]
- 41.Lambert G., Krempf M., Costet P. 2006. PCSK9: a promising therapeutic target for dyslipidemias? Trends Endocrinol. Metab. 17: 79–81 [DOI] [PubMed] [Google Scholar]
- 42.Frank-Kamenetsky M., Grefhorst A., Anderson N. N., Racie T. S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., et al. 2008. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA. 105: 11915–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maione D., Wiznerowicz M., Delmastro P., Cortese R., Ciliberto G., La Monica N., Savino R. 2000. Prolonged expression and effective readministration of erythropoietin delivered with a fully deleted adenoviral vector. Hum. Gene Ther. 11: 859–868 [DOI] [PubMed] [Google Scholar]
- 44.De Benedetti F., Pignatti P., Vivarelli M., Meazza C., Ciliberto G., Savino R., Martini A. 2001. In vivo neutralization of human IL-6 (hIL-6) achieved by immunization of hIL-6-transgenic mice with a hIL-6 receptor antagonist. J. Immunol. 166: 4334–4340 [DOI] [PubMed] [Google Scholar]