

Abstract
Adipocytes and fat cells play critical roles in the regulation of energy homeostasis. Adipogenesis (adipocyte differentiation) is regulated via a complex process including coordinated changes in hormone sensitivity and gene expression. PPARγ is a ligand-dependent transcription factor and important in adipogenesis, as it enhances the expression of numerous adipogenic and lipogenic genes in adipocytes. Prostaglandins (PGs), which are lipid mediators, are associated with the regulation of PPARγ function in adipocytes. Prostacyclin promotes the differentiation of adipocyte-precursor cells to adipose cells via activation of the expression of C/EBPβ and δ. These proteins are important transcription factors in the activation of the early phase of adipogenesis, and they activate the expression of PPARγ, which event precedes the maturation of adipocytes. PGE2 and PGF2α strongly suppress the early phase of adipocyte differentiation by enhancing their own production via receptor-mediated elevation of the expression of cycloxygenase-2, and they also suppress the function of PPARγ. In contrast, PGD2 and its non-enzymatic metabolite, Δ12-PGJ2, activate the middle-late phase of adipocyte differentiation through both DP2 receptors and PPARγ. This paper focuses on potential roles of PGs as PPARγ modulators in adipogenesis and regulators of obesity.
1. Introduction
Obesity is a major health concern worldwide [1] and is associated with the development of a number of pathological disorders such as type 2 diabetes, hypertension, and cardiovascular disease [2–4]. Excess adipose tissue can be the consequence of both an increased number (hyperplasia) and an enlarged size (hypertrophy) of adipose cells. A major role of adipocytes is to store large amounts of triglycerides during periods of energy excess and to mobilize these depots during periods of nutritional deprivation [2–4]. Moreover, adipocytes are highly specialized cells that secrete various adipocytokines, whose release largely reflects the amounts of stored triglyceride [2, 5–8].
The regulation of adipocyte differentiation (adipogenesis) is complex and this process includes alteration of the sensitivity to hormones and the expression of a number of genes in response to various stimuli including lipid mediators. Peroxisome proliferator-activated receptor (PPAR) γ and CCAAT/enhancer-binding proteins (C/EBPs) are the most important transcription factors involved in the activation of adipogenesis, and they induce the expression of a number of adipogenic and lipogenic genes that participate in the control of adipogenesis [9, 10].
PPARs are members of the nuclear receptor superfamily and play critical roles in the regulation of storage and catabolism of lipids [11, 12]. To date, three types of PPAR subtypes have been identified, that is, PPARα, PPARβ/δ, and PPARγ [11, 12]. PPARs increase the expression of a variety of genes in various cells through heterodimerization with retinoic acid receptors or retinoid X receptors in a ligand-dependent manner [12–16]. Among them, PPARγ is expressed predominantly in adipose tissue and macrophages, is closely related to the regulation of lipid and glucose metabolisms, and is associated with the control of obesity and related diseases [11, 12]. Until now, many natural and synthetic ligands for PPARγ have been identified [17–19]. 15-Deoxy-Δ12,14-prostaglandin (PG) J2 (15d-PGJ2) was the first identified endogenous ligand for PPARγ, and it activates adipogenesis in cultured cells [20, 21]. Moreover, fatty acids such as lauric acid (C12:0) and petroselinic acid (C18:1) of the saturated fatty acids [22], linolenic acid (C18:3), eicosapentaenoic acid (C20:5), and docosahexaenoic acid (C22:6) of the ω3 (n-3) family [23], arachidonic acid of the ω6 (n-6) family [22, 23], and very-long chain fatty acids [24] were later identified as other endogenous PPARγ ligands that activate PPARγ functions. In addition, 9-hydroxy and 13-hydroxy octadecadienoic acids (HODE), the components of oxidized low-density lipoprotein (ox-LDL), were also identified as endogenous ligands for PPARγ [25, 26]. However, whether these natural molecules can function as physiological ligands of PPARγ in vivo remains unknown. In addition to natural ligands, many synthetic ligands have been identified. For example, thiazolidinediones (TZDs) such as Troglitazone, Rosiglitazone, Ciglitazone, and Pioglitazone are used for the treatment of type 2 diabetes mellitus; and these ligands affect insulin resistance and glucose homeostasis by activating PPARγ functions [12, 18]. However, these TZDs increase hepatic toxicity and cardiovascular risk. Finally, Troglitazone was withdrawn from the market [27]. It is still unknown whether the toxicities associated with TZDs are derived from the binding with PPARγ.
PGs are lipid mediators that play a number of physiological roles in a variety of cells. PGs are synthesized through the following three enzymatic steps (Figure 1). First, arachidonic acid is liberated from the membrane phospholipids by the action of cytosolic phospholipase A2 (cPLA2) [28]. Second, arachidonic acid is converted to PGH2, which is a common precursor of all prostanoids, by either cyclooxygenase- (COX-) 1 or COX-2 [29]. The activity of these enzymes is critical to determine the production rate of PGs. Third, PGH2 is metabolized to various PGs, that is, PGD2, PGE2, PGF2α, prostacyclin (PGI2), and thromboxane A2 (TXA2), by the action of specific PG synthases [29]. PGs exert a wide range of actions through their binding to specific PG receptors that belong to the G protein-coupled receptors (GPCRs) gene family [30]. GPCRs span cell membranes via seven transmembrane-spanning segments and are the most important therapeutic targets. In this decade, the functions of PGs in the regulation of adipogenesis have been extensively investigated. Elucidation of the molecular mechanisms underlying adipogenesis may provide strategies for reducing the prevalence of obesity. This paper focuses on the recent advances in our understanding of the function of PGs as modulators of PPARγ in the regulation of adipogenesis.
Figure 1.
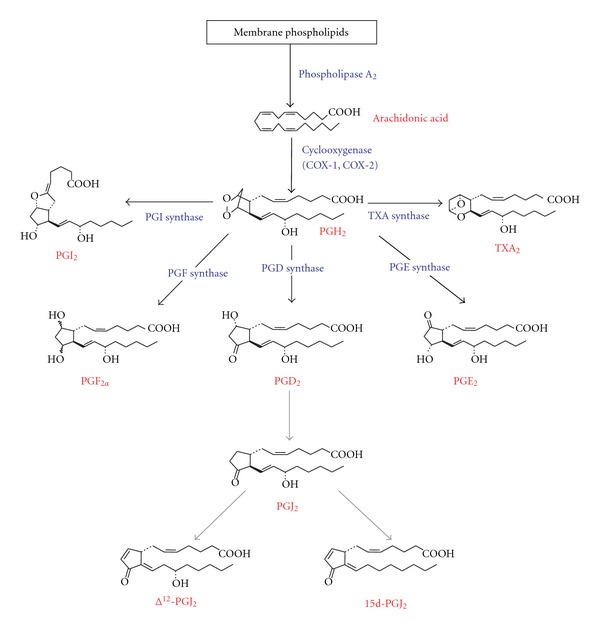
Biosynthetic pathway of prostaglandins. PGJ2, Δ12-PGJ2, and 15d-PGJ2 are converted from PGD2 by nonenzymatic dehydrations.
2. Roles of COXs in Adipocytes
COX consists of two isozymes, COX-1 and COX-2, and is the rate-limiting enzyme in the PG biosynthesis [29]. COX-1 is constitutively expressed in most cells including adipocytes, whereas COX-2 expression is induced by various stimuli [29] and transiently activated in the early phase of adipogenesis, followed by lowered expression during adipogenesis [31]. There have been a number of reports regarding the contribution of COX isozymes to the regulation of adipocyte differentiation. However, the roles that COX-2 plays during adipogenesis are still controversial.
In cell-based studies, Yan et al. demonstrated that inhibition of COX activities by their selective inhibitors, for example, SC-560 for COX-1, and NS-398 and Celecoxib for COX-2, enhances adipocyte differentiation via an increase in the mRNA levels of PPARγ and C/EBPα. Thus, both COX-1 and COX-2 participate in the regulation of adipogenesis [32]. Moreover, in 3T3-L1 cells stably expressing COX-2 in the antisense direction, lipid accumulation is enhanced during adipogenesis with elevated expression of adipogenic genes such as PPARγ and C/EBPα. In addition, this enhancement of lipid accumulation in antisense COX-2-expressing cells can be reversed by cotreatment with either antiadipogenic PGE2 or PGF2α [33].
In contrast, when 3T3-L1 cells are pretreated before the initiation of adipocyte differentiation or treated during the clonal expansion phase with SC-58236, a selective COX-2 inhibitor, and then caused to differentiate into adipocytes, lipid accumulation is reduced along with repressed expression of the adipogenic fatty acid-binding protein 4 (FABP4, also called aP2) gene [34]. In contrast, a selective COX-1 inhibitor, SC-58560 does not have any effect on adipogenesis. Additionally, when 3T3-L1 cells are caused to differentiate into adipocyte in a medium containing each of two selective COX-1 and COX-2 inhibitors that are added after the clonal expansion phase, adipogenesis is not affected. Thus, inhibition of COX-2 activity suppresses adipocyte differentiation by repressing the clonal expansion phase [34].
In in vivo studies, overexpression of COX-2 in white adipose tissue (WAT) increases de novo recruitment of brown adipose tissue (BAT) and energy expenditure, while suppressing the high fat diet-induced gain in body weight [35]. Also, Ghoshal et al. reported that in COX-2 gene-knock-out mice, their total body weight is significantly lower than that of wild-type mice, along with reduced expression of adipogenic genes such as those of PPARγ and lipoprotein lipase [36]. In addition, PGD2 and 15d-PGJ2 levels in cells prepared from adipose tissues of COX-2 gene-knock-out mice and placed in primary culture are reduced as compared with those in wild-type mice [36]. Thus, further studies are needed to elucidate the precise functions of COXs in the regulation of adipogenesis.
3. Repression of the Early Phase of Adipogenesis by PGF2 α
PGF2α and PGE2 suppress the differentiation of adipocytes and exert their functions as antiadipogenic agents exert by acting through their specific FP [37–41] and EP4 [42, 43] receptors, respectively.
PGF2α is synthesized by a variety of PGF synthase (PGFS) activity-carrying enzymes [44], for example, aldoketo reductase (AKR) 1B3 [45], AKR1B7 [46], and prostamide/PGFS [47] in mice. In humans, AKR1C3 acts as a PGFS in adipocytes and is associated with the suppression of adipogenesis through inhibition of PPARγ function [48]. Although PGFS has never been identified in adipocytes, we and another group identified AKR1B3 [31] and AKR1B7 [49] as being PGFSs in adipocytes.
AKR1B3-produced PGF2α is detected in preadipocytes and its level is enhanced with a peak at 3 h after the initiation of adipogenesis and then decreases [50], indicating that PGF2α suppresses an early phase of adipogenesis. Fluprostenol, an FP receptor agonist, clearly reduces the expression of PPARγ and its target genes [31, 50]. Moreover, this Fluprostenol-mediated suppression of the gene expression is cleared by cotreatment with AL8810, an FP receptor antagonist, indicating that PGF2α inhibits adipocyte differentiation of 3T3-L1 cells by acting through an FP receptor.
AKR1B7 gene-knock-out mice display excessive adiposity resulting from adipocyte hyperplasia/hypertrophy and exhibit high sensitivity to diet-induced obesity. Treatment of 3T3-L1 cells or AKR1B7 gene-knock-out mice with Cloprostenol, an FP receptor agonist, decreases adipocyte size and inhibits the expression of lipogenic genes [49].
The precise molecular mechanism of PGF2α-mediated suppression of adipogenesis has been investigated. PGF2α represses the function of PPARγ by causing its phosphorylation via FP receptors [50]. In addition, Fluprostenol enhances the expression of COX-2 via activation of the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) 1/2 pathway. Moreover, promoter-luciferase and chromatin immunoprecipitation assays demonstrated that PGF2α-derived COX-2 expression is activated by the binding of cAMP-responsive element binding protein (CREB) to the promoter region of the COX-2 gene in 3T3-L1 cells [50]. Thus, the MEK/ERK-CREB cascade forms a positive feedback loop, one that probably plays a critical role in the suppression of the early phase of adipogenesis by elevating the de novo production of antiadipogenic PGF2α.
4. Suppression of the Early Phase of Adipogenesis by PGE2
PGE2 is also known to suppress adipogenesis through suppression of PPARγ function. PGE2 and an EP4 agonist, AE1-329, increase the intracellular cAMP levels in preadipocytes in a dose-dependent manner [42]. Moreover, AE1-329 decreases the expression of adipogenic genes such as PPARγ and C/EBPα [51]. The inhibitory effect of PGE2, but not that of Fluprostenol, is reversed by the addition of an EP4 antagonist, AE3-208 [42], indicating that PGE2 suppresses adipogenesis through the EP4 receptor. Although the functions of PGE2 and the expression of the functions of PGE2 and the expression of PGESs have been investigated in adipocytes [27, 52, 53], the PGE2-producing enzyme in adipocytes has never been identified. To date, three major PGESs have been identified [54, 55]. Microsomal PGES-1 (mPGES-1) is a member of the membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG) protein family [56] and produces PGE2 in response to various stimuli [57]. Microsomal PGES-2 (mPGES-2) has also been identified and its expression is high in the heart and brain [58]. Cytosolic PGES (cPGES) is constitutively and ubiquitously expressed in various cells [59].
PGE2 production is detected in preadipocytes and increases during the early phase of adipogenesis with a peak at 3 h after the initiation of adipogenesis; and mPGES-1 is expressed in these cells, with its mRNA and protein levels being consistently detected during adipogenesis. Finally, we found that mPGES-1 is responsible for the production of PGE2 in adipocytes [60]. This result is consistent with results showing that treatment of mouse embryonic fibroblast (MEF) cells with PGE2 for the first two days of adipocyte differentiation is enough to suppress adipocyte differentiation, with reduced expression of the PPARγ2 gene and reduced accumulation of intracellular lipids [43].
In wild-type mouse MEF cells, inhibition of endogenous PG synthesis by indomethacin enhances adipocyte differentiation, and this enhancement is reversed by the addition of PGE2. In MEF cells prepared from EP4 receptor gene-knock-out mice, adipocyte differentiation is elevated, and no more enhancement of adipocyte differentiation is observed following treatment with indomethacin. Thus, PGE2-EP4 receptor signaling suppresses the early phase of adipocyte differentiation in MEF cells [43].
5. Synergistic Suppression of Early Phase of Adipogenesis by PGF2 α and PGE2
Both PGF2α and PGE2 suppress the early phase of adipogenesis, and so we investigated the synergistic regulation of these PGs in 3T3-L1 cells. The increased production of PGF2α and PGE2 in the early phase of adipogenesis is a consequence of the elevated expression of the COX-2 gene [61]. PGF2α forms a positive feedback loop that coordinately suppresses the early phase of adipogenesis through the increased production of antiadipogenic PGF2α and PGE2, both of which inhibit PPARγ function. In addition, PGE2 also enhances the production of PGF2α and itself through the elevation of the expression of the COX-2 gene in an EP4 receptor-mediated fashion. Moreover, when the cells are caused to differentiate into adipocytes in medium containing both PGF2α and PGE2, the expression of the adipogenic genes is decreased to a greater extent than when the cells are cultured in a medium containing either of them. Thus, PGE2 and PGF2α synergistically suppress the early phase of adipogenesis through a self-amplifying loop, triggered by PGF2α-FP receptor and PGE2-EP4 receptor couplings and activation of the COX-2 gene expression in 3T3-L1 cells [61].
However, Inazumi et al. demonstrated that the differentiation of MEF cells prepared from FP receptor gene-knock-out mice is almost the same as that in these cells from wild-type mice and still shows sensitivity to indomethacin, indicating that FP receptor-mediated suppression is not directly associated with the regulation of adipocyte differentiation in MEF cells [43]. Therefore, the regulation of suppression of adipogenesis by PGE2 and PGF2α might occur in a cell-type-dependent manner.
6. Acceleration of Adipocyte Differentiation by PGD2 and Its Metabolites
PGD2 acts as an allergic and inflammatory mediators and is produced in a variety of cells such as mast cells, macrophages, and adipose cells [62, 63]. PGD2 is produced from PGH2 by the action of PGD synthases (PGDSs), enzymes that catalyze the isomerization of the 9,11-endoperoxide group of PGH2 to PGD2. Two distinct types of PGDSs have been identified. One is hematopoietic PGDS (H-PGDS), which is abundantly expressed in mast cells and Th2 cells [64]. The other is L-PGDS, which is detected abundantly in the brain, male genital organs, and heart [62, 63].
PGD2 has been considered a candidate for a molecule that acts as an endogenous inducer of adipogenesis, basically because 15d-PGJ2, one of its metabolites, has been identified as a ligand for PPARγ and activates adipogenesis in vitro [20, 21]. PGD2 is nonenzymatically metabolized to PGs of the J series, that is, PGJ2, Δ12-PGJ2, and 15d-PGJ2. However, the concentrations of 15d-PGJ2 required for the activation of PPARγ reported in most of the literature are much higher (2.5–100 μmol/L) than those of conventional PGs (pmol/L range); and 15d-PGJ2 is present in vivo at a low level that is insufficient for activation of adipocyte differentiation [65], whose finding is consistent with our current results indicating that 15d-PGJ2 is not detectable in adipocytes [60]. Recently, we identified Δ12-PGJ2 as being the dominant PGD2 metabolite in differentiated adipocytes [60], in good agreement with recent results showing that Δ12-PGJ2 is produced in adipocytes and activates the expression of adipogenic genes in 3T3-L1 cells [66].
PGD2 is synthesized by the action of L-PGDS in adipocytes [67]. However, another PGDS, H-PGDS may not be involved in the production of PGD2 in adipocytes, because the expression level of H-PGDS is very low during adipogenesis. Although the function of PGD2 or L-PGDS in vitro has been extensively investigated, the in vivo function is still controversial. Ragolia et al. reported that adipose size is increased in L-PGDS gene-knock-out mice under normal and high-fat diet feeding. Moreover, L-PGDS gene-knock-out mice become glucose intolerant and insulinresistant. Also the serum adiponectin level is decreased in such mice [68]. Adipocytes isolated from L-PGDS gene-knock-out mice are significantly less sensitive to insulin-stimulated glucose transport. Thus, L-PGDS is an important mediator of muscle and adipose glucose transport which is modulated by glycemic conditions and plays a significant role in the glucose intolerance associated with type 2 diabetes [69]. Furthermore, Tanaka et al. showed that L-PGDS gene-knock-out mice have a significantly increased body weight when fed high-fat diet and the size of adipocytes in the subcutaneous and visceral fat tissues is significantly enlarged [70].
In contrast, Fujitani et al. demonstrated that transgenic mice overexpressing human H-PGDS, which produce plenty of PGD2 in every tissue including adipose, become obese under high-fat diet feeding but that obesity is not observed under normal diet feeding [71]. Serum leptin, insulin, and adiponectin levels are increased in these PGD2-overproducing mice. Moreover, their triglyceride level is decreased by about 50% as compared with that in WT mice. Moreover, the PGD2-overproducing mice show increased insulin sensitivity [71]. Furthermore, the epididymal adipose tissue mass of COX-2 gene-knock-out mice is decreased. PGD2 and the levels of PGD2 metabolites are also decreased in the adipose tissue of these mice. Thus, reduced adiposity in COX-2 gene-knock-out mice results from the inhibition of the production of PGD2 and its metabolites required for PPARγ activation [36]. This discrepancy may be derived from a variety of physiological functions of PGD2 in the body. Therefore, the adipocyte-specific function of PGD2 and/or L-PGDS in the regulation of obesity should be further clarified.
7. Activation of Adipogenesis in Adipose-Precursor Cells by PGI2
PGI2 activates the protein kinase A (PKA) pathway by binding to its IP receptor and enhances the differentiation of adipose precursor cells [72, 73]. The activation of IP receptors upregulates the expression of C/EBPβ and C/EBPδ, both of which are critical for the progression of the early phase of adipogenesis and directly activate the expression of the PPARγ and C/EBPα genes for maturation of adipocytes [9, 10]. Moreover, IP receptor gene-knock-out mice fed a high-fat diet do not show any changes in body weight, fat mass, or adipose size [74, 75]. Therefore, PGI2 activates the progression of adipogenesis in the adipose precursor cells through the enhancement of the expression C/EBPβ and C/EBPδ via the cAMP-PKA pathway.
8. Conclusion
PGs are involved in the regulation of adipogenesis and act as modulators of PPARγ functions. The regulation of adipogenesis by PGs is very complex, because PGs regulate adipogenesis both positively and negatively. In the early phase of adipogenesis, PGF2α and PGE2 suppress the progression of adipogenesis, and their receptor-mediated mechanisms leading to suppressed PPARγ function have been well elucidated. In contrast, PGD2 and its metabolites activate the middle-late phase of adipogenesis (Figure 2). In addition, recently we found that PGD2 and its metabolite Δ12-PGJ2 accelerate adipogenesis by acting through DP2 (CRTH2; chemoattractant receptor homologous molecule of Th2 cells) receptors and PPARγ, thus, indicating that when elucidating the function of a given PG, the roles of not only it but also those of its metabolites should be considered.
Figure 2.
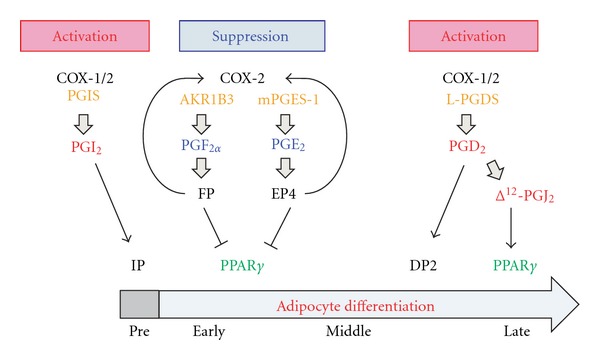
Regulation of adipogenesis by prostaglandins. “Pre” indicates adipocyte precursor cells. “Early,” “Middle,” and “Late” mean early, middle, and late phases of adipogenesis, respectively.
All PGs function through their specific G protein-coupled receptors and PPARγ. Although their receptor agonists and antagonists are functional in the cultured adipocytes (in vitro), in vivo studies do not show clear effects of PGs in the regulation of obesity. Moreover, PG receptor gene-knock-out mice are not affected like the cells observed in in vitro studies. The explanation of the problems is quite difficult. As PGs have a variety of physiological functions, studies using gene-knock-out mice might not be appropriate to elucidate the functions of PGs in obesity. The precise in vivo functions of PGs especially those of 15d-PGJ2 required further clarification. Tissue- (cell-)specific gene-knock-out mice might be a powerful tool to identify the in vivo function of PGs. Understanding of the mechanisms of PG-mediated regulation of adipogenesis may lead to a novel therapeutic strategy for the treatment of obesity.
Acknowledgments
The author's studies cited in this paper were supported in part by Grants from the programs of theGrant-in-Aid for Scientific Research and Scientific Research on Innovative Areas of the Ministry of Education, Culture, Sports, Science and Technology of Japan and Technology of Japan (MEXT), and Research for Promoting Technological Seeds of Japan Science and Technology Agency (JST) and by Grants from the Suzuken Memorial Foundation, the Sumitomo Foundation, the Japanese Biochemical Society, the Japan Foundation for Applied Enzymology, the Takeda Science Foundation, the Naito Foundation, the Research Foundation for Pharmaceutical Sciences, and the Daiwa Securities Health Foundation.
References
- 1.Friedman JM. Modern science versus the stigma of obesity. Nature Medicine. 2004;10(6):563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- 2.Attie AD, Scherer PE. Adipocyte metabolism and obesity. Journal of Lipid Research. 2009;50:S395–S399. doi: 10.1194/jlr.R800057-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornier MA, Dabelea D, Hernandez TL, et al. The metabolic syndrome. Endocrine Reviews. 2008;29(7):777–822. doi: 10.1210/er.2008-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pi-Sunyer X. The medical risks of obesity. Postgraduate Medicine. 2009;121(6):21–33. doi: 10.3810/pgm.2009.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Molecular and Cellular Endocrinology. 2010;316(2):129–139. doi: 10.1016/j.mce.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. Journal of Clinical Endocrinology and Metabolism. 2008;93(11):s64–s73. doi: 10.1210/jc.2008-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes and Metabolism. 2008;34(1):2–11. doi: 10.1016/j.diabet.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Current Pharmaceutical Design. 2010;16(17):1896–1901. doi: 10.2174/138161210791208893. [DOI] [PubMed] [Google Scholar]
- 9.Lefterova MI, Lazar MA. New developments in adipogenesis. Trends in Endocrinology and Metabolism. 2009;20(3):107–114. doi: 10.1016/j.tem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Rosen E, Eguchi J, Xu Z. Transcriptional targets in adipocyte biology. Expert Opinion on Therapeutic Targets. 2009;13(8):975–986. doi: 10.1517/14728220903039706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anghel SI, Wahli W. Fat poetry: a kingdom for PPARγ . Cell Research. 2007;17(6):486–511. doi: 10.1038/cr.2007.48. [DOI] [PubMed] [Google Scholar]
- 12.Christodoulides C, Vidal-Puig A. PPARs and adipocyte function. Molecular and Cellular Endocrinology. 2010;318(1-2):61–68. doi: 10.1016/j.mce.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 13.White UA, Stephens JM. Transcriptional factors that promote formation of white adipose tissue. Molecular and Cellular Endocrinology. 2010;318(1-2):10–14. doi: 10.1016/j.mce.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siersbæk R, Nielsen R, Mandrup S. PPARγ in adipocyte differentiation and metabolism—novel insights from genome-wide studies. FEBS Letters. 2010;584(15):3242–3249. doi: 10.1016/j.febslet.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ . Annual Review of Biochemistry. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 16.Sharma AM, Staels B. Review: peroxisome proliferator-activated receptor γ and adipose tissue—understanding obesity-related changes in regulation of lipid and glucose metabolism. Journal of Clinical Endocrinology and Metabolism. 2007;92(2):386–395. doi: 10.1210/jc.2006-1268. [DOI] [PubMed] [Google Scholar]
- 17.Tabe Y, Konopleva M, Andreeff M, Ohsaka A. Effects of PPARγ ligands on leukemia. PPAR Research. 2012;2012:8 pages. doi: 10.1155/2012/483656.483656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends in Endocrinology and Metabolism. 2012;23(5):205–215. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Penumetcha M, Santanam N. Nutraceuticals as ligands of PPARγ . PPAR Research. 2012;2012:7 pages. doi: 10.1155/2012/858352.858352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ . Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 21.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 22.Kliewer SA, Sundseth SS, Jones SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ . Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krey G, Braissant O, L’Horset F, et al. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Molecular Endocrinology. 1997;11(6):779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi T, Fujimori K. Very long chain-fatty acids enhance adipogenesis through co-regulation of Elovl3 and PPARγ in 3T3-L1 cells. American Journal of Physiology, Endocrinology and Metabolism. 2012;302(12):E1461–E1471. doi: 10.1152/ajpendo.00623.2011. [DOI] [PubMed] [Google Scholar]
- 25.Nagy L, Tontonoz P, Alvarez JGA, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ . Cell. 1998;93(2):229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 26.Tontonoz P, Nagy L, Alvarez JGA, Thomazy VA, Evans RM. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 27.Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR) Toxicological Sciences. 2006;90(2):269–295. doi: 10.1093/toxsci/kfj062. [DOI] [PubMed] [Google Scholar]
- 28.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Progress in Lipid Research. 2011;50(2):152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Smith WL, Urade Y, Jakobsson PJ. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chemical Reviews. 2011;111(10):5821–5865. doi: 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narumiya S, Furuyashiki T. Fever, inflammation, pain and beyond: prostanoid receptor research during these 25 years. FASEB Journal. 2011;25(3):813–818. doi: 10.1096/fj.11-0302ufm. [DOI] [PubMed] [Google Scholar]
- 31.Fujimori K, Ueno T, Nagata N, et al. Suppression of adipocyte differentiation by aldo-keto reductase 1B3 acting as prostaglandin F2α synthase. Journal of Biological Chemistry. 2010;285(12):8880–8886. doi: 10.1074/jbc.M109.077164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan H, Kermouni A, Abdel-Hafez M, Lau DCW. Role of cyclooxygenases COX-1 and COX-2 in modulating adipogenesis in 3T3-L1 cells. Journal of Lipid Research. 2003;44(2):424–429. doi: 10.1194/jlr.M200357-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Chu X, Nishimura K, Jisaka M, Nagaya T, Shono F, Yokota K. Up-regulation of adipogenesis in adipocytes expressing stably cyclooxygenase-2 in the antisense direction. Prostaglandins and Other Lipid Mediators. 2010;91(1-2):1–9. doi: 10.1016/j.prostaglandins.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Fajas L, Miard S, Briggs MR, Auwerx J. Selective cyclo-oxygenase-2 inhibitors impair adipocyte differentiation through inhibition of the clonal expansion phase. Journal of Lipid Research. 2003;44(9):1652–1659. doi: 10.1194/jlr.M300248-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Vegiopoulos A, Müller-Decker K, Strzoda D, et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science. 2010;328(5982):1158–1161. doi: 10.1126/science.1186034. [DOI] [PubMed] [Google Scholar]
- 36.Ghoshal S, Trivedi DB, Graf GA, Loftin CD. Cyclooxygenase-2 deficiency attenuates adipose tissue differentiation and inflammation in mice. Journal of Biological Chemistry. 2011;286(1):889–898. doi: 10.1074/jbc.M110.139139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casimir DA, Miller CW, Ntambi JM. Preadipocyte differentiation blocked by prostaglandin stimulation of prostanoid FP2 receptor in murine 3T3-L1 cells. Differentiation. 1996;60(4):203–210. doi: 10.1046/j.1432-0436.1996.6040203.x. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Clipstone NA. Prostaglandin F2α inhibits adipocyte differentiation via a Gαq-calcium-calcineurin-dependent signaling pathway. Journal of Cellular Biochemistry. 2007;100(1):161–173. doi: 10.1002/jcb.21044. [DOI] [PubMed] [Google Scholar]
- 39.Miller CW, Casimir DA, Ntambi JM. The mechanism of inhibition of 3T3-L1 preadipocyte differentiation by prostaglandin F2α . Endocrinology. 1996;137(12):5641–5650. doi: 10.1210/endo.137.12.8940395. [DOI] [PubMed] [Google Scholar]
- 40.Serrero G, Lepak NM. Prostaglandin F2α receptor (FP receptor) agonists are potent adipose differentiation inhibitors for primary culture of adipocyte precursors in defined medium. Biochemical and Biophysical Research Communications. 1997;233(1):200–202. doi: 10.1006/bbrc.1997.6433. [DOI] [PubMed] [Google Scholar]
- 41.Reginato MJ, Krakow SL, Bailey ST, Lazar MA. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ . Journal of Biological Chemistry. 1998;273(4):1855–1858. doi: 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- 42.Tsuboi H, Sugimoto Y, Kainoh T, Ichikawa A. Prostanoid EP4 receptor is involved in suppression of 3T3-L1 adipocyte differentiation. Biochemical and Biophysical Research Communications. 2004;322(3):1066–1072. doi: 10.1016/j.bbrc.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 43.Inazumi T, Shirata N, Morimoto K, Takano H, Segi-Nishida E, Sugimoto Y. Prostaglandin E2-EP4 signaling suppresses adipocyte differentiation in mouse embryonic fibroblasts via an autocrine mechanism. Journal of Lipid Research. 2011;52(8):1500–1508. doi: 10.1194/jlr.M013615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe K. Recent reports about enzymes related to the synthesis of prostaglandin (PG) F2 (PGF2α and 9α, 11β-PGF2 . Journal of Biochemistry. 2011;150(6):593–596. doi: 10.1093/jb/mvr116. [DOI] [PubMed] [Google Scholar]
- 45.Kabututu Z, Manin M, Pointud JC, et al. Prostaglandin F2α synthase activities of aldo-keto reductase 1B1, 1B3 and 1B7. Journal of Biochemistry. 2009;145(2):161–168. doi: 10.1093/jb/mvn152. [DOI] [PubMed] [Google Scholar]
- 46.Tirard J, Gout J, Lefrançois-Martinez AM, Martinez A, Begeot M, Naville D. A novel inhibitory protein in adipose tissue, the aldo-keto reductase AKR1B7: its role in adipogenesis. Endocrinology. 2007;148(5):1996–2005. doi: 10.1210/en.2006-1707. [DOI] [PubMed] [Google Scholar]
- 47.Moriuchi H, Koda N, Okuda-Ashitaka E, et al. Molecular characterization of a novel type of prostamide/prostaglandin F synthase, belonging to the thioredoxin-like superfamily. Journal of Biological Chemistry. 2008;283(2):792–801. doi: 10.1074/jbc.M705638200. [DOI] [PubMed] [Google Scholar]
- 48.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. Journal of Steroid Biochemistry and Molecular Biology. 2011;125(1-2):95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volat FE, Pointud JC, Pastel E, et al. Depressed levels of prostaglandin F2α in mice lacking akr1b7 increase basal adiposity and predispose to diet-Induced obesity. Diabetes. 2012;61(11):2796–2806. doi: 10.2337/db11-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ueno T, Fujimori K. Novel suppression mechanism operating in early phase of adipogenesis by positive feedback loop for enhancement of cyclooxygenase-2 expression through prostaglandin F2α receptor mediated activation of MEK/ERK-CREB cascade. FEBS Journal. 2011;278(16):2901–2912. doi: 10.1111/j.1742-4658.2011.08213.x. [DOI] [PubMed] [Google Scholar]
- 51.Sugimoto Y, Tsuboi H, Okuno Y, et al. Microarray evaluation of EP4 receptor-mediated prostaglandin E2 suppression of 3T3-L1 adipocyte differentiation. Biochemical and Biophysical Research Communications. 2004;322(3):911–917. doi: 10.1016/j.bbrc.2004.07.194. [DOI] [PubMed] [Google Scholar]
- 52.Xie Y, Kang X, Ackerman WE, et al. Differentiation-dependent regulation of the cyclooxygenase cascade during adipogenesis suggests a complex role for prostaglandins. Diabetes, Obesity and Metabolism. 2006;8(1):83–93. doi: 10.1111/j.1463-1326.2005.00472.x. [DOI] [PubMed] [Google Scholar]
- 53.Hétu PO, Riendeau D. Down-regulation of microsomal prostaglandin E2 synthase-1 in adipose tissue by high-fat feeding. Obesity. 2007;15(1):60–68. doi: 10.1038/oby.2007.514. [DOI] [PubMed] [Google Scholar]
- 54.Hara S, Kamei D, Sasaki Y, Tanemoto A, Nakatani Y, Murakami M. Prostaglandin E synthases: understanding their pathophysiological roles through mouse genetic models. Biochimie. 2010;92(6):651–659. doi: 10.1016/j.biochi.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. International Journal of Biochemistry and Cell Biology. 2010;42(2):198–201. doi: 10.1016/j.biocel.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 56.Jakobsson PJ, Thorén S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(13):7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacological Reviews. 2007;59(3):207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 58.Tanikawa N, Ohmiya Y, Ohkubo H, et al. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochemical and Biophysical Research Communications. 2002;291(4):884–889. doi: 10.1006/bbrc.2002.6531. [DOI] [PubMed] [Google Scholar]
- 59.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. Journal of Biological Chemistry. 2000;275(42):32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- 60.Fujimori K, Maruyama T, Kamauchi S, Urade Y. Activation of adipogenesis by lipocalin-type prostaglandin D synthase-generated Δ12-PGJ2 acting through PPARγ-dependent and independent pathways. Gene. 2012;505(1):46–52. doi: 10.1016/j.gene.2012.05.052. [DOI] [PubMed] [Google Scholar]
- 61.Fujimori K, Yano M, Ueno T. Synergistic suppression of early phase of adipogenesis by microsomal PGE synthase-1 (PTGES1)-produced PGE2 and aldo-keto reductase 1B3-produced PGF2α . PloS ONE. 2012;7(9) doi: 10.1371/journal.pone.0044698.e44698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urade Y, Hayaishi O. Prostaglandin D synthase: structure and function. Vitamins and Hormones. 2000;58:89–120. doi: 10.1016/s0083-6729(00)58022-4. [DOI] [PubMed] [Google Scholar]
- 63.Urade Y, Hayaishi O. Biochemical, structural, genetic, physiological, and pathophysiological features of lipocalin-type prostaglandin D synthase. Biochimica et Biophysica Acta. 2000;1482(1-2):259–271. doi: 10.1016/s0167-4838(00)00161-8. [DOI] [PubMed] [Google Scholar]
- 64.Kanaoka Y, Urade Y. Hematopoietic prostaglandin D synthase. Prostaglandins Leukotrienes and Essential Fatty Acids. 2003;69(2-3):163–167. doi: 10.1016/s0952-3278(03)00077-2. [DOI] [PubMed] [Google Scholar]
- 65.Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-Δ12,14-PGJ2 and the ligation of PPARγ . Journal of Clinical Investigation. 2003;112(6):945–955. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hossain MS, Chowdhury AA, Rahman MS, et al. Development of enzyme-linked immunosorbent assay for Δ12- prostaglandin J2 and its application to the measurement of the endogenous product generated by cultured adipocytes during the maturation phase. Prostaglandins and Other Lipid Mediators. 2011;94(3-4):73–80. doi: 10.1016/j.prostaglandins.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 67.Fujimori K, Aritake K, Urade Y. A novel pathway to enhance adipocyte differentiation of 3T3-L1 cells by up-regulation of lipocalin-type prostaglandin D synthase mediated by liver X receptor-activated sterol regulatory element-binding protein-1c. Journal of Biological Chemistry. 2007;282(25):18458–18466. doi: 10.1074/jbc.M701141200. [DOI] [PubMed] [Google Scholar]
- 68.Ragolia L, Palaia T, Hall CE, Maesaka JK, Eguchi N, Urade Y. Accelerated glucose intolerance, nephropathy, and atherosclerosis in prostaglandin D2 synthase knock-out mice. Journal of Biological Chemistry. 2005;280(33):29946–29955. doi: 10.1074/jbc.M502927200. [DOI] [PubMed] [Google Scholar]
- 69.Ragolia L, Hall CE, Palaia T. Lipocalin-type prostaglandin D2 synthase stimulates glucose transport via enhanced GLUT4 translocation. Prostaglandins and Other Lipid Mediators. 2008;87(1–4):34–41. doi: 10.1016/j.prostaglandins.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Tanaka R, Miwa Y, Mou K, et al. Knockout of the l-pgds gene aggravates obesity and atherosclerosis in mice. Biochemical and Biophysical Research Communications. 2009;378(4):851–856. doi: 10.1016/j.bbrc.2008.11.152. [DOI] [PubMed] [Google Scholar]
- 71.Fujitani Y, Aritake K, Kanaoka Y, et al. Pronounced adipogenesis and increased insulin sensitivity caused by overproduction of prostaglandin D2 in vivo. FEBS Journal. 2010;277(6):1410–1419. doi: 10.1111/j.1742-4658.2010.07565.x. [DOI] [PubMed] [Google Scholar]
- 72.Vassaux G, Gaillard D, Ailhaud G, Negrel R. Prostacyclin is a specific effector of adipose cell differentiation. Its dual role as a cAMP- and Ca2+-elevating agent. Journal of Biological Chemistry. 1992;267(16):11092–11097. [PubMed] [Google Scholar]
- 73.Vassaux G, Gaillard D, Darimont C, Ailhaud G, Negrel R. Differential response of preadipocytes and adipocytes to prostacyclin and prostaglandin E2: physiological implications. Endocrinology. 1992;131(5):2393–2398. doi: 10.1210/endo.131.5.1330499. [DOI] [PubMed] [Google Scholar]
- 74.Aubert J, Saint-Marc P, Belmonte N, Dani C, Négrel R, Ailhaud G. Prostacyclin IP receptor up-regulates the early expression of C/EBPβ and C/EBPδ in preadipose cells. Molecular and Cellular Endocrinology. 2000;160(1-2):149–156. doi: 10.1016/s0303-7207(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 75.Massiera F, Saint-Marc P, Seydoux J, et al. Arachidonic acid and prostacyclin signaling promote adipose tissue development: a human health concern? Journal of Lipid Research. 2003;44(2):271–279. doi: 10.1194/jlr.M200346-JLR200. [DOI] [PubMed] [Google Scholar]