

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a vascular dysplasia caused by mutations in endoglin (ENG; HHT1) or activin receptor-like kinase (ALK1; HHT2) genes, coding for transforming growth factor-β (TGF-β) superfamily receptors. We demonstrated previously that endoglin and ALK1 interact with endothelial NO synthase (eNOS) and affect its activation. Endothelial cells deficient in endoglin or ALK1 proteins show eNOS uncoupling, reduced NO, and increased reactive oxygen species (ROS) production. In this study, we measured NO and H2O2 levels in several organs of adult Eng and Alk1 heterozygous mice, to ascertain whether decreased NO and increased ROS production is a generalized manifestation of HHT. A significant reduction in NO and increase in ROS production were found in several organs, known to be affected in patients. ROS overproduction in mutant mice was attributed to eNOS, as it was L-NAME inhibitable. Mitochondrial ROS contribution, blocked by antimycin, was highest in liver while NADPH oxidase, inhibited by apocynin, was a major source of ROS in the other tissues. However, there was no difference in antimycin- and apocynin-inhibitable ROS production between mutant and control mice. Our results indicate that eNOS-derived ROS contributes to endothelial dysfunction and likely predisposes to disease manifestations in several organs of HHT patients.
1. Introduction
HHT (Rendu-Osler-Weber-syndrome) is inherited in an autosomal dominant fashion and has a prevalence of about 1 in 5–8,000 individuals, worldwide. Mutations in endoglin (ENG) and activin receptor-like kinase (ACVRL1 or ALK1) are responsible for HHT1 and HHT2, respectively, and account for approximately 85% of cases [1]. Both endoglin and ALK1 are components of the transforming growth factor-β (TGF-β) superfamily of receptors, predominantly expressed in the vascular endothelium. The receptor complex for TGF-β1 and -β3 isoforms contains the coreceptor endoglin associated with type II (TβRII) and type I receptors ALK1 or ALK5 [2]. Endoglin and ALK1 directly bind to the bone morphogenetic proteins BMP-9 and BMP-10 and mediate their effects in conjunction with BMPRII [3]. ALK1 activation leads to phosphorylation of Smads1/5/8, while ALK5 phosphorylates Smads2/3. Phosphorylated Smads form a heteromeric complex with the common Smad4, accumulate in the nucleus, and regulate gene transcription. MADH4 mutations coding for Smad4 account for about 3%–5% of cases of HHT, those often associated with juvenile polyposis [4]. Two other loci are in linkage disequilibrium with HHT, on chromosomes 5 and 7 and representing HHT3 and HHT4, respectively [5, 6]. The identification of these genes may increase our understanding of the molecular defects responsible for this disease. HHT is characterized by epistaxis, mucocutaneous and gastrointestinal telangiectases, and arteriovenous malformations (AVMs) in visceral organs [1]. Although genetic testing is required to determine the genotype of a patient, the incidence of pulmonary and cerebral AVMs is generally higher in HHT1 while hepatic AVMs and intestinal complications occur more often in HHT2 [7, 8]. The underlying mechanism of disease is haploinsufficiency, indicating that mutations lead to lower levels of functional endoglin or ALK1 rather than to a dominant negative effect by the mutant protein [9, 10].
Mice totally deficient in endoglin or ALK1 are characterized by an embryonic lethal phenotype due to cardiovascular defects [11, 12]. Endoglin heterozygous (Eng +/−) and ALK1 heterozygous (Alk1 +/−) mice have been used as models for HHT1 and HHT2, respectively, allowing us to dissect the underlying mechanisms of disease [11, 12]. Our earlier studies have revealed that nitric-oxide- (NO-) mediated vasodilation is impaired in Eng +/− mice and that endoglin is important in endothelial NO synthase (eNOS) activation [13, 14]. Moreover, endoglin and ALK1 associate with and stabilize the eNOS activation complex leading to NO production. In endothelial cells deficient in endoglin or ALK1, eNOS becomes uncoupled and produces more superoxide than NO, leading to tissue damage and impaired vascular tone [13–16].
Interestingly, our recent work also revealed that adult Eng +/− and Alk1 +/− mice show signs of pulmonary arterial hypertension (PAH) including rarefaction of the distal pulmonary vasculature, muscularization of small arteries, increased right ventricular (RV) systolic pressure, and RV hypertrophy [15, 16]. These disease manifestations were attributable to uncoupled eNOS activity and increased superoxide (•O2 −) production, as antioxidant treatment could prevent the onset of PAH [15, 16].
It is known that increased bioavailability of reactive oxygen species (ROS) and dysregulated redox signalling together with decreased NO production can contribute to endothelial injury in vascular diseases [17]. The major enzymatic sources of ROS production in endothelial cells are mitochondrial respiratory enzymes, NADPH oxidases, and uncoupled eNOS [17]. Excessive ROS production may therefore have an important role in the pathogenesis of endothelial dysfunction leading to diseases such as HHT and PAH. We therefore investigated with the help of inhibitors, the contribution of NO and ROS producing enzymes in various organs of adult Eng +/− and Alk1 +/− mice. We report that NO generation is decreased while oxidative stress is increased in several tissues of the mutant mice. Mitochondria were the major source of ROS in liver, while NADPH oxidase produced most of the ROS in other tissues, both in mutant and control mice. We identified eNOS as the main source of ROS overproduction in Eng and Alk1 heterozygous mice. Our findings suggest that oxidative stress may predispose the vascular endothelium of patients with ENG and ALK1 haploinsufficiency to disease manifestations. Antioxidant therapy should be considered for these patients.
2. Materials and Methods
2.1. Chemicals and Reagents
Phosphate buffered saline, (PBS) pH 7.4, was from Life Technologies Inc., Burlington, ON, Canada; S-Nitroso-N-acetylpenicillamine (SNAP50) from World precision Instruments Inc. Sarasota, FL, USA; copper sulfate from Fisher Scientific, Ottawa, ON, Canada; L-Arginine, NG-nitro-L-arginine methyl ester (L-NAME), Antimycin A, Apocynin, and HEPES from Sigma-Aldrich, Oakville, ON, Canada; EDTA from Ambion, Life Technologies Inc; protease inhibitor cocktail from Fermantas, Burlington, ON, Canada. The Amplex Red assay kit including horseradish peroxidase was from Molecular Probes, Eugene, OR, USA.
2.2. Experimental Animals
All protocols were approved by the Hospital for Sick Children and the Toronto Centre for Phenogenomics Animal Care Committees (AUP#0067) in accordance with the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996; A5047-01). N28–N32 Eng +/− and Eng +/+ C57BL/6 mice were generated by successive backcrosses and genotyped as previously described [11]. Alk1 +/− and Alk1 +/+ (N13–17) were generated and genotyped as described [18]. Mice ranging from 14 to 18 weeks of age were used for all measurements.
2.3. Tissue Collection
Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). For lung tissue collection, mice were perfused with PBS through the right ventricle. For all other organ dissections, PBS perfusion was done through the left ventricle. Organs were kept in cold PBS and used immediately for NO or Amplex red detection.
2.4. NO Measurement
Excised cardiac, pulmonary, and hepatic tissues were incubated in Krebs-Henseleit buffer for 2 hours at 37°C in the presence or absence of L-NAME. NO production was quantified using an NO microsensor ISO-NOP700 attached to an Apollo 4000 Free Radical Analyzer. The microsensor was calibrated using SNAP50 in the presence of copper sulfate.
2.5. H2O2 Measurement
H2O2 levels were estimated using the Amplex Red assay kit in accordance with manufacturer's instructions. Carefully dissected tissues were flushed with PBS and homogenized in 25 mM HEPES buffer, pH 7.4, plus 1 mM EDTA and protease inhibitors. After centrifugation at 6000 g for 5 minutes at 4°C, the supernatants were incubated in a microtiter plate at 37°C for 1 hour with Amplex red, horseradish peroxidase, and L-Arginine (NOS substrate, 1 mM). The effects of 1 mM L-NAME (NOS inhibitor), 50 μM Antimycin (respiratory chain reaction inhibitor), and 100 μM Apocynin (nicotinamide-adenine dinucleotide phosphate, NADPH oxidase inhibitor) on the reaction were tested. Fluorescence was quantified using excitation at 544 nm and emission at 590 nm; data were normalized for protein content.
2.6. Lipid Peroxidation Measurement in Lung Tissue
The concentration of the lipid peroxidation product 8-iso PGF2α was determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described [19].
2.7. Statistical Analysis
Comparisons were performed by appropriate ANOVA test, and significant differences were evaluated post hoc using the Newman-Keuls test. Results are expressed as the mean ± SEM, with P < 0.05 representing significance.
3. Results
3.1. Decreased NO Production in Tissues of Eng and Alk1 Heterozygous Mice
NO levels were decreased in pulmonary, hepatic, and cardiac tissues of Eng +/− and Alk1 +/− mice, when compared to respective littermate controls (Figures 1(a) and 1(b)). L-NAME inhibited NO production in heart, lungs, and liver of wild-type mice, indicating NOS involvement. As endoglin and ALK1 are mostly expressed in endothelial cells, eNOS is the enzyme most likely involved. In Eng +/− and Alk1 +/− mice, L-NAME did not inhibit NO production, suggesting abnormal eNOS activation and potential uncoupling.
Figure 1.
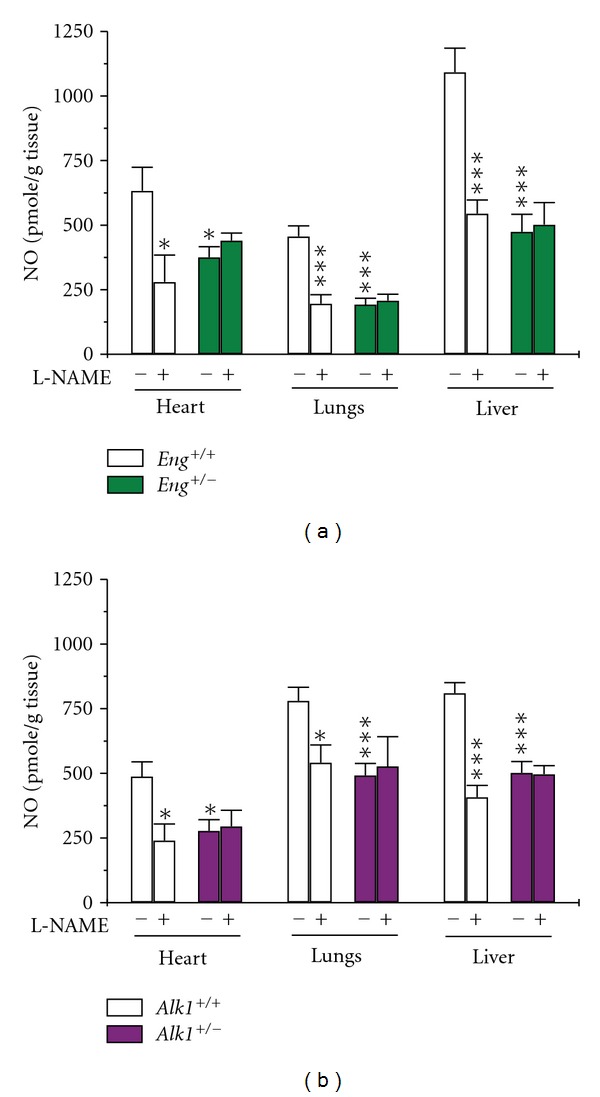
Decreased NO production in tissues of Eng and Alk1 heterozygous mice. NO levels were measured using a microsensor in the presence or absence of the NOS inhibitor L-NAME, 1 mM. NO levels were significantly reduced in heart, lungs, and liver of (a) Eng +/− and (b) Alk1 +/− mice compared to their control littermates. Nitric oxide production was inhibited by L-NAME in all tissues of control mice but not in tissues of (a) Eng +/− or (b) Alk1 +/− mice. *P < 0.05, and ***P < 0.001 versus +/+ untreated mice; N = 6–8/group for Eng mice and 6–10/group for Alk1 mice.
3.2. ROS Production Is Increased in Tissues of Eng and Alk1 Mutant Mice
The Amplex Red assay revealed a substantial increase in H2O2 production in lungs, liver, and colon of Eng +/− and Alk1 +/− mice, compared to corresponding wild-type mice (Figures 2(a) and 2(b)). This increase in H2O2 likely reflects a rise in ROS production. The highest levels of H2O2 were observed in highly vascularized tissues of the heterozygous mice, such as liver, and lungs, suggesting that endothelial cells are the major producers of ROS. This might also explain why despite reduced NO levels, ROS levels were not different between mutant and control mice in cardiac tissue, where cardiomyocytes represent ~75% of total tissue mass.
Figure 2.
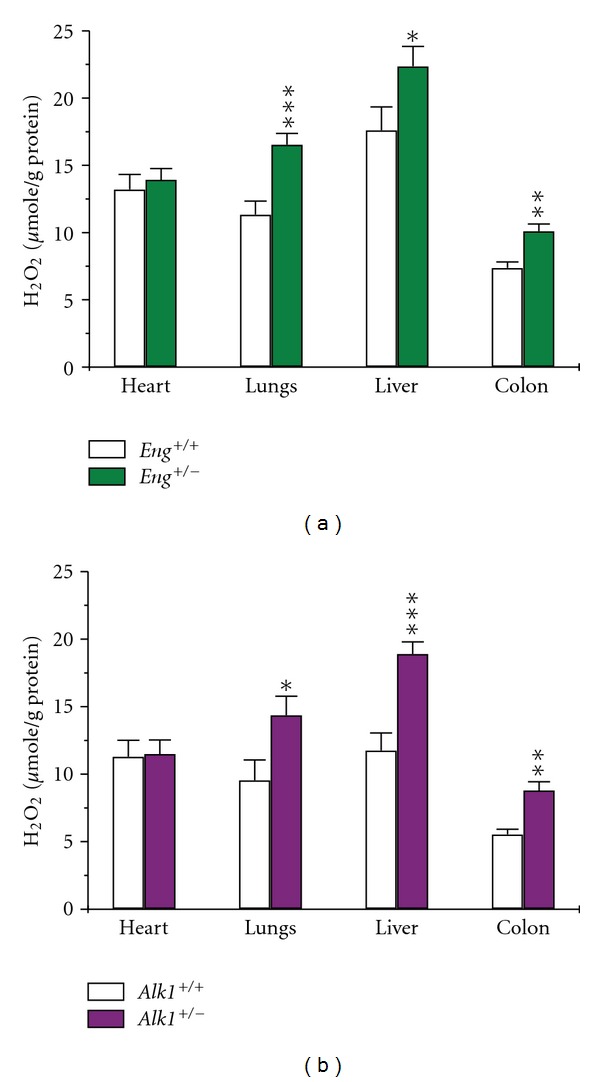
ROS production is increased in tissues of mutant mice. H2O2 production was measured by Amplex red, to estimate tissue ROS production. H2O2 generation was increased in pulmonary, hepatic, and colonic tissues of (a) Eng +/− and (b) Alk1 +/− mice compared to those of corresponding wild-type mice. Cardiac ROS production was similar between mutant and control mice. *P < 0.05, **P < 0.01, and ***P < 0.001 versus +/+ mice; N = 10–15/group for Eng mice and 7–20/group for Alk1 mice.
To confirm that H2O2 levels reflect ROS production, we estimated the amount of the lipid peroxidation product 8-iso-PGF2α in lungs of heterozygous mice. We found higher levels of this product in Eng +/− lungs (0.43 ± 0.05 ng/g of tissue) compared to littermate controls (0.28 ± 0.04 ng/g of tissue; N = 8, P < 0.05). We also observed increased levels of pulmonary 8-iso-PGF2α in Alk1 +/− mice as reported previously [16]. These results confirm the higher oxidative stress in lungs of Eng +/− and Alk1 +/− mice compared to littermate controls and validate the use of Amplex red assay as a means of assessing ROS.
3.3. L-NAME Decreases ROS Production in Tissues of Eng and Alk1 Heterozygous Mice
To ascertain the source of increased ROS production in Eng +/− and Alk1 +/− mice, we compared the ability of various inhibitors to affect H2O2 levels. We showed that the NOS inhibitor L-NAME, significantly reduced H2O2 generation in lungs, liver, and colon of Eng +/− and Alk1 +/− mice, but had no effect on wild-type mice (Figures 3(a) and 3(b)). This result suggests that eNOS is indeed uncoupled in the heterozygous mice leading to ROS production whereas in control mice, eNOS is normal and produces NO and not ROS.
Figure 3.
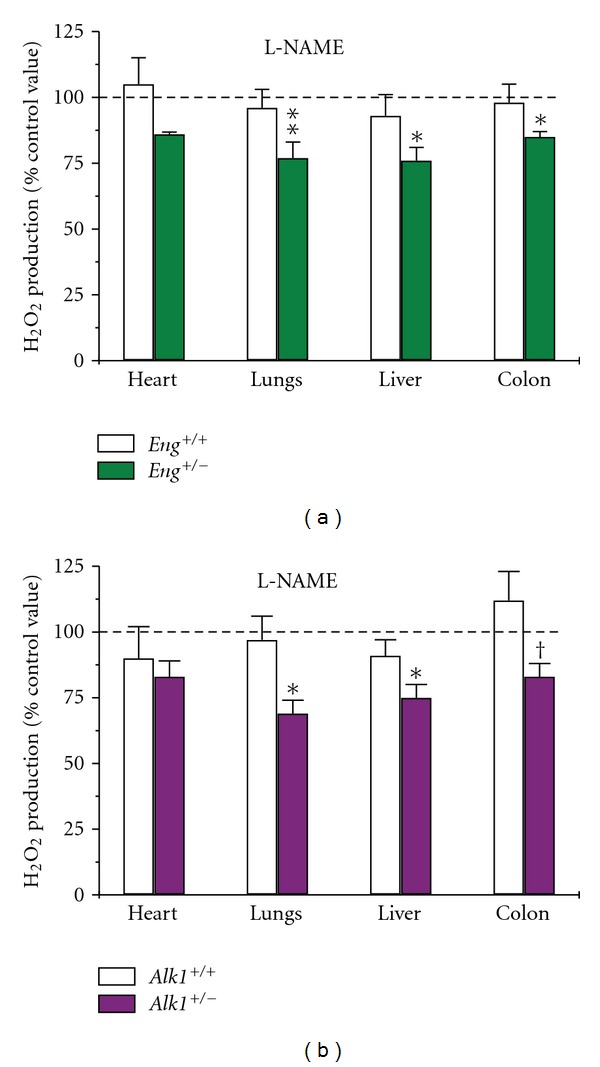
ROS production is inhibited by L-NAME in tissues of mutant mice. H2O2 production was measured by Amplex red with and without the NOS inhibitor L-NAME. Results are expressed as a percentage of the respective control values (without inhibitors). L-NAME significantly inhibited H2O2 production in pulmonary, hepatic, and colonic tissues of (a) Eng +/− and (b) Alk1 +/− mice but had no effect on tissues of control mice. In cardiac tissue, L-NAME effect was not significantly different between mutant and control mice. *P < 0.05, **P < 0.01, and † P < 0.1 versus samples from corresponding control values; N = 6–13/group for both Eng and Alk1 mice.
3.4. Mitochondrial and NADPH Oxidase-Mediated ROS Production Is Similar in Tissues of Mutant and Control Mice
Antimycin, a respiratory chain inhibitor, decreased ROS production to the same extent in tissues of Eng +/−, Alk1 +/− mice and their respective wild-type controls (Figures 4(a) and 4(b)). The strongest effect was seen in liver, where 50%–75% of ROS was antimycin-inhibitable followed by lungs (25%–50%), heart (25%–30%), and colon (5%–25%) (Figures 4(a) and 4(b)). However, the absolute levels of H2O2 measured in antimycin-treated samples were still significantly higher in lungs and liver of Eng +/− (P < 0.01) and Alk1 +/− (P < 0.05) mice compared to their respective wild-type controls, indicating higher overall ROS production. Apocynin, an NADPH oxidase inhibitor, blocked half of ROS production in tissues of both mutant and wild-type mice (Figures 4(c) and 4(d)). The biggest inhibitory effect of apocynin was observed in colon, followed by lungs, heart, and liver. There was no significant difference in NADPH-dependent H2O2 production between heterozygous and wild-type mice.
Figure 4.
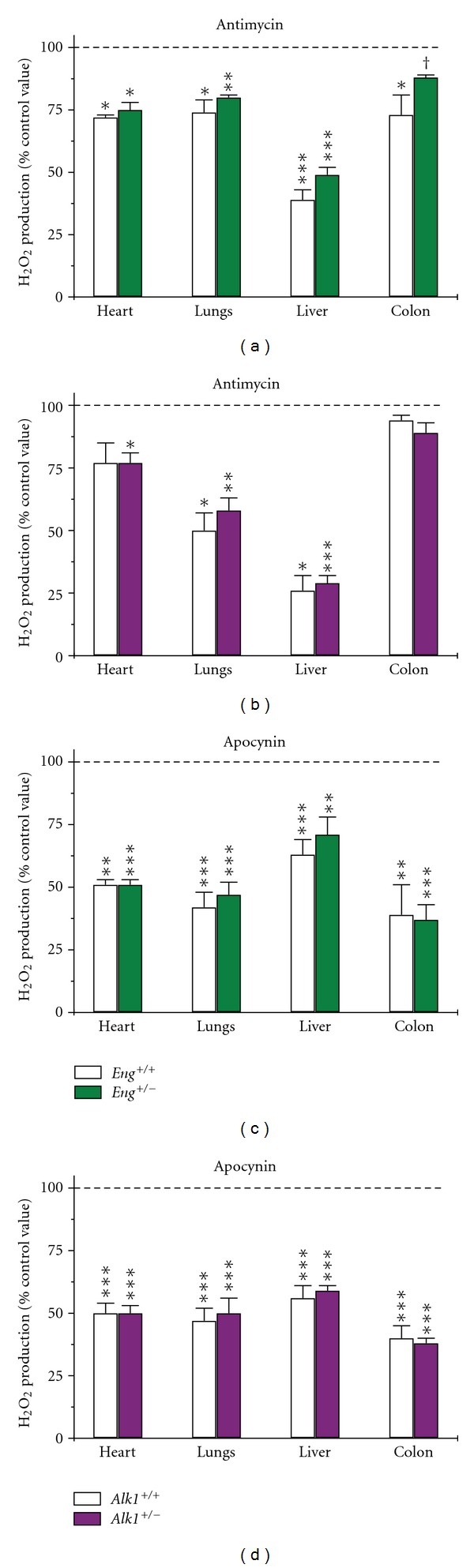
Mitochondrial and NADPH-dependent ROS production does not differ between mutant and wild-type mice. Results are expressed as a percentage of the respective control values in the absence of inhibitor. (a) and (b) Antimycin, 50 μM, inhibited mitochondrial ROS production to the same level in tissues of both Eng +/−, Alk1 +/− and respective wild-type mice. N = 5–11/group for Eng mice and 5–15/group for Alk1 mice, except for liver samples from Alk1 +/+ mice, where N = 4. (c) and (d) Apocynin, 100 μM, inhibited NADPH-oxidase dependent H2O2 production in tissues of both mutant and wild-type mice. N = 5–8/group for both Eng and Alk1 mice, except for heart samples from both groups of mice, where N = 4. *P < 0.05, **P < 0.01, ***P < 0.001, and † P < 0.1 versus corresponding control values (samples without inhibitor).
4. Discussion
We report that Eng and Alk1 heterozygous mice have decreased NO production and increased ROS levels in several organs, indicating that oxidative stress may lead to endothelial dysfunction. This is the first report comparing the levels of ROS produced in different tissues of these mice and establishing that decreased NO and increased ROS generation can be generalized to several organs. The major source of ROS overproduction in Eng +/− and Alk1 +/− mice is NO synthase, suggesting uncoupled eNOS as a major mechanism of increased oxidative stress in lungs, liver, and colon of Eng +/− and Alk1 +/− mice. We also showed that in liver, mitochondria contribute most ROS while NADPH oxidase is the major source in other tissues. However, the percentage of ROS produced by these two sources is similar in HHT and control mice.
Reactive oxygen species have been recognized as important players in both physiological and pathophysiological conditions. A small amount of ROS is necessary and serves as a physiological regulator of intracellular signalling pathways [20]. However, an overproduction of ROS (such as superoxide, hydrogen peroxide, hydroxyl radicals, and peroxynitrite) contributes to the pathogenesis of different diseases [21, 22]. The most prominent role of oxidative stress is in the pathologic mechanisms underlying endothelial dysfunction in cardiovascular disorders such as hypertension, hyperlipidemia, diabetes, arteriosclerosis, vasculitis, and myocarditis, to name a few. Although multiorgan vascular dysplasia and vascular stress are hallmarks of HHT [23], there are few studies investigating ROS involvement in the pathogenesis of this disease. We previously reported that increased H2O2 generation in lungs of Eng +/− mice accounts for the heightened pulmonary vasorelaxation [24]. We also showed that the onset of PAH in Eng +/− and Alk1 +/− adult mice is attributable to increased endothelial oxidative stress, as it could be prevented by antioxidant treatment [15, 16]. These findings are highly relevant as individuals with ALK1 and ENG mutations can develop PAH, with or without HHT [25–27]. Also, mutations in the gene encoding the BMP receptor type II (BMPR2) underlie the majority of cases of familial PAH and up to 40% of idiopathic cases [28]. In addition, increased oxidative stress has been observed in patients with PAH [29, 30]. These findings suggest a functional role for endoglin, ALK1, and BMPR2 in the regulation of vascular function and oxidative stress.
To our knowledge, this is the first report showing increased ROS production in several major organs of Eng +/− and Alk1 +/− mice, including lungs, liver, and colon, which are affected in HHT. We estimated ROS levels by measuring H2O2 by the Amplex red assay. In a previous paper [16] we observed a good correlation between Amplex red assay results and ROS levels estimated using fluorescent probes (H2DCFDA and DHE), direct H2O2 measurements by ultrasensitive probe, and the lipid peroxidation product 8-iso-PGF2α.
We identified NOS as a major source of ROS overproduction in tissues of Eng +/− and Alk1 +/− mice. Our previous work showed eNOS uncoupling in endothelial cells from HHT patients and from Eng and Alk1 heterozygous mice [14, 16]. eNOS is closely associated with caveolae/lipid rafts in endothelial cells, where endoglin and ALK1 colocalize with eNOS serving as scaffolding molecules for eNOS/Hsp90 association and eNOS activation [14, 15, 31]. In the mutant mice, eNOS uncoupling renders the enzyme refractory to regulation by TGF-β/BMP signaling and represents a critical event leading to excessive oxidative stress. Uncoupled eNOS forms monomers and generates ROS rather than NO, as shown in our proposed model (Figure 5). Neuronal NOS was not observed, and inducible NOS was barely detectable in mouse tissues [13, 16]; therefore, we conclude that the observed increase in L-NAME inhibitable H2O2 in tissues of HHT mice (Figure 3) is eNOS-derived. Indeed, eNOS uncoupling has been suggested as one of the main pathomechanisms in a broad range of cardiovascular and pulmonary disorders [32]. Our results strongly suggest that this phenomenon occurs in multiple organs of the mutant mice and can explain the decrease in NO and the increase in ROS production (Figures 1 and 2). We also previously reported a systemic decrease in NO bioavailability as determined by lower plasma nitrate levels in Eng +/− mice [13, 14]. NOS-dependent (L-NAME inhibitable) H2O2 production was also the only source of ROS that significantly differed between mutant and control mice.
Figure 5.
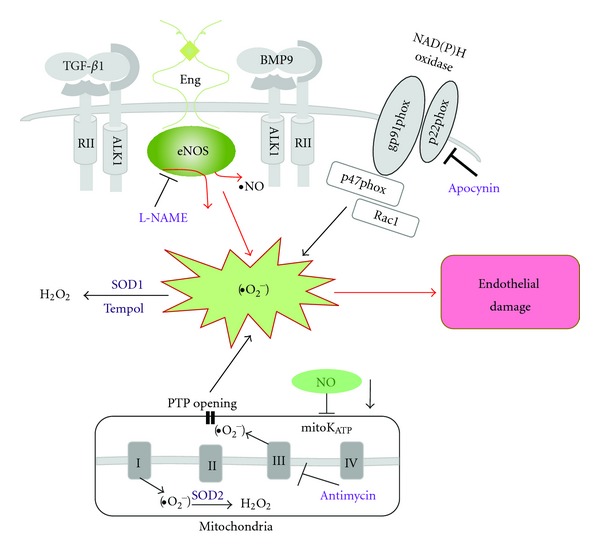
Model of oxidative stress in mutant mice leading to vascular endothelial damage. In Eng +/− and Alk1 +/− mice, altered eNOS activation renders the enzyme refractory to regulation by TGF-β/BMP signaling and represents a critical event leading to excessive oxidative stress. Uncoupled eNOS produces low amounts of NO and high levels of oxygen radicals (•O2 −). The NOS inhibitor, L-NAME, inhibits ROS production in tissues of mutant mice. Superoxide dismutase (SOD) and the SOD mimetic compound Tempol convert •O2 − into less harmful hydrogen peroxide (H2O2). A large portion of ROS is produced by mitochondria (Antimycin-inhibitable) and NADPH oxidases (Apocynin-inhibitable) however that percentage does not differ between mutant and control mice. Low NO cellular level may also inhibit mitochondrial KATP channel (mitoKATP) opening, trigger permeability transition pores (PTP) opening, and further increase the oxidative stress caused by mitochondrial ROS release.
Mitochondria have long been known as an important source of ROS. The role of small quantities of mitochondrial ROS in cellular signaling was recently recognized [33], but it is generally assumed that an increase in mitochondrial ROS is involved in the pathobiology of many vascular and neurodegenerative disorders [34]. Experiments in isolated mitochondria estimate a 2%-3% leakage of electrons to form •O2 − [35] that is largely dismutated to H2O2 by manganese superoxide dismutase (SOD2) in the mitochondrial matrix (Figure 5). The importance of mitochondrial ROS and their removal is emphasized by the observation that SOD2 null mice exhibit perinatal lethality due to cardiac dysfunction [36]. We found that antimycin, a mitochondrial complex III inhibitor [37], significantly inhibited H2O2 production in tissues of mutant and control mice (Figures 4(a) and 4(b)). Despite the fact that the antimycin-inhibitable ROS portion differed between organs, it was similar between heterozygous and control mice. However, despite a similar percentage of inhibition, the absolute amount of H2O2 estimated in antimycin-treated samples was higher in lungs and liver of mutant mice. This might be explained by NO reduction indirectly inhibiting the mitochondrial KATP channel opening (Figure 5), triggering the opening of the permeability transition pores and further increasing oxidative stress caused by mitochondrial ROS release [38, 39].
The NADPH oxidases have emerged as relevant enzymatic sources. In fact, NADPH oxidase is a major ROS generator in endothelial cells [40]. The NADPH oxidase enzyme complex consists of two membrane-bound components, gp91phox (also known as Nox2) and p22phox, and several cytosolic regulatory subunits, including p47phox, p67phox, and the small GTPase Rac (Figure 5). In endothelial cells Nox1, Nox2, Nox4, and Nox5 are expressed [40]. Upon enzyme activation, the cytoplasmic subunits translocate to the cell membrane and the resulting complex transfers electrons from NAD(P)H to molecular oxygen to form •O2 − [41]. The small amount of ROS produced in endothelial cells by NADPH oxidase may help maintain the critical balance between superoxide and NO levels in vessels. Recently it has been shown that endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived ROS [42]. Excessive amounts of ROS produced by these enzymes contribute to the development of cardiovascular diseases including hypertension and atherosclerosis [41]. However, the use of NADPH oxidase antagonists has been limited to animal studies because the majority of these compounds, including apocynin used in this study and diphenylene iodonium, may possess nonspecific off-target effects. New triazolo pyrimidines, such as VAS3947, are specific for NADPH oxidases but their possible interference with alternative sources of ROS, such as the mitochondrial electron chain, cannot be excluded [43]. On the other hand, it is known that beneficial effects of some antihypertensive drugs (i.e., angiotensin-converting enzyme inhibitors and AT1 receptor antagonists) are partly due to their ability to inhibit NADPH oxidase activation and expression [41]. Results of our study have confirmed that NADPH oxidase is the major source of ROS production in heart, lungs, and colon but not in liver. However, the apocynin-dependent H2O2 production was similar in heterozygous and control mice indicating that the portion of NADPH-dependent ROS is not different in tissues of mutant and control mice (Figures 4(c) and 4(d)).
It is well known that intracellular ROS levels are regulated by the balance between ROS generating enzymes and antioxidant enzymes that include SOD (Figure 5) and catalase. In previous studies, we have shown that expression of SOD1, 2, and 3 and catalase is similar in lungs of Eng +/− [24] and Alk1 +/− [16] mice when compared to control mice.
In summary, our study demonstrates increased oxidative stress in several organs of Eng +/− and Alk1 +/− mice and suggests eNOS uncoupling as the major source of ROS overproduction. This excessive ROS generation may play a pivotal role in the pathobiology of endothelial dysfunction in HHT. Our results also suggest that HHT patients may benefit from antioxidant treatment. We showed previously that the antioxidant Tempol was able to prevent the onset of PAH in Eng +/− and Alk1 +/− mice [15, 16]. In HHT patients, the only trial carried out to date was a pilot study in which the antioxidant N-acetylcysteine was given to patients for twelve weeks [44]. The results of this study look promising, with a significant decrease in the frequency and severity of epistaxis in treated HHT patients, improving their quality of life. However, antioxidant treatments used in some clinical studies to ameliorate cardiovascular disease have showed little or no improvement [45]. General strategies with antioxidants used for different diseases may not be very effective because ROS production differs between organs. Effective antioxidant treatment should be tailored differently for various conditions. Our results suggest that antioxidants are promising therapeutic tools in HHT and encourage their use in experimental and clinical studies that will help determine most effective therapeutic strategies.
5. Conclusions
Our study has demonstrated that Eng and Alk1 heterozygous mice show decreased NO and increased eNOS-dependent ROS production in several organs suggesting the role of oxidative stress in endothelial dysfunction in these mice. Results of our studies also suggest that HHT patients may benefit from antioxidant treatment.
Conflict of Interests
The authors declare that they have no conflict of intersts.
Acknowledgments
The authors thank M. Post, D. Reynaud, and M. Leadley from The Hospital for Sick Children, Toronto, for 8-iso-PGF2α measurements and expertise and comments. they thank S.P. Oh, University of Florida, for giving them Alk1 heterozygous mice. This work was supported by the Heart and Stroke Foundation of Canada (Grant no. T5598) to M. Letarte.
References
- 1.McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genetics in Medicine. 2011;13(7):607–616. doi: 10.1097/GIM.0b013e3182136d32. [DOI] [PubMed] [Google Scholar]
- 2.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-β superfamily. Journal of Biological Chemistry. 1999;274(2):584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- 3.David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(5):1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 4.Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) The Lancet. 2004;363(9412):852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 5.Cole SG, Begbie ME, Wallace GMF, Shovlin CLL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. Journal of Medical Genetics. 2005;42(7):577–582. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. American Journal of Medical Genetics A. 2006;140(20):2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 7.Berg J, Porteous M, Reinhardt D, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. Journal of Medical Genetics. 2003;40(8):585–590. doi: 10.1136/jmg.40.8.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Letteboer TGW, Mager JJ, Snijder RJ, et al. Genotype-phenotype relation in hereditary hemorrhagic teleangiectasia. Journal of Medical Genetics. 2006;43(4):371–377. doi: 10.1136/jmg.2005.035451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pece-Barbara N, Cymerman U, Vera S, Marchuk DA, Letarte M. Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Human Molecular Genetics. 1999;8(12):2171–2181. doi: 10.1093/hmg/8.12.2171. [DOI] [PubMed] [Google Scholar]
- 10.Ricard N, Bidart M, Mallet C, et al. Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: a diagnostic tool for novel ACVRL1 mutations. Blood. 2010;116(9):1604–1612. doi: 10.1182/blood-2010-03-276881. [DOI] [PubMed] [Google Scholar]
- 11.Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. Journal of Clinical Investigation. 1999;104(10):1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasan S, Hanes MA, Dickens T, et al. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Human Molecular Genetics. 2003;12(5):473–482. doi: 10.1093/hmg/ddg050. [DOI] [PubMed] [Google Scholar]
- 13.Jerkic M, Rivas-Elena JV, Prieto M, et al. Endoglin regulates nitric oxide-dependent vasodilatation. The FASEB Journal. 2004;18(3):609–611. doi: 10.1096/fj.03-0197fje. [DOI] [PubMed] [Google Scholar]
- 14.Toporsian M, Gros R, Kabir MG, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circulation Research. 2005;96(6):684–692. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- 15.Toporsian M, Jerkic M, Zhou YQ, et al. Spontaneous adult-onset pulmonary arterial hypertension attributable to increased endothelial oxidative stress in a murine model of hereditary hemorrhagic telangiectasia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(3):509–517. doi: 10.1161/ATVBAHA.109.200121. [DOI] [PubMed] [Google Scholar]
- 16.Jerkic M, Kabir MG, Davies A, et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovascular Research. 2011;92(3):375–384. doi: 10.1093/cvr/cvr232. [DOI] [PubMed] [Google Scholar]
- 17.Montezano AC, Touyz RM. Reactive oxygen species and endothelial function - role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic and Clinical Pharmacology and Toxicology. 2012;110(1):87–94. doi: 10.1111/j.1742-7843.2011.00785.x. [DOI] [PubMed] [Google Scholar]
- 18.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circulation Research. 2003;93(7):682–689. doi: 10.1161/01.RES.0000095246.40391.3B. [DOI] [PubMed] [Google Scholar]
- 19.Copland IB, Reynaud D, Pace-Asciak C, Post M. Mechanotransduction of stretch-induced prostanoid release by fetal lung epithelial cells. American Journal of Physiology. 2006;291(3):L487–L495. doi: 10.1152/ajplung.00510.2005. [DOI] [PubMed] [Google Scholar]
- 20.Finkel T. Signal transduction by reactive oxygen species. Journal of Cellular Biology. 2011;194(1):7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovascular Research. 1999;43(3):562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 22.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49(4):717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. Journal of Medical Genetics. 2006;43(2):97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belik J, Jerkic M, McIntyre BAS, et al. Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. American Journal of Physiology. 2009;297(6):L1170–L1178. doi: 10.1152/ajplung.00168.2009. [DOI] [PubMed] [Google Scholar]
- 25.Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. The New England Journal of Medicine. 2001;345(5):325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 26.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. American Journal of Respiratory and Critical Care Medicine. 2010;181(8):851–861. doi: 10.1164/rccm.200908-1284OC. [DOI] [PubMed] [Google Scholar]
- 27.Mache CJ, Gamillscheg A, Popper HH, Haworth SG. Early-life pulmonary arterial hypertension with subsequent development of diffuse pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia type 1. Thorax. 2008;63(1):85–86. doi: 10.1136/thx.2007.076109. [DOI] [PubMed] [Google Scholar]
- 28.Machado RD, Aldred MA, James V, et al. Mutations of the TGF-b Type II receptor BMPR2 in pulmonary arterial hypertension. Human Mutation. 2006;27(2):121–132. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 29.Cracowski JL, Cracowski C, Bessard G, et al. Increased lipid peroxidation in patients with pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine. 2001;164(6):1038–1042. doi: 10.1164/ajrccm.164.6.2104033. [DOI] [PubMed] [Google Scholar]
- 30.Bowers R, Cool C, Murphy RC, et al. Oxidative stress in severe pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine. 2004;169(6):764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 31.Santibanez JF, Blanco FJ, Garrido-Martin EM, Sanz-Rodriguez F, Del Pozo MA, Bernabeu C. Caveolin-1 interacts and cooperates with the transforming growth factor-β type I receptor ALK1 in endothelial caveolae. Cardiovascular Research. 2008;77(4):791–799. doi: 10.1093/cvr/cvm097. [DOI] [PubMed] [Google Scholar]
- 32.Gielis JF, Lin JY, Wingler K, Van Schil PEY, Schmidt HH, Moens AL. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radical Biology and Medicine. 2011;50(7):765–776. doi: 10.1016/j.freeradbiomed.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 33.Bell EL, Klimova TA, Eisenbart J, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. Journal of Cell Biology. 2007;177(6):1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Lisa F, Kaludercic N, Carpi A, Menabò R, Giorgio M. Mitochondria and vascular pathology. Pharmacological Reports. 2009;61(1):123–130. doi: 10.1016/s1734-1140(09)70014-3. [DOI] [PubMed] [Google Scholar]
- 35.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 37.Henderson JR, Swalwell H, Boulton S, Manning P, McNeil CJ, Birch-Machin MA. Direct, real-time monitoring of superoxide generation in isolated mitochondria. Free Radical Research. 2009;43(9):796–802. doi: 10.1080/10715760903062895. [DOI] [PubMed] [Google Scholar]
- 38.Wajima T, Shimizu S, Hiroi T, Ishii M, Kiuchi Y. Reduction of myocardial infarct size by tetrahydrobiopterin: possible involvement of mitochondrial KATP channels activation through nitric oxide production. Journal of Cardiovascular Pharmacology. 2006;47(2):243–249. doi: 10.1097/01.fjc.0000201360.71813.8a. [DOI] [PubMed] [Google Scholar]
- 39.Chang JC, Kou SJ, Lin WT, Liu CS. Regulatory role of mitochondria in oxidative stress and atherosclerosis. World Journal of Cardiology. 2010;2(6):150–159. doi: 10.4330/wjc.v2.i6.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxidants and Redox Signaling. 2009;11(4):791–810. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schramm A, Matusik P, Osmenda G, Guzik TJ. Targeting NADPH oxidases in vascular pharmacology. Vascular Pharmacology. 2012;56(5-6):216–231. doi: 10.1016/j.vph.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng J, Damrauer SM, Lee M, Sellke FW, Ferran C, Abid MR. Endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived reactive oxygen species. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(9):1703–1710. doi: 10.1161/ATVBAHA.110.209726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wind S, Beuerlein K, Eucker T, et al. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. British Journal of Pharmacology. 2010;161(4):885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Gussem EM, Snijder RJ, Disch FJ, Zanen P, Westermann CJJ, Mager JJ. The effect of N-acetylcysteine on epistaxis and quality of life in patients with HHT: a pilot study. Rhinology. 2009;47(1):85–88. [PubMed] [Google Scholar]
- 45.Sugamura K, Keaney JF., Jr. Reactive oxygen species in cardiovascular disease. Free Radical Biology and Medicine. 2011;51(5):978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]