

Abstract
Acute lymphoblastic leukemia is still an incurable disease with resistance to therapy developing in the majority of patients. We investigated the effect of TPEN, an intracellular zinc chelator, in Jurkat and in ex vivo acute lymphoblastic leukemia (ALL) cells resistant to chemotherapy. Changes of nuclei morphology, reactive oxygen species generation, presence of hypodiploid cells, phosphatidylserine translocation, mitochondrial membrane depolarization, immunohistochemical identification of cell death signalling molecules, and pharmacological inhibition were assayed to detect the apoptotic cell death pathways. We found that TPEN induces apoptosis in both types of cells by a molecular oxidative stress pathway involving O2 •− > H2O2 ≫ NF-κB (JNK/c-Jun) >p53> loss ΔΨm> caspase-3, AIF > chromatin condensation/DNA fragmentation. Interestingly, TPEN induced apoptosis independently of glucose; leukemic cells are therefore devoid of survival capacity by metabolic resistance to treatment. Most importantly, TPEN cytotoxic effect can eventually be regulated by the antioxidant N-acetyl-cysteine and zinc ions. Our data suggest that TPEN can be used as a potential therapeutic prooxidant agent against refractory leukemia. These data contribute to understanding the importance of oxidative stress in the treatment of ALL.
1. Introduction
Leukemia is a malignancy of hematopoietic cell populations responsible of at least 37,520 new cases and 15,200 people deaths in the United States [1]. Although the cause of leukemia is still unknown, several cellular, genetic, and biochemical alterations are the most probable mechanisms of cause [2–5]. Apoptosis is a controlled and regulated form of programmed cell death defined by specific morphological and biochemical features [6–8]. Reactivation of the apoptotic cell death appears as a major goal to eliminate cancer cells [9, 10]. Unfortunately, secondary therapy-related leukemia (e.g., acute lymphocytic leukemia, ALL) might emerge following chemotherapy, radiotherapy, and/or terminal differentiation for primary malignancies [11–13]. Consequently, leukemia is still an incurable disease with resistance to therapy developing in the majority of patients. Therefore, it is necessary to investigate therapies to either achieve maximal cancer cell death or cell terminal differentiation. Given the complexity of death/differentiation pathways within a cell [8, 14–16], placing these pathways in the proper relationship to the drug trigger is challenging. Since metal dysregulation (e.g., zinc) has been shown in patients with leukemia [17, 18] and it is essential cofactor for many proteins and transcription factors [19, 20], it is reasonable to think that the use of chelators might be a potential class of pharmaceutical agents to battle different types of cancer [21, 22]. Therefore, zinc depletion [19, 20] in ALL patients might be a realistic goal.
TPEN (N,N,N,N-Tetrakis(2-pyridylmethyl)-ethylenediamine) is a lipid-soluble zinc metal chelator and has been shown to induce apoptosis in several cancer cells [23–27] through caspase family of proteases [25, 28–33], transcription factor p53 [28, 33], and X-linked inhibitor of apoptosis protein (XIAP [27]). Yet, the complete molecular mechanism of cell death signaling induced by TPEN in a single cell has not yet fully established. Interestingly, the antioxidant N-acetyl-L-cysteine (NAC) inhibited the cytotoxic effect of TPEN, indicating that oxidative stress (OS) is the likely mediator of Zn-deficiency-related cell death [25]. Remarkably, molecules that generated OS induce a minimal completeness of cell death signaling pathway as a mechanistic explanation of cancer cell demise [34]. We hypothesize that TPEN might induce apoptosis by OS in leukemia cells.
To test this assumption, we sought (i) to determine whether TPEN treatment induces OS through H2O2, and activation of the proapoptotic transcription factors, kinases, and caspase-3 in Jurkat cells, used as a model of human ALL. We also wanted (ii) to determine the role played by the apoptosis-inducing factor (AIF) in TPEN treatment. Glucose appears to be critical pathophysiology resistance against OS in cancer cells [35, 36]. We evaluated therefore whether glucose might alter the survival response to TPEN in Jurkat cells. To validate this in vitro data, ALL cells from one patient resistant to chemotherapy and radiotherapy were challenged with TPEN. ALL cells were similarly evaluated for nuclei morphological changes, loss ΔΨm, p53, and caspase-3 activation or AIF analysis. Understanding the mechanism of OS may provide insight into more effective anticancer therapy.
2. Materials and Methods
3,3′-dihexyloxacarbocyanine iodide (DiOC6(3), cat # D-273), ammonium pyrrolidinedithiocarbamate (PDTC, cat. # 548000), and 1,9-pyrazoloanthrone (SP600125, cat # 420119) were acquired from Calbiochem. Propidium Iodide and 7-AAD BD via-probe cell viability was all purchased from BD Bioscience (San Jose, CA). Annexin V-Phycoerythrin (PE) apoptosis detection kit was purchased from BD Pharmingen (San Diego, CA). Dichlorofluorescein diacetate (DCFH2-DA) was obtained from Invitrogen. All other reagents were from Sigma-Aldrich.
2.1. Jurkat T Leukemia Cell Culture
Jurkat clone E6-1 (ATCC Catalog no. TIB-152) were cultured according to supplier's indications.
2.2. Experiments with Jurkat Leukemia Cell Line
2.2.1. Morphological Assessment of Cell Death by Fluorescence Microscopy and Flow Cytometry Analysis
The cell suspension (1 mL, final volume) was exposed to increasing TPEN (0.1–5 μM) concentrations freshly prepared in RPMI-1640 medium either with glucose (G11, G55; Gibco/Invitrogen) or absence (G0) in the absence or presence different products of interest for 24 h at 37°C. Fluorescent microscopy analysis and quantification of apoptotic morphology was performed according to [37]. The apoptotic indexes were assessed 3 times in independent experiments blind to experimenter. For annexin V/7-AAD flow cytometry analysis, cells were evaluated according to supplier's protocol (BD Pharmingen, San Diego, CA) with a flow cytometer FACSCanto II, Becton Dickinson (San José, CA). The flow cytometry apoptosis was assessed 3 times in independent experiments.
2.2.2. Determination of DNA Fragmentation by Flow Cytometry
DNA fragmentation was determined by using a hypotonic solution of propidium iodide (PI). Cells entering the sub-G1 phase were used as the marker for apoptosis. Cell suspensions were analyzed in a FACScanto III flow cytometer (Beckton Dickinson). 20,000 events were assessed. Determination of DNA fragmentation was assessed 3 times in independent experiments.
2.2.3. Evaluation of Intracellular Reactive Oxygen Species (ROS)
Superoxide anion radical (O2 •−) was examined for formation of formazan positive cells and quantification was evaluated according to [38]. The assessment was repeated 3 times in independent experiments.
Hydrogen peroxide (H2O2) was determine with 2′,7′-dichlorofluorescein diacetate (5 μM, DCFH2-DA) according to supplier's protocol using a flow cytometer FACSCanto II, Becton Dickinson (San José, CA). The assessment was repeated 3 times in independent experiments.
2.2.4. Analysis of Mitochondrial Membrane Potential ( ΔΨm) by Flow Cytometry
Jurkat cell line was treated as described above. Then, cells (1 × 105) were incubated for 20 min at room temperature in the dark with cationic lipophilic DiOC6(3) (10 nM, final concentration) and intercalating agent propidium iodide (PI, 12.5 ng/mL, final concentration) according to standard protocol. Cells were analyzed using a flow cytometer FACSCanto II, Becton Dickinson (San José, CA). The assessment was repeated 3 times in independent experiments.
2.3. Immunocytochemistry Detection of Transcription Factor NF-κB, p53, c-Jun, Caspase-3, and Apoptosis-Inducing Factor (AIF)
The Santa Cruz Biotechnology supplier protocol (goat ABC staining System: cat # sc-2023) was followed for the immunocytochemistry using primary goat polyclonal antibodies NF-κB p65 (C-20)-G (cat # sc-372-G), p53 (FL-393) (cat # sc-6243-G), p-(Ser73)-c-Jun (cat # sc-7981), caspase-3 (cat # sc-22171), and AIF (cat # sc-9417). The cells were immune-stained and diaminobenzidine positive (DAB+) cells were quantified according to [37].
2.4. Protection and Rescue Experiments
The Jurkat cell suspension (1 mL, final volume) was treated with (3 μM) TPEN alone immediately (for protection experiments) or left for 6 h (for rescue experiments). Then after, cells were coincubated with antioxidant and inhibitor reagents at the concentration listed in Table 1 at 37°C for up to 24 h. After this time, cells were evaluated for apoptotic features by either fluorescence microscopy or flow cytometry, as described in previous section. The assessment was repeated 3 times in independent experiments.
Table 1.
NF-κB, c-Jun, p53, and caspase-3 signalling molecules and oxidative stress are involved in TPEN-induced apoptosis in Jurkat cells. Jurkat cells (1 × 106 cells/mL) were exposed to TPEN (3 μM) in absence or presence of PDTC (10 nM), pifithrin-α (PFT, 50 nM), SP600125 (1 μM), and NSCI (10 μM) signaling inhibitors, and ZnSO4 (10 μM) and NAC (1 mM) at 37°C for 24 h. Cells were then evaluated for apoptotic nuclei morphology, plasma membrane integrity, and mitochondrial membrane potential, as described in Material and Methods section. The percentage of positive AO/EB/Hoechst staining, AV+/7-AAD−/+ and PI−/+/DiOC6(3)− of Jurkat cells treated with or without TPEN expressed as a mean percentage ± S.D. from three independent experiments. **P < 0.05.
| Treatment/assay | AO/EB/Hoechst (%) | AV+/7-AAD−/+ (%) | PI−/+/DiOC6(3)− (%) |
|---|---|---|---|
| Untreated | <1 ± 0 | 4 ± 1 | 3 ± 1 |
| TPEN (3 μM) | 100 | 82 ± 2 | 80 ± 1 |
|
| |||
| PDTC (10 nM) | <1 ± 0 | 2 ± 1 | 3 ± 2 |
| PDTC (10 nM) + TPEN (3 μM) | 5 ± 1** | 18 ± 1** | 24 ± 2** |
| SP600125 (1 μM) | <1 ± 1 | 3 ± 1 | 2 ± 1 |
| SP600125 (1 μM) + TPEN (3 μM) | 3 ± 1** | 10 ± 1** | 39 ± 2** |
| PFT (50 nM) | <1 ± 0 | 2 ± 1 | 3 ± 1 |
| PFT (50 nM) + TPEN (3 μM) | 6 ± 1** | 28 ± 1** | 20 ± 1** |
| NSCI (10 μM) | <1 ± 0 | 3 ± 1 | 2 ± 2 |
| NSCI (10 μM) + TPEN (3 μM) | 8 ± 1** | 33 ± 1** | 35 ± 1** |
| ZnSO4 (10 μM) | <1 ± 0 | 1 ± 1 | 1 ± 1 |
| ZnSO4 (10 μM) + TPEN (3 μM) | 3 ± 2** | 6 ± 1** | 6 ± 1** |
| Zn (10 μM)/TPEN (3 μM) complex | 6 ± 2** | 5 ± 1** | 3 ± 1** |
|
| |||
| NAC (1 mM) | <1 ± 0 | 3 ± 2 | 2 ± 2 |
| NAC (1 mM) + TPEN (3 μM) | 2 ± 1** | 10 ± 3** | 15 ± 2** |
2.5. Patient Blood Cells Culture Conditions
After obtaining informed consent, peripheral blood was collected from a chemotherapy and radiotherapy resistant ALL cells from a patient (female, 53 years old) diagnosed according to classical morphological, cytogenetic, and immunophenotypical criteria. The Institutional Ethics Committee at University of Antioquia approved this study. Whole blood sample (1 mL) from an ALL patient was incubated with 0, 5, 10, 15, 20 μM TPEN at 37°C for 24 h. A smear of blood (30 μL) was stained with Giemsa and an aliquot (100 μL) was stained with AO/EB/Hoechst dye for nuclei morphological changes analysis. After hemolysis of red blood cells in the remaining sample volume, cells were analyzed for ΔΨm under identical conditions by flow cytometry. Results represent a mean ± S.D. of two replicas from one experiment. Lymphoblastic cells (LC) purified by Ficoll-Hypaque (Lymphoprep, Bio-Whittaker Products, Verviers, Belgium) were washed thrice with PBS (10 mM sodium phosphate, 160 mM NaCl, pH 7.4) and finally suspended in RPMI-1640 medium (GIBCO laboratories, NY, USA) under standard culture conditions. The LC were plated in 24-wells (1 × 106 cells/mL/well) and treated with similar TPEN concentrations as described above at 37°C for 24 h. LC were used for immunohistochemistry analysis.
2.6. Statistical Analysis
Data are means ± S.D. of three independent experiments. One-way ANOVA analyses with Bonferroni or Games-Howell post-hoc comparison were calculated with SPSS 18 software. A P value of * < 0.05 and ** < 0.001 was considered significant.
3. Results
3.1. TPEN Induces Apoptosis in Jurkat Cells in a Concentration- and Time-Dependent Fashion but Independent of Reactive Oxygen Species
As shown in Figure 1, TPEN induces typical morphology of apoptosis (Figure 1(b)) compared to untreated cells (Figure 1(a)) in a concentration-dependent manner (Figure 1(c)). Since we found that 3–5 μM TPEN induces 100% AO/EB/Hoechst positive nuclei staining, we used 3 μM TPEN for further experiments. At this concentration, kinetic analysis shows that TPEN induces production of O2 •− (Figure 2(a)) in a belt-like shape with a maximum generation at 6 h (Figure 2(c)) and H2O2 (Figure 2(b)) in a time-independent fashion (Figure 2(c)), according to NBT staining and flow cytometry assay, respectively. Additionally, TPEN provokes DNA fragmentation (Figure 3(a)), externalization of phosphatidylserine, (PtdSer, Figure 3(b)), and loss ΔΨm (Figure 3(c)) in a time-dependent fashion, as determined by flow cytometry assay.
Figure 1.
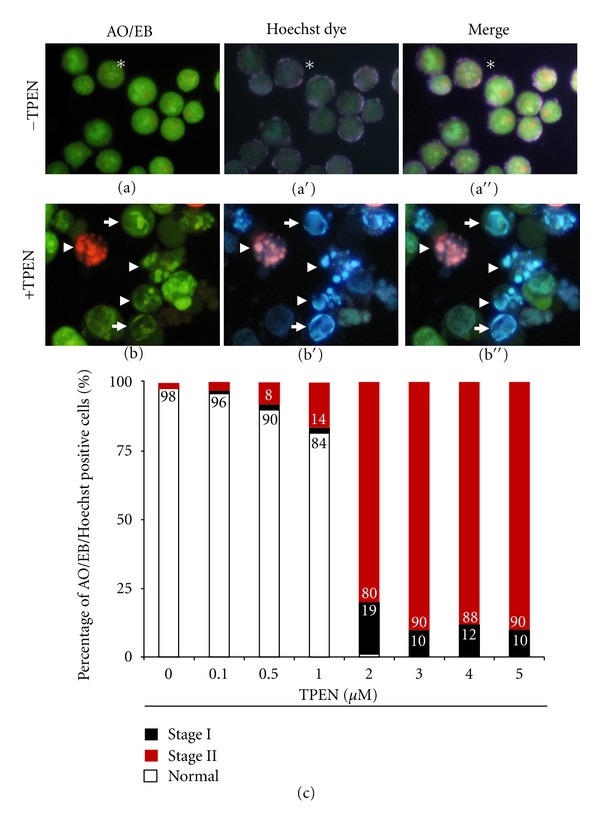
TPEN induces chromatin condensation and DNA fragmentation in Jurkat cells in a concentration-dependent fashion. Representative fluorescent photomicrography showing untreated (−TPEN) cells with normal nuclei (asterisk), or treated cells (+TPEN, 3 μM) with condensed chromatin (arrows, stage I nuclei morphology) and DNA fragmentation (arrowheads, stage II nuclei morphology) analyzed by either AO/EB ((a), (b)) (ex. 450–490 nm, em. 515 nm), Hoechst staining ((a′), (b′)) (ex. 354 nm, em. 442 nm), or merge image ((a′′), (b′′)). (c) The percentage of positive AO/EB/Hoechst staining of Jurkat cells treated with increasing TPEN concentrations with normal, stage I, and stage II nuclei morphology are expressed as a mean percentage ± S.D. from three independent experiments. Magnification ((a), (b)) 800x. Photomicrographs shown in (a)-(b) and Figures 2(a), 4(a)–4(f), 6(a)–6(f) and 7(a)–7(d) were taken using a Zeiss (Axiostart 50) microscope equipped with a Canon PowerShot G5 digital camera. Fiji (Fiji-win32) image processing package was used for composite image ((a′′)-(b′′)).
Figure 2.
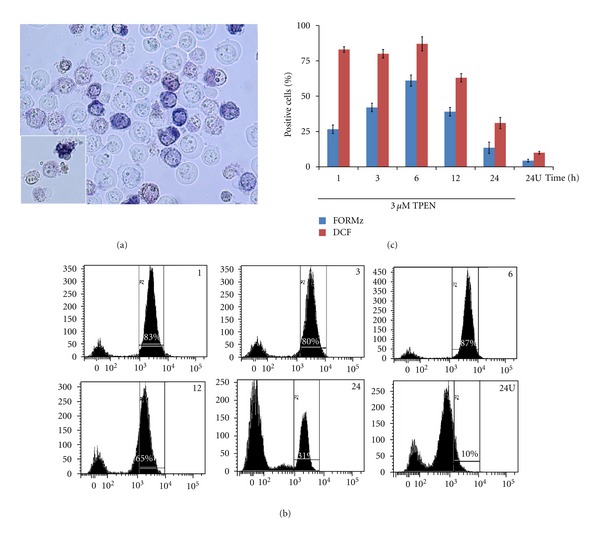
TPEN induces production of O2 •− and H2O2 in a time-independent fashion. Jurkat cells (1 × 106 cells/mL) were either exposed to TPEN (3 μM) at 37°C for 1, 3, 6, 12, and 24 h or untreated for 24h (24U) (a) Representative light photomicrography illustrating formazan (FORMz) precipitation, as indicative of O2 •− generation in Jurkat cells incubated with TPEN (3 μM) at 37°C for 3 h. (b) Representative histograms showing the percentage of DFC+ cells under TPEN (3 μM) exposure assessed at indicate interval of time, according to Material and Methods section (c) The percentage of positive FORMz and dichlorofluorescein (DCF) cells after each interval are expressed as a mean percentage ± S.D. from three independent experiments.
Figure 3.
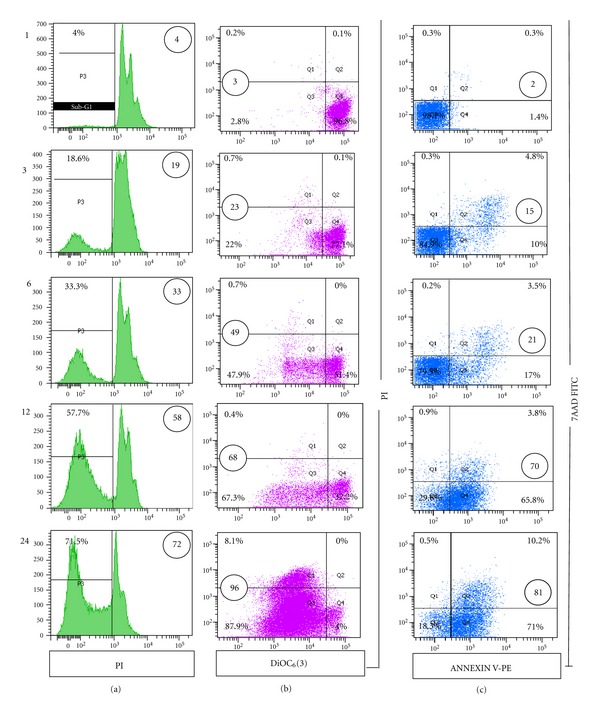
TPEN induces mitochondrial depolarization, plasma membrane damage, and chromatin fragmentation in Jurkat cells. ((a)–(c)) Jurkat cells (1 × 106 cells/mL) were incubated with TPEN (3 μM) at 37°C for 1, 3, 6, 12, and 24 h. The cells were then used for several assays. (a) Representative histograms showing SubG1 cell population indicative of DNA fragmentation after TPEN treatment, as described in according to Material and Methods section (b) Representative density (dot) plots showing PI/DiOC6(3) percentage of four different cell populations under TPEN treatment, as described in Material and Methods section. (c) Representative density (dot) plots showing annexin V/7-AAD percentage of four different cell populations under TPEN exposure, as described Material and Methods section. Circled number is the mean percentage of three independent experiments. S.D. values were below ±5% (also for Figures 5 and 6) of the mean values and are not shown.
3.2. TPEN Induces Transcription Factors, Caspase-3 Activation, and AIF Nuclei Translocation
Next, we investigated whether TPEN was able to activate transcription factors, JNK kinase, caspase-3 [34], and AIF. As shown in Figure 4, compared to untreated cells (e.g., ~2%, Figure 4(a)), TPEN effectively induced NF-κB (e.g., 38 ± 1%, Figure 4(b)), p53 (e.g., 42 ± 2%, Figure 4(c)), c-Jun (e.g., 57 ± 2%, Figure 4(d)), caspase-3 (e.g., 48 ± 3%, Figure 4(e)), and AIF (e.g., 55 ± 2%, Figure 4(f)) at 24 h. These observations were further confirmed by incubating Jurkat cells in the presence of pharmacologic inhibitors (Table 1), according to AO/EB/Hoechst staining and flow cytometry technique. Moreover, cells pre-exposed to TPEN for 6 h and then co-incubated with inhibitor compounds were able to rescue Jurkat cells from apoptosis (Table 2).
Figure 4.
TPEN induces activation of NF-κB, p53 and c-Jun transcription factors, caspase-3, and AIF in Jurkat T cells. Jurkat cells (1 × 106 cells/mL) were incubated with TPEN (3 μM) at 37°C for 24 hours. Untreated (a) and TPEN-treated cells were stained with anti-NF-κB-p65 (b), anti-p53 (c), anti-c-Jun (d), anti-caspase-3 (e), and anti-AIF (f) antibodies according to protocol described in according to Material and Methods section Notice that compared to control, NF-κB, p53, c-Jun, caspase-3, and AIF positive nuclei (dark brown color) reflect their nuclear translocation/activation and appear to correlate with the apoptotic nuclear morphology. Notice also that (a) represents the control for each antibody used in ((b)–(f)). Magnification 700x ((a)–(f)).
Table 2.
Signaling inhibitors, ZnSO4 and NAC rescue Jurkat cells from TPEN-induced apoptosis. Jurkat cells (1 × 106 cells/mL) were preexposed to TPEN (3 μM) for 6 hours. After this time, either PDTC (10 nM), pifithrin-α (PFT, 50 nM), SP600125 (1 μM), NSCI (10 μM), ZnSO4 (10 μM) or NAC (1 mM) were supplemented for upto 24 h at the same experimental conditions, as mentioned above. Cells were then evaluated for apoptotic nuclei morphology, plasma membrane integrity, and mitochondrial membrane potential, as described in Material and Methods section. The percentage of positive AO/EB/Hoechst staining, AV+/7-AAD−/+ and PI−/+/DiOC6(3)− of Jurkat cells treated with or without TPEN are expressed as a mean percentage ± S.D. from three independent experiments. **P < 0.05.
| Treatment/assay | AO/EB/Hoechst (%) | AV+/7-AAD−/+ (%) | PI−/+/DiOC6(3)− (%) |
|---|---|---|---|
| Untreated | <1 ± 0 | 4 ± 1 | 3 ± 1 |
| TPEN (3 μM) | 100 | 82 ± 2 | 80 ± 1 |
|
| |||
| PDTC (10 nM) | <1 ± 0 | 2 ± 1 | 3 ± 2 |
| PDTC (10 nM) + TPEN (3 μM) | 21 ± 1** | 31 ± 1** | 17 ± 1** |
| SP600125 (1 μM) | <1 ± 1 | 3 ± 1 | 2 ± 1 |
| SP600125 (1 μM) + TPEN (3 μM) | 11 ± 1** | 9 ± 1** | 16 ± 1** |
| PFT (50 nM) | <1 ± 0 | 2 ± 1 | 3 ± 1 |
| PFT (50 nM) + TPEN (3 μM) | 30 ± 1** | 39 ± 1** | 38 ± 2** |
| NSCI (10 μM) | <1 ± 0 | 3 ± 1 | 2 ± 2 |
| NSCI (10 μM) + TPEN (3 μM) | 32 ± 1** | 45 ± 2** | 30 ± 1** |
| ZnSO4 (10 μM) | <1 ± 0 | 1 ± 1 | 1 ± 1 |
| ZnSO4 (10 μM) + TPEN (3 μM) | 12 ± 2** | 29 ± 2** | 15 ± 1** |
| Zn (10 μM)/TPEN (3 μM) complex | 10 ± 2** | 21 ± 2** | 9 ± 1** |
|
| |||
| NAC (1 mM) | <1 ± 0 | 3 ± 2 | 2 ± 2 |
| NAC (1 mM) + TPEN (3 μM) | 5 ± 1** | 7 ± 3** | 20 ± 2** |
3.3. TPEN Induces Apoptosis in Jurkat Cells Independent of Glucose Concentration but Its Toxic Effect Is Diminished by N-Acetyl-Cysteine
As depicted in Figure 5, TPEN induces apoptosis (Figure 5(a)), loss of ΔΨm (Figure 5(b)), and DNA fragmentation (Figure 5(c)) in Jurkat cells in a glucose-and time-independent manner compared to cells cultured in glucose alone. In contrast, antioxidant compounds such as NAC (1 mM) and zinc ions (10 μM) clearly protects (Table 1) and rescues (Table 2) TPEN-induced apoptosis effect in Jurkat cells. To ascertain that TPEN induces apoptosis meanly via OS, cells were exposed to Zn(SO4)/TPEN complex. No significant difference in nuclei morphology, plasma membrane damage, and loss ΔΨm were detected in Jurkat cells treated with either the Zn/TPEN complex or treated with Zn plus TPEN (Tables 1 and 2). Furthermore, no significant difference in NBT+ (e.g., 17 ± 3%) and DCF+(e.g., 13 ± 3%) percentages were observed between cells treated with Zn/TPEN complex and untreated (Figure 2) or zinc alone (e.g., 3 ± 1% NBT+, 12 ± 2% DCF+) for 24 h, as indication of reduced O2 •− and H2O2 production.
Figure 5.
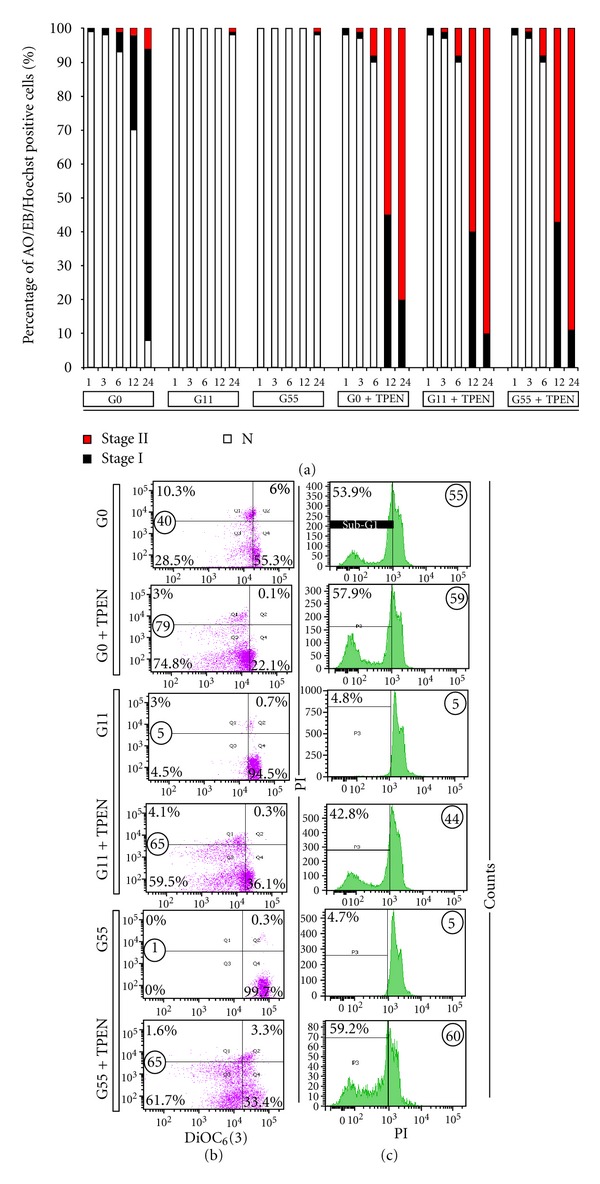
Effect of glucose on Jurkat cells exposed to TPEN. (a) The percentage of positive AO/EB/Hoechst staining of Jurkat cells (1 × 106 cells/mL) incubated under glucose-starvation (GS), glucose 11 mM (G11), or glucose 55 mM (G55) medium in absence or presence of TPEN (3 μM) at 37°C for 1, 3, 6, 12, and 24 h with normal, stage I, and stage II nuclei morphology are expressed as a mean percentage ± S.D. from three independent experiments. (b) Representative density (dot) plots showing PI/DiOC6(3) percentage of four different cell populations at 12 h of incubation in GS, G11, and G55 medium with or without TPEN (3 μM), as described in Material and Methods section. (c) Representative histograms showing SubG1 cell populations at 12 hours in GS, G11, and G55 medium with or without TPEN (3 μM), as described in according to Material and Methods section.
3.4. TPEN Induces Apoptosis in ALL Cells via Mitochondria Caspase-Dependent and -Independent Pathways
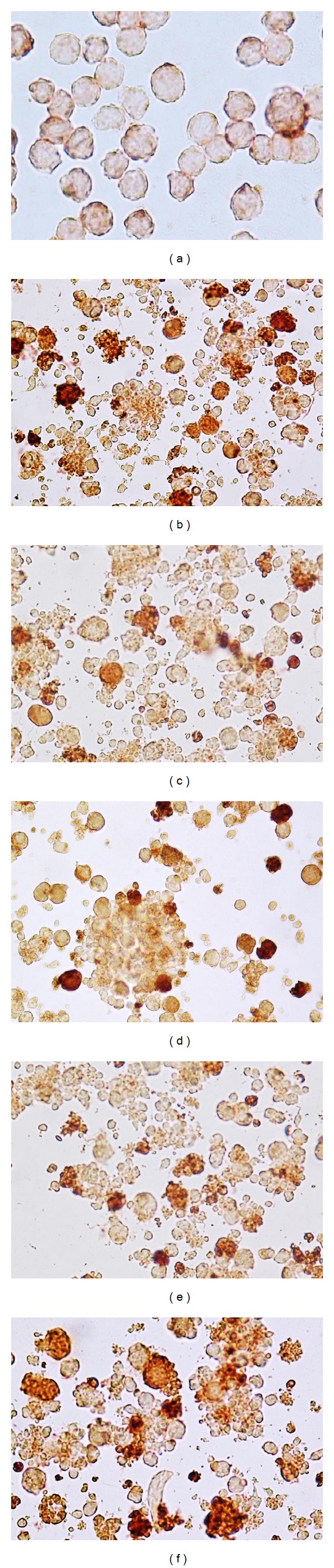
As shown in Figures 6(b)–6(f), treatment of ALL cells from whole blood sample with TPEN displayed nuclei changes typical of apoptosis compared to untreated cells (Figure 6(a) and inset). Noticeably, these morphologies are comparable to those displayed by Jurkat cells treated with TPEN (Figure 1(b)). Likewise, TPEN induced mitochondrial depolarization (Figure 6(g) versus Figure 6(h)). Interestingly, TPEN induces both phenomena in a concentration dependent fashion (Figure 6(i)). As shown in Figure 7, TPEN significantly induces activation of p53 (Figure 7(b)), caspase-3 (Figure 7(c)), and translocation of AIF to nuclei (Figure 7(d)) compared to cells in absence of zinc chelator (Figure 7(a)) similar to TPEN treated Jurkat cells. Clearly, not only the amount of DAB+nuclei in ALL cells treated with TPEN was significantly different to DAB+nuclei in untreated ALL cells, but also the number of AO/EB/Hoechst cells and ΔΨm markers were significantly different compared to untreated cells (Table 3).
Figure 6.
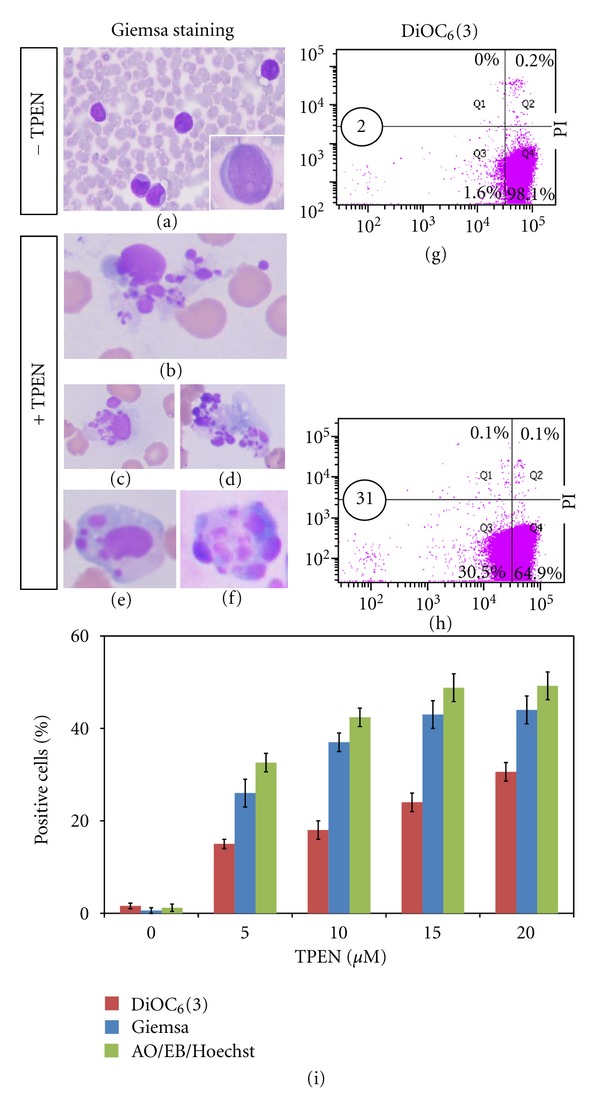
TPEN induces chromatin condensation, nuclei fragmentation, and mitochondrial depolarization in whole blood ALL cells. ALL cells were incubated with TPEN (0, 5, 10, 15, and 20 μM) at 37°C for 24 hours. The cells were then used for several assays. Representative photomicrography ((a)–(f)) illustrates untreated ALL with normal chromatin/nuclei (a) and treated ALL cells with TPEN (20 μM) with condensed chromatin and nuclear fragmentation ((b)–(f)), as determined by Giemsa staining. ((g)–(h)) Representative density (dot) plots showing PI+/−/DiOC6(3)Low percentage of four different cell populations in untreated cells ((g)) and cells treated with TPEN (20 μM, (h)) from patient. (i) The percentage of positive condensed/fragmented nuclei evaluated by AO/EB and Giemsa staining and PI+/−/DiOC6(3)Low cells in ALL cells exposed to increasing concentration of TPEN are expressed as a mean percentage ± S.D. of two replicas from one independent experiment.
Figure 7.
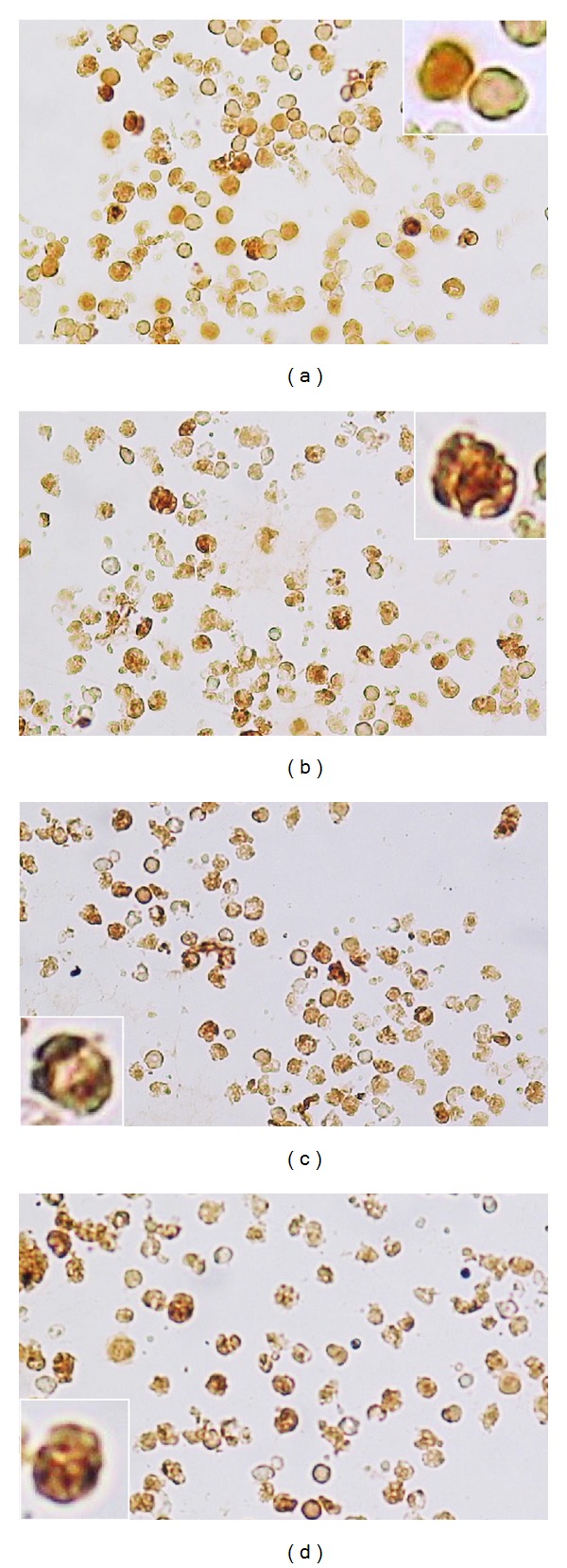
TPEN induces activation of p53, caspase-3, and AIF in isolated ALL cells. ALL cells (1 × 106 cells/mL) were incubated with TPEN (5 μM) at 37°C for 24 hours. Untreated (a) and TPEN-treated cells were stained with anti-p53 (b), anti-caspase-3 (c), and anti-AIF (d) antibodies according to protocol described in Material and Methods section. Notice that compared to control, p53, caspase-3 and AIF positive-nuclei (dark brown color) reflect their nuclear translocation/activation and appear to correlate with the apoptotic nuclear morphology. Notice also that (a) represents the control for each antibody used in ((b)-(d)). Magnification 360x ((a)–(d)), insets 1400x.
Table 3.
TPEN induces nuclei fragmentation, mitochondrial membrane depolarization, activation of p53, caspase-3, and AIF in isolated ALL cells. ALL cells were left untreated or exposed to TPEN (5 μM) for 24 h. After this time, cells were evaluated for fluorescence apoptotic nuclei morphology by AO/EB/Hoechst staining, mitochondrial potential damage by PI/DiOC6(3) flow cytometry, and p53, caspase-3, and AIF detection by immunohistochemistry, as described in Material and Methods section. Percentage of positive staining markers of untreated or treated ALL cells with TPEN is expressed as a mean percentage ± S.D. of two replicas from one independent experiment.
| Treatment | Assay | ||||
|---|---|---|---|---|---|
| AO/EB/Hoechst (%) | PI−/+/DiOC6(3) − (%) | p53 (%) | Caspase-3 (%) | AIF (%) | |
| Untreated |
5 ± 2 | 9 ± 3 | 9 ± 2 | 13 ± 2 | 16 ± 2 |
| TPEN (5 μM) |
97 ± 1 | 79 ± 2 | 74 ± 3 | 90 ± 3 | 82 ± 3 |
4. Discussion
In the present investigation, we report for the first time in vitro evidence of a cause and effect mechanism of apoptosis induced by the zinc chelator TPEN in Jurkat cells, and in ALL cells from a leukemic patient. The TPEN-induced apoptosis complies with the model of minimal completeness of cell death signaling induced by OS [34]. Most importantly, we demonstrated that TPEN induced cell death independently of cellular energy requirement (i.e., glucose [39]) in the culture milieu. The model provides therefore a mechanistic explanation of leukemia cell demise. Specifically, we showed that TPEN induces apoptosis primarily via OS. We found that NAC not only significantly protect but also rescue Jurkat cells against TPEN exposure [25]. Moreover, when cells were exposed to Zn/TPEN complex, comparable O2 •−/H2O2 values to untreated cell were detected; consequently, TPEN-induced apoptosis was significantly reduced. This data implies that zinc disabled TPEN to generate ROS, thereby turning down cell signalling. This observation therefore suggests that TPEN induces apoptosis independently of its metal chelator function. However, the source of TPEN generated O2 •− is unknown. Whatever the mechanism of generation may be, we have shown that TPEN induces sustained generation of O2 •− up to six hours of incubation and then slowly declined until complete incubation time (24 h). Overproduction of O2 •− leads to production of H2O2 through either enzymatic or nonenzymatic reactions. Effectively, TPEN induced a dramatic increase of H2O2 from the first hours up to 12 h (~80%-60%) and then declined to ~30% at 24 h. These H2O2 production values are aberrantly high when compared to base line in untreated cells (~10%). It is accepted that high H2O2 may serves as oxidant signal which might activate critical signalling molecules such as NF-κB [40]. It has been shown that H2O2 indirectly activates the transcription factor NF-κB via several kinases [41–45]. Accordingly, TPEN induces activation of NF-κB transcription factor in Jurkat cells showing fragmentation and condensation, typical indication of apoptotic morphology. Pharmacological inhibition of NF-κB with PDTC significantly inhibited TPEN-induced apoptosis. These observations comply with the notion that NF-κB is involved in apoptosis [46, 47] in Jurkat cells exposed to TPEN. In line with this notion, NF-κB transcribes proapoptotic genes such as p53 [48]. TPEN induces p53 activation and translocation to nuclei. TPEN activates p53 not only in Jurkat cells (this work) but also in neuronal cells [28, 33]. It is perhaps not surprising that activated p53 triggers apoptosis by altering mitochondria function [49], suppressing anti-oxidant genes [50] and regulating metabolic genes [51]. These data suggest that p53 acts both as crucial node downstream of diverse stress signals and as a stress sensor. However, p53-induced apoptosis can be stopped and reverse. Specific inhibitor PFT is able to protect and rescue TPEN-induced apoptosis in Jurkat cells. This observation further reinforces the notion that p53 is susceptible to regulation process, thereby potentially directed to specifically destroy malignant cells. P53 has therefore become an excellent candidate to therapeutic approaches against leukaemia cells [52–54]. In contrast to Ak and Levine [55], we conclude that both NF-κB and p53 have evolved to respond to OS and that they can function in the same cell at the same time.
TPEN/H2O2 stress provoked loss of ΔΨm concomitantly with externalization of PtdSer in a time-dependent fashion, as typical marker of apoptosis. However, the flow cytometer values for loss ΔΨm and PtdSer were significantly different. This observation suggests that either drop ΔΨm occurs previous to externalization of PtdSer or Annexin V assay unsuccessfully label all apoptotic cells [56]. In accordance with others [25, 28, 30–33], we found caspase-3 activation in treated cells. The participation of caspase-3 in TPEN toxicity was confirmed by using NSCI inhibitor, which not only protects but also rescues cells from TPEN toxicity. Caspase-3 activation therefore constitutes a critical protease and a marker of cell death induced by OS. We report for the first time that TPEN provokes AIF to translocate to the nucleus and induces chromatin condensation. In contrast to others [57], AIF is primarily involved in apoptosis in Jurkat cells exposed to TPEN, at least under the present experimental conditions. In support of this view, Stambolsky et al. [58] have shown that p53 regulates expression of AIF. We conclude that depending on the cell type and stress stimuli, AIF might be involved in apoptosis or necrosis [57, 59]. It is concluded that TPEN induces apoptosis in Jurkat cells mainly mitochondrial-mediated pathway [60] by two complimentary but independent cell death subroutes: AIF- and caspase-3 dependent mechanism. However, we found that the proportion of nuclei in stage II detected by fluorescence microscopy was higher than the proportion of nuclei in stage I. One possible explanation is that TPEN chelates zinc from caspase-3, increasing its catalytic activity [32] and thereby overpassing the mitochondrial caspase activation process. Furthermore, TPEN also destroy XIAP, a natural caspase-3 inhibitor [27]. It is therefore not surprising that zinc, being a potent inhibitor of caspase-3 [61], protects and rescues Jurkat cells from TPEN toxic effects. Therefore, AIF and caspase 3 should be used as regular markers of cell death. Additionally, TPEN induces apoptosis via activation of c-Jun and JNK kinase and their activation might be mediated by H2O2 [62]. It has been reported that JNK phosphorylated p53 [63]. In view of the present data and those reported by Yin et al. [64] comply with the notion that NF-κB, JNK and p53 pathways are involved in OS in HepG2 cells and Jurkat cells (this work).
We found that TPEN-induced nuclei fragmentation morphology and loss ΔΨm in a concentration-dependent manner in whole blood cells from ALL patient. Treatment of isolated ALL cells with (5μM) TPEN displayed p53, caspase-3, and AIF DAB+ compared to untreated cells. These data suggest that TPEN induces apoptosis in ALL leukemic cells by a similar mechanism as the one induced by TPEN in Jurkat cells (Figure 8). However, we noticed that almost fivefold concentration of TPEN was necessary on whole blood ALL cells to reach a comparable value of apoptosis in Jurkat cells treated with TPEN. Since TPEN was able to fully induce apoptosis in ALL via activation of cell death signaling mechanism, it is concluded that the resistance of the ALL cells to chemotherapy is unrelated to malfunction of the cells' death machinery elements, at least, in the death markers assessed in this work, but most probably by other genetic alterations [65, 66]. It is therefore proposed that TPEN might be explored as potential therapeutic agent molecule against ALL leukemia.
Figure 8.
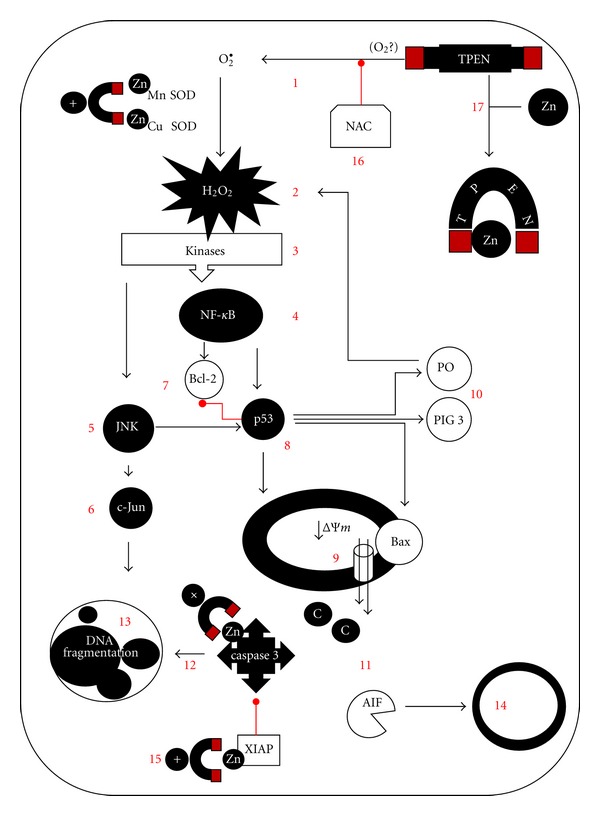
Proposed scheme of TPEN-induced apoptosis in Jurkat cell line and ALL cells. TPEN generates O2 •− (1, by a yet unknown mechanism), which in turn dismutates into H2O2 (2). This last compound serves as signal molecule. Indeed, it indirectly activates NF-κB (4) and c-Jun (6) transcription factors through activation of kinases (v.gr. ASK1, MEKK1, SHIP-1 (3) and JNK (5)). Once NF-κB (4) through phosphorylation of its inhibitor IκBα, it translocates into the nucleus and transcribes several antiapoptotic genes (7) and proapoptotic genes, amongst them the p53 (8). This transcription factors balance the cell decision to death by several actions: p53 either directly impact mitochondria or transcribes proapoptotic genes such as Bax contributing to the permeabilization of the outer mitochondrial membrane by antagonizing antiapoptotic proteins (9). Simultaneously, p53 induces expression of prooxidant genes (e.g., p53-induced gene-3 (PIG3), proline oxidase (PO), (10), which generate more H2O2 (2), and represses the transcription of antioxidant genes. Thus, H2O2 over-production and further activation of NF-κB induce upregulation of proapoptotic genes (e.g., p53), which in turn amplify the initial H2O2-induced cell death signal. Mitochondrial damage allows the release of apoptogenic factors (11) such as AIF, and cytochrome C, which together with dATP, pro-caspase-9, and apoptotic protease activating factor-1; that is, the apoptosome complex triggers activation of caspase-3 protease (12). This protease in turn activates the endonuclease DFF40/CAD, by cutting the nuclease's inhibitor DFF45/ICAD. Finally, DFF40/CAD causes nuclear chromatin fragmentation (stage II nuclei morphology, (13), typical of apoptosis. Alternatively, AIF translocates into the nucleus to generate chromatin fragmentation (stage I nuclei morphology (14)). Noticeably, zinc chelator TPEN further warrants killing of the cell by either directly activating caspase-3 (15) or by releasing caspase-3 from X-linked inhibitor of apoptosis (XIAP, 15). Interestingly, TPEN toxicity can be blocked by the antioxidant N-acetyl-cysteine (NAC) (16) and zinc ions (17), probably by changing its tridimensional structure.
Conflict of Interests
The authors report no conflict of interests.
Acknowledgments
This work was supported by Colciencias grants nos.1115-408-20525 to C. Velez-Pardo and M. Jimenez-Del-Rio. M. Mendivil-Perez is an associated researcher funded by Colciencias Contract no. 8790-2514-2010 and funded from a Young Research Program Award by Colciencias contract no. 8790-016-2011. The authors thank Dr. F. Cuellar, MD (Hematologist) from Leon XIII Clinic and the ALL patient for blood cell sample donation. They thank Catalina Burbano-Arciniegas from the Flow Cytometry Unit, GICIG-SIU-UdeA for technical assistance.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics 2012. A Cancer Journal for Clinicians. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Pasternak G, Hochhaus A, Schultheis B, Hehlmann R. Chronic myelogenous leukemia: molecular and cellular aspects. Journal of Cancer Research and Clinical Oncology. 1998;124(12):643–660. doi: 10.1007/s004320050228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pui CH, Relling MV, Downing JR. Mechanisms of disease: acute lymphoblastic leukemia. The New England Journal of Medicine. 2004;350(15):1535–1548. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Tennant DA, Durán RV, Boulahbel H, Gottlieb E. Metabolic transformation in cancer. Carcinogenesis. 2009;30(8):1269–1280. doi: 10.1093/carcin/bgp070. [DOI] [PubMed] [Google Scholar]
- 6.Kerr JFR, Gobé GC, Winterford CM, Harmon BV. Chapter 1 anatomical methods in cell death. Methods in Cell Biology C. 1995;46:1–27. doi: 10.1016/s0091-679x(08)61921-4. [DOI] [PubMed] [Google Scholar]
- 7.Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death and Differentiation. 2009;16(1):3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death and Differentiation. 2011;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113(16):3655–3665. doi: 10.1182/blood-2009-01-198911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly GL, Strasser A. The essential role of evasion from cell death in cancer. Advances in Cancer Research. 2011;111:39–96. doi: 10.1016/B978-0-12-385524-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Wang E, Lu Y, Gaal KK, Huang Q. Therapy-related acute lymphoblastic leukemia without 11q23 abnormality: report of six cases and a literature review. American Journal of Clinical Pathology. 2010;133(1):75–82. doi: 10.1309/AJCPYWC6AQC7BAVJ. [DOI] [PubMed] [Google Scholar]
- 12.Szotkowski T, Rohon P, Zapletalova L, Sicova K, Hubacek J, Indrak K. Secondary acute myeloid leukemia—a single center experience. Neoplasma. 2010;57(2):170–178. doi: 10.4149/neo_2010_02_170. [DOI] [PubMed] [Google Scholar]
- 13.Kelaidi C, Adès L, Fenaux P. Treatment of acute promyelocytic leukemia with high white cell blood counts. Mediterranean Journal of Hematology and Infectious Diseases. 2011;3 doi: 10.4084/MJHID.2011.038.e2011038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed JC. Warner-Lambert/Parke Davis award lecture: mechanisms of apoptosis. American Journal of Pathology. 2000;157(5):1415–1430. doi: 10.1016/S0002-9440(10)64779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. The KEGG (Kyoto Encyclopedia of Genes and Genomes) PATHWAY Database, (Apoptosis), http://www.genome.jp/kegg/pathway/hsa/hsa04210.html.
- 16.Dang TP. Notch apoptosis and cancer. Advances in Experimental Medicine and Biology. 2012;727:199–209. doi: 10.1007/978-1-4614-0899-4_15. [DOI] [PubMed] [Google Scholar]
- 17.Zuo XL, Chen JM, Zhou X, Li XZ, Mei GY. Levels of selenium, zinc, copper, and antioxidant enzyme activity in patients with leukemia. Biological Trace Element Research. 2006;114(1–3):41–53. doi: 10.1385/BTER:114:1:41. [DOI] [PubMed] [Google Scholar]
- 18.Ciarcia R, D’Angelo D, Pacilio C, et al. Dysregulated calcium homeostasis and oxidative stress in chronic myeloid leukemia (CML) cells. Journal of Cellular Physiology. 2010;224(2):443–453. doi: 10.1002/jcp.22140. [DOI] [PubMed] [Google Scholar]
- 19.Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiological Reviews. 1993;73(1):79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 20.Plum LM, Rink L, Hajo H. The essential toxin: impact of zinc on human health. International Journal of Environmental Research and Public Health. 2010;7(4):1342–1365. doi: 10.3390/ijerph7041342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao R, Planalp RP, Ma R, et al. Role of zinc and iron chelation in apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Biochemical Pharmacology. 2004;67(9):1677–1688. doi: 10.1016/j.bcp.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 22.Khan G, Merajver S. Copper chelation in cancer therapy using tetrathiomolybdate: an evolving paradigm. Expert Opinion on Investigational Drugs. 2009;18(4):541–548. doi: 10.1517/13543780902845622. [DOI] [PubMed] [Google Scholar]
- 23.Adler M, Shafer H, Hamilton T, Petrali JP. Cytotoxic actions of the heavy metal chelator TPEN on NG108-15 neuroblastoma-glioma cells. NeuroToxicology. 1999;20(4):571–582. [PubMed] [Google Scholar]
- 24.Donadelli M, Pozza ED, Costanzo C, Scupoli MT, Scarpa A, Palmieri M. Zinc depletion efficiently inhibits pancreatic cancer cell growth by increasing the ratio of antiproliferative/proliferative genes. Journal of Cellular Biochemistry. 2008;104(1):202–212. doi: 10.1002/jcb.21613. [DOI] [PubMed] [Google Scholar]
- 25.Hashemi M, Ghavami S, Eshraghi M, Booy EP, Los M. Cytotoxic effects of intra and extracellular zinc chelation on human breast cancer cells. European Journal of Pharmacology. 2007;557(1):9–19. doi: 10.1016/j.ejphar.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Jiang S, Zhivotovsky B, Burgess DH, Gahm A, Chow SC, Orrenius S. The role of proteolysis in T cell apoptosis triggered by chelation of intracellular Zn2+ . Cell Death and Differentiation. 1997;4(1):39–50. doi: 10.1038/sj.cdd.4400204. [DOI] [PubMed] [Google Scholar]
- 27.Makhov P, Golovine K, Uzzo RG, et al. Zinc chelation induces rapid depletion of the X-linked inhibitor of apoptosis and sensitizes prostate cancer cells to TRAIL-mediated apoptosis. Cell Death and Differentiation. 2008;15(11):1745–1751. doi: 10.1038/cdd.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corniola RS, Tassabehji NM, Hare J, Sharma G, Levenson CW. Zinc deficiency impairs neuronal precursor cell proliferation and induces apoptosis via p53-mediated mechanisms. Brain Research C. 2008;1237:52–61. doi: 10.1016/j.brainres.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 29.Kolenko VM, Uzzo RG, Dulin N, Hauzman E, Bukowski R, Finke JH. Mechanism of apoptosis induced by zinc deficiency in peripheral blood T lymphocytes. Apoptosis. 2001;6(6):419–429. doi: 10.1023/a:1012497926537. [DOI] [PubMed] [Google Scholar]
- 30.Lee JM, Kim YJ, Ra H, et al. The involvement of caspase-11 in TPEN-induced apoptosis. FEBS Letters. 2008;582(13):1871–1876. doi: 10.1016/j.febslet.2008.04.056. [DOI] [PubMed] [Google Scholar]
- 31.López JJ, Redondo PC, Salido GM, Pariente JA, Rosado JA. N,N,N',N'-tetrakis(2-pyridylmethyl)ethylenediamine induces apoptosis through the activation of caspases-3 and -8 in human platelets. A role for endoplasmic reticulum stress. Journal of Thrombosis and Haemostasis. 2009;7(6):992–999. doi: 10.1111/j.1538-7836.2009.03431.x. [DOI] [PubMed] [Google Scholar]
- 32.Marini M, Frabetti F, Canaider S, Dini L, Falcieri E, Poirier GG. Modulation of caspase-3 activity by zinc ions and by the cell redox state. Experimental Cell Research. 2001;266(2):323–332. doi: 10.1006/excr.2001.5222. [DOI] [PubMed] [Google Scholar]
- 33.Ra H, Kim HL, Lee HW, Kim YH. Essential role of p53 in TPEN-induced neuronal apoptosis. FEBS Letters. 2009;583(9):1516–1520. doi: 10.1016/j.febslet.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez-Del-Rio M, Velez-Pardo C. The bad the good and the ugly about oxidative stress. Oxidative Medicine and Cellular Longevity. 2012;2012:13 pages. doi: 10.1155/2012/163913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Goffe C, Vallette G, Charrier L, et al. Metabolic control of resistance of human epithelial cells to H2O2 and NO stresses. Biochemical Journal. 2002;364(2):349–359. doi: 10.1042/BJ20011856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamanaka RB, Chandel NS. Targeting glucose metabolism for cancer therapy. The Journal of Experimental Medicine. 2012;209:211–215. doi: 10.1084/jem.20120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonilla-Porras AR, Jimenez-Del-Rio M, Velez-Pardo C. Vitamin K3 and vitamin C alone or in combination induced apoptosis in leukemia cells by a similar oxidative stress signalling mechanism. Cancer Cell International. 2011;11, article 19 doi: 10.1186/1475-2867-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melinn M, McLaughlin H. Nitroblue tetrazolium reduction in lymphocytes. Journal of Leukocyte Biology. 1987;41(4):325–329. doi: 10.1002/jlb.41.4.325. [DOI] [PubMed] [Google Scholar]
- 39.Annibaldi A, Widmann C. Glucose metabolism in cancer cells. Current Opinion in Clinical Nutrition and Metabolic Care. 2010;13(4):466–470. doi: 10.1097/MCO.0b013e32833a5577. [DOI] [PubMed] [Google Scholar]
- 40.Brigelius-Flohé R, Flohé L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxidants and Redox Signaling. 2011;15:2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gloire G, Charlier E, Rahmouni S, et al. Restoration of SHIP-1 activity in human leukemic cells modifies NF-κB activation pathway and cellular survival upon oxidative stress. Oncogene. 2006;25(40):5485–5494. doi: 10.1038/sj.onc.1209542. [DOI] [PubMed] [Google Scholar]
- 42.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 43.Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IκBα kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88(2):213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 44.Schoonbroodt S, Ferreira V, Best-Belpomme M, et al. Crucial role of the amino-terminal tyrosine residue 42 and the carboxylterminal PEST domain of IκBα in NF-κB activation by an oxidative stress. Journal of Immunology. 2000;164(8):4292–4300. doi: 10.4049/jimmunol.164.8.4292. [DOI] [PubMed] [Google Scholar]
- 45.Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65. Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. Journal of Biological Chemistry. 2003;278(26):24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 46.Fan Y, Dutta J, Gupta N, Fan G, Gélinas C. Regulation of programmed cell death by NF-κB and its role in tumorigenesis and therapy. Advances in Experimental Medicine and Biology. 2008;615:223–250. doi: 10.1007/978-1-4020-6554-5_11. [DOI] [PubMed] [Google Scholar]
- 47.Radhakrishnan SK, Kamalakaran S. Pro-apoptotic role of NF-κB: implications for cancer therapy. Biochimica et Biophysica Acta. 2006;1766(1):53–62. doi: 10.1016/j.bbcan.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Wu H, Lozano G. NF-κB activation of p53. A potential mechanism for suppressing cell growth in response to stress. Journal of Biological Chemistry. 1994;269(31):20067–20074. [PubMed] [Google Scholar]
- 49.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochimica et Biophysica Acta. 2009;1787(5):414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olovnikov IA, Kravchenko JE, Chumakov PM. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Seminars in Cancer Biology. 2009;19(1):32–41. doi: 10.1016/j.semcancer.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang XD, Qin ZH, Wang J. The role of p53 in cell metabolism. Acta Pharmacologica Sinica. 2010;31(9):1208–1212. doi: 10.1038/aps.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCormack E, Haaland I, Venas G, et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia. 2012;26:910–907. doi: 10.1038/leu.2011.315. [DOI] [PubMed] [Google Scholar]
- 53.Saha MN, Micallef J, Qiu L, Chang H. Pharmacological activation of the p53 pathway in haematological malignancies. Journal of Clinical Pathology. 2010;63(3):204–209. doi: 10.1136/jcp.2009.070961. [DOI] [PubMed] [Google Scholar]
- 54.Tyner JW, Jemal AM, Thayer M, Druker BJ, Chang BH. Targeting surviving and p53 in pediatric acute lymphoblastic leukemia. Leukemia. 2012;26:623–632. doi: 10.1038/leu.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ak P, Levine AJ. p53 and NF-κB: different strategies for responding to stress lead to a functional antagonism. The FASEB Journal. 2010;24(10):3643–3652. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 56.Willingham MC. Cytochemical methods for the detection of apoptosis. Journal of Histochemistry and Cytochemistry. 1999;47(9):1101–1110. doi: 10.1177/002215549904700901. [DOI] [PubMed] [Google Scholar]
- 57.Boujrad H, Gubkina O, Robert N, Krantic S, Susin SA. AIF-mediated programmed necrosis: a highly regulated way to die. Cell Cycle. 2007;6(21):2612–2619. doi: 10.4161/cc.6.21.4842. [DOI] [PubMed] [Google Scholar]
- 58.Stambolsky P, Weisz L, Shats I, et al. Regulation of AIF expression by p53. Cell Death and Differentiation. 2006;13(12):2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- 59.Perez-Alvarez S, Iglesias-Guimarais V, Solesio ME, et al. Methadone induces CAD degradation and AIF-mediated necrotic-like cell death in neuroblastoma cells. Pharmacological Research. 2011;63(4):352–360. doi: 10.1016/j.phrs.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 60.Guo B, Yang M, Liang D, Yang L, Cao J, Zhang L. Cell apoptosis induced by zinc deficiency in osteoblastic MC3T3-E1 cells via a mitochondrial-mediated pathway. Molecular and Cellular Biochemistry. 2012;361:209–216. doi: 10.1007/s11010-011-1105-x. [DOI] [PubMed] [Google Scholar]
- 61.Perry DK, Smyth MJ, Stennicke HR, et al. Zinc is a potent inhibitor of the apoptotic protease, caspase-3: a novel target for zinc in the inhibition of apoptosis. Journal of Biological Chemistry. 1997;272(30):18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 62.Son YO, Jang YS, Shi X, Lee JC. Activation of JNK and c-Jun is involved in glucose oxidase-mediated cell death of human lymphoma cells. Molecules and Cells. 2009;28(6):545–551. doi: 10.1007/s10059-009-0149-1. [DOI] [PubMed] [Google Scholar]
- 63.Buschmann T, Potapova O, Bar-Shira A, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Molecular and Cellular Biology. 2001;21(8):2743–2754. doi: 10.1128/MCB.21.8.2743-2754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin Y, Chen W, Tang C, et al. NF-κB JNK and p53 pathways are involved in tubeimoside-1-induced apoptosis in HepG2 cells with oxidative stress and G2/M cell cycle arrest. Food and Chemical Toxicology. 2011;49:3046–3054. doi: 10.1016/j.fct.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 65.Palmi C, Vendramini E, Silvestri D, et al. Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia. 26(10):2245–2253. doi: 10.1038/leu.2012.101. [DOI] [PubMed] [Google Scholar]
- 66.Schrappe M, Hunger SP, Pui CH, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. The New England Journal of Medicine. 2012;366:1371–1381. doi: 10.1056/NEJMoa1110169. [DOI] [PMC free article] [PubMed] [Google Scholar]