

Abstract
Disassembly and phagocytic removal of dying cells is critical to maintain immune homeostasis. The factors regulating fragmentation and uptake of dying lymphocytes are not well understood. Degradation of fodrin, a cytoskeletal linker molecule that attaches CD45 to the actin cytoskeleton, has been described in apoptotic cells, although no specific initiator of fodrin degradation has been identified. CD45 is a glycoprotein receptor for galectin-1, an endogenous lectin that can trigger lymphocyte apoptosis. CD45 is not required for membrane changes or DNA degradation during galectin-1 death. However, here we show that fodrin degradation occurs during galectin-1 T cell death, and CD45 is essential for fodrin degradation to occur. In the absence of CD45 and fodrin degradation, cell death is not accompanied by membrane blebbing, indicating that fodrin degradation occurs via a distinct pathway compared to the pathway that initiates apoptotic membrane changes and DNA degradation. Moreover, there is slower phagocytic uptake of cells in which fodrin degradation is blocked relative to cells in which CD45-mediated fodrin degradation occurs. These studies identify a novel role for CD45 in regulating cellular disassembly and promoting phagocytic clearance during galectin-1 induced T cell death.
Apoptosis is a complex process that culminates in the disassembly and removal of dying cells. Clearance of apoptotic cells is critical for proper tissue development and homeostasis. Efficient phagocytic clearance of dying cells has also been proposed to prevent inflammation triggered by release of intracellular contents, as well as avoiding exposure of sequestered self-antigens that could provoke autoimmune disease (1–4).
Several apoptotic triggers for T cells have been described. Galectin-1 is a mammalian lectin expressed by many cell types that induces death of specific subsets of thymocytes and peripheral T cells. Galectin-1 regulates thresholds of positive and negative selection in the thymus, and selectively kills CD4 Th1 vs. Th2 cells, due to expression of preferred glycan ligands on the former (5–8). The galectin-1 death pathway is unique, in that galectin-1 T cell death requires CD7 as a galectin-1 receptor, appears to be caspase-independent, and involves a unique pattern of mitochondrial events, with selective release of endonuclease G (Endo G) that moves to the nucleus to degrade cellular DNA (9–11).
Galectin-1 also binds to CD45 on the T cell surface, although CD45 expression is not absolutely required for galectin-1 death of T cell lines (9,12). However, CD45 can both positively and negatively regulate susceptibility to galectin-1; if CD45 is expressed, the extracellular domain of CD45 must be appropriately glycosylated for galectin-1 binding and signaling to occur (8,12). The CD45 cytoplasmic region has two domains, a tyrosine phosphatase domain and a homologous domain that lacks enzymatic activity. The second domain interacts with the cytoskeletal linker protein fodrin (αII-spectrin); CD45 interaction with fodrin is critical for CD45 delivery to the plasma membrane, and also regulates enzymatic activity of the tyrosine phosphatase domain (13–15).
Fodrin is the major spectrin family member expressed in non-erythroid cells. Fodrin degradation accompanies apoptosis triggered by many death signals in many cell types, such as T cell death initiated by Fas ligation, B cell death initiated by TGF-β, and neural cell death initiated by staurosporine (16–18), although different proteases, including caspases and calpains, appear to be responsible for fodrin degradation in different apoptotic pathways (19–22). Fodrin degradation is proposed to contribute to apoptotic cell blebbing and other morphologic changes that occur during breakdown of dying cells (16,19,20). We found that, during galectin-1 T cell death, CD45-associated fodrin undergoes proteolytic degradation, in a parallel but separate process from the intracellular death pathway that results in phophatidylserine externalization and nuclear DNA cleavage. Fodrin degradation was required for the full spectrum of morphologic changes observed during galectin-1 T cell death. Moreover, fodrin degradation enhanced the rate of macrophage engulfment of galectin-1 treated T cells.
RESULTS AND DISCUSSION
CD45-associated fodrin degradation during galectin-1 induced T cell death
Treatment of Jurkat T cells with galectin-1 resulted in cleavage of fodrin from the 240 kD intact molecule into 150 kD and 120 kD fragments (Fig. 1A). These fodrin fragments have been described during apoptosis of several cell types treated with a variety of apoptotic triggers (16,18,22,23). The 150 kD fragment, in particular, can be generated by cellular proteases after cell lysis, even in control treated cells, so several protease inhibitors are included in lysis buffers (22), although a faint band at 150 kD can sometimes be detected in control treated cells. However, substantial fodrin cleavage was clearly seen in galectin-1 treated cells, with abundant 150 and 120 kD fragments generated. Fodrin that was cleaved during galectin-1 death was associated with CD45, as immunoprecipitation of CD45 after galectin-1 treatment demonstrated reduced full-length fodrin, and the presence of 150 kD and 120 kD fragments (Fig. 1B).
Figure 1.
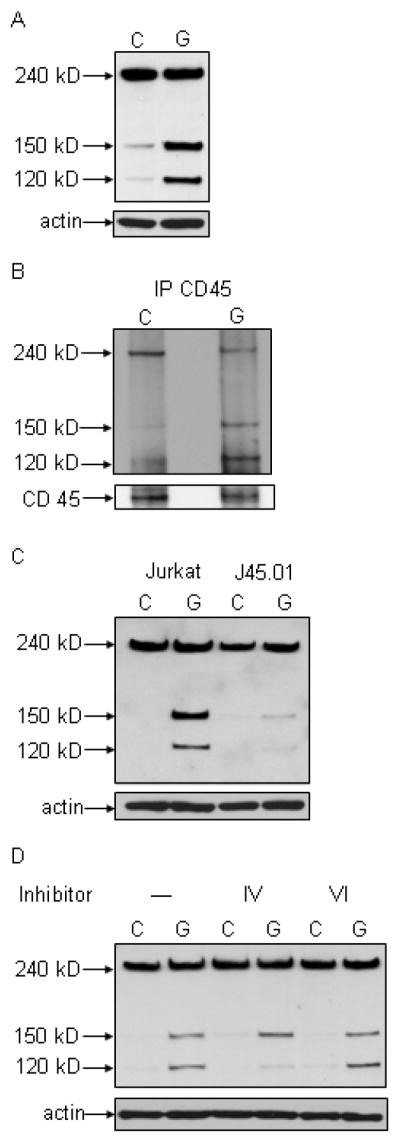
Galectin-1 binding to T cells results in fodrin degradation. A) Fodrin cleavage products of 150 kD and 120 kD are abundant in galectin-1 (G) Jurkat treated cells, compared to control (C) cells. B) Fodrin associates with CD45 before and after cleavage. CD45 was immunoprecipitated from control (C) and galectin-1 (G) treated Jurkat cells, and fodrin detected by immunoblotting. Full length fodrin associates with CD45 in control cells, while the 150 and 120 kD cleavage products associate with CD45 in galectin-1 treated cells. C) Fodrin cleavage does not occur in cells that lack CD45. Jurkat and J45.01 cells were treated with buffer control or galectin-1. Fodrin cleavage products are abundant in galectin-1 Jurkat cells, but not in J45.01 cells. D) Fodrin degradation was reduced in galectin-1 treated Jurkat cells in the presence of calpain inhibitor IV that inhibits m-calpain, while calpain inhibitor VI that inhibits μ-calpain had no effect on fodrin cleavage. Data are representative of three or more independent experiments for each panel.
CD45 expression appeared to be required for galectin-1 induced fodrin degradation, as J45.01 cells that do not express CD45 demonstrated no fodrin cleavage after galectin-1 treatment (Fig. 1C) compared to parental Jurkat cells. A requirement for CD45 for fodrin degradation appears to be specific for galectin-1 cell death, as we observed equivalent fodrin degradation in Jurkat and J45.01 cells after Fas ligation (data not shown).
Several proteases can participate in fodrin degradation in different cell death pathways, including caspases and m- and μ-calpains (19–22). Fodrin degradation during galectin-1 death, especially production of the 120 kD fragment, was reduced by an m-calpain inhibitor but not by a μ-calpain inhibitor (Fig. 1D), while a panel of caspase inhibitors had no effect on fodrin cleavage in galectin-1 treated cells (data not shown). Thus, fodrin cleavage occurs at least in part via m-calpain-mediated proteolysis during galectin-1 death.
These data demonstrate that fodrin degradation occurs in a CD45-mediated process during galectin-1 T cell death. While fodrin degradation occurs in many cell types in many death pathways, no specific cell surface receptor required for this process has been proposed. Similarly, no role for CD45 in cytoskeletal disassembly during apoptosis has been previously described.
Fodrin degradation occurs by a separate and parallel pathway from other apoptotic events in galectin-1 treated T cells
As shown in Fig. 1C, we observed no fodrin degradation in J45.01 cells. However, while CD45 is a major receptor for galectin-1 on T cells and CD45 can regulate T cell susceptibility to galectin-1, CD45 expression is not absolutely required for galectin-1 T cell death (12,24). This suggests that fodrin degradation occurs through a separate and parallel pathway from other events in galectin-1 T cell death; this is in contrast to other cell death pathways, in which fodrin degradation is tightly coupled to other events in the pathways (16,20,23).
To address this, we examined other markers of cell death in Jurkat and J45.01 cells, and Jurkat cells treated with calpain inhibitors, during galectin-1 cell death. As shown in Fig. 2A, phosphatidylserine externalization was virtually identical in Jurkat and J45.01 cells treated with galectin-1, although we observed minimal fodrin cleavage in J45.01 cells (Fig. 1C). Similarly, phosphatidylserine externalization was virtually identical in Jurkat cells treated with or without calpain inhibitors (Fig. 2B), although m-calpain inhibitor IV reduced fodrin degradation (Fig. 1D). Thus, inhibition of fodrin degradation either by absence of CD45 or by m-calpain inhibition did not affect the early event of phosphatidylserine externalization during cell death.
Figure 2.

Fodrin cleavage is not required for other hallmarks of cell death in galectin-1 treated Jurkat T cells. A) Annexin V binding to Jurkat and J45.01 cells treated with buffer control (thin line) or galectin-1 (thick line). B) Annexin V binding to Jurkat cells treated with buffer control (thin line) or galectin-1 (thick line) in the absence of presence of the indicated calpain inhibitors. C) DNA degradation detected by TUNEL in control or galectin-1 treated cells in the absence or presence of the indicated calpain inhibitors. Data are representative (A and B) or mean +/− SD of triplicate samples (C) from one of three independent experiments.
We also examined DNA degradation using the TUNEL assay. We observed no effect of the m-calpain inhibitor on the extent of TUNEL labeling of apoptotic cells (Fig. 2C), indicating that DNA degradation proceeded normally despite m-calpain inhibition and reduced fodrin degradation. Thus, fodrin degradation is not required upstream of DNA degradation in galectin-1 mediated T cell death. We also asked if inhibition of DNA cleavage affected fodrin degradation. Blocking mitochondrial release of Endo G reduced TUNEL labeling, but did not affect fodrin degradation in galectin-1 treated cells (data not shown). Thus, fodrin proteolysis and the events leading to plasma membrane changes and DNA degradation appear to occur via separate pathways during galectin-1 T cell death.
Fodrin degradation is important for CD45 clustering and morphologic changes during galectin-1 T cell death
During galectin-1 induced T cell death, CD45 clusters on membrane blebs and co-localizes with externalized phosphatidylserine on the blebs; this is in contrast to the uniform phosphatidylserine externalization seen on T cells during Fas-induced cell death (8,9,12). To ask if fodrin degradation is important for membrane blebbing and other morphologic changes that occur during galectin-1 T cell death, we examined cells treated with galectin-1 with or without m-calpain inhibitor IV.
Cells treated with galectin-1 displayed massive membrane blebbing typical of apoptotic cells (Fig. 3A). In contrast, cells treated with galectin-1 in the presence of m-calpain inhibitor IV displayed minimal blebbing or other morphologic changes detected by light microscopy. In control treated cells, CD45 and fodrin were uniformly distributed at the plasma membrane. After galectin-1 treatment, CD45 clustered primarily on apoptotic blebs. Moreover, the continuous ring of fodrin staining seen in control cells was disrupted in galectin-1 treated cells, with pronounced fodrin accumulation near apoptotic blebs. As the fodrin antibody does not distinguish intact from degraded fodrin by immunofluorescence, it is not clear whether fodrin that associates with CD45 in membrane blebs is intact or degraded, but the continuous fodrin staining observed in control cells was disrupted after galectin-1 binding.
Figure 3.

Fodrin cleavage promotes CD45 clustering and membrane blebbing during galectin-1 T cell death. A) (left) Jurkat cells were treated with buffer control (C) or galectin-1 (G) for 60 min in the absence or presence of calpain inhibitor IV. CD45 (green) and fodrin (red) were detected by immunofluorescence confocal microscopy. (right) CD45 (green) and annexin V (A5) binding were detected on cells treated as above. CD45 clustering, fodrin redistribution, and membrane blebbing were reduced in the presence of calpain inhibitor IV, compared to cells treated with galectin-1 alone. Annexin V binding was evident on cells treated with calpain inhibitor IV, but clustering on blebs was reduced compared to cells treated with galectin-1 alone. B) Jurkat and J45.01 cells were treated with buffer control (C) or galectin-1 (G), and CD45 and fodrin (left), or CD45 and annexin V binding (right), detected as in (A). No CD45 staining was detected on J45.01 cells. Fodrin redistribution, membrane blebbing, and annexin V clustering were markedly reduced in J45.01 cells compared to Jurkat cells. Data are representative of three independent experiments.
In the presence of m-calpain inhibitor IV, there was minimal blebbing of galectin-1 treated cells, and CD45 was present in small patches evenly distributed on the cell surface, in contrast to the large CD45 clusters on membrane blebs on cells treated with galectin-1 alone. Similarly, in the presence of m-calpain inhibitor IV, fodrin co-localized with CD45 on galectin-1 treated cells, but remained relatively evenly distributed around the cell perimeter, in contrast to the pronounced clustering of fodrin seen in cells treated with galectin-1 alone. Annexin V binding demonstrated that phosphatidylserine externalization occurred on cells in the absence or presence of the m-calpain inhibitor. However, clustered Annexin V staining on apoptotic blebs on cells treated with galectin-1 alone was not seen when cells were treated with galectin-1 plus m-calpain inhibitor IV.
While CD45 expression is not essential for phosphatidylserine externalization and TUNEL labeling during galectin-1 death, we have not previously examined membrane blebbing on CD45− cells treated with galectin-1. Galectin-1 treatment of J45.01 cells resulted in phosphatidylserine externalization, detected by annexin V binding, but we did not observe the membrane blebs on J45.01 cells that are typically seen on galectin-1 treated CD45+ cells (Fig. 3B). Moreover, as seen with m-calpain inhibitor IV (Fig. 3A), fodrin remained relatively evenly distributed around the cell perimeter in J45.01 cells after galectin-1 treatment, in contrast to the fodrin clustering seen on Jurkat cells. Thus, reducing fodrin degradation, either by loss of CD45 expression or by m-calpain inhibition, decreased membrane blebbing during galectin-1 death.
Reconstitution of CD45 expression restores fodrin degradation, CD45 clustering and membrane blebbing during galectin-1 T cell death
To directly ask if CD45 is required for fodrin degradation during galectin-1 cell death, we examined fodrin degradation in J45.01 cells reconstituted with full-length CD45 (RABC) or CD45 lacking the D1 tyrosine phosphatase domain (D1−) (25). J45.01 cells transfected with vector alone demonstrated no significant fodrin degradation after galectin-1 treatment, while reconstitution with full-length CD45RABC restored fodrin degradation after galectin-1 treatment (Fig. 4A). However, expression of CD45 lacking the D1 domain was not sufficient to restore fodrin cleavage in D1− cells treated with galectin-1. All four cell lines demonstrated other hallmarks of cell death after galectin-1 treatment, including annexin V binding and increased membrane permeability (data not shown). In addition, fodrin co-immunoprecipitated with both full length CD45 in the RABC cells and with mutant CD45 in the D1− cells (data not shown), demonstrating that lack of fodrin degradation in the D1− cells did not result from loss of fodrin association with CD45.
Figure 4.

Expression of CD45 is sufficient to restore fodrin cleavage after galectin-1 binding. J45.01 T cells were transfected with vector alone (Vector), full-length murine CD45(RABC) or CD45 lacking the intracellular D1 phosphatase domain (D1−). A) Fodrin cleavage was not detected in J45.01 cells transfected with vector alone. Expression of CD45RABC restored fodrin cleavage after galectin-1 binding, while no fodrin cleavage was detected in D1− cells. B) CD45 (green) and fodrin (red) localization was examined by immunofluorescence confocal microscopy. Cells expressing CD45RABC demonstrated membrane blebbing, CD45 clustering and fodrin redistribution after galectin-1 treatment, comparable to that observed for Jurkat cells, while these changes were not seen in D1− cells. C) Jurkat cells were treated with indicated concentrations of the tyrosine phosphatase inhibitor bpV(phen) prior to galectin-1. Inhibitor treatment abrogated fodrin degradation. D) Fodrin was precipitated from RABC and D1− cells, and immunoblots probed for phosphotyrosine and fodrin. The ratio of phosphorylated fodrin/total fodrin in RABC cells was reduced compared to D1− cells. Data are representative of three independent experiments.
We then examined morphologic changes in these cells during galectin-1 induced cell death (Fig. 4B). J45.01 cells transfected with vector alone did not display the massive blebbing seen with Jurkat cells, nor did fodrin cluster in these cells, after galectin-1 treatment. In contrast, cells reconstituted with full length CD45RABC, demonstrated membrane blebbing and co-localization of clustered CD45 with fodrin after galectin-1 treatment. However, cells transfected with the D1− CD45 construct had no apparent membrane blebbing after galectin-1 treatment, and cells had patchy but continuous CD45 and fodrin localization around the plasma membrane. These results suggested that the phosphatase domain of CD45 is essential for fodrin cleavage.
To ask if inhibiting tyrosine phosphatase activity affected fodrin degradation, we treated cells with the tyrosine phosphatase inhibitor bpV(phen) and examined fodrin degradation after galectin-1 binding. As previously described (12), bpV(phen) did not inhibit galectin-1 death of the cells, detected by annexin V binding and membrane permeability (data not shown). However, inhibition of tyrosine phosphatase activity markedly reduced fodrin degradation after galectin-1 treatment, indicating that tyrosine de-phosphorylation is important for fodrin cleavage (Fig. 4C). This is consistent with previous reports documenting that phosphorylation of a specific tyrosine residue in fodrin confers resistance to calpain-mediated cleavage (21,22).
A specific phosphatase responsible for fodrin dephosphorylation in mammalian cells has not been identified; however, fodrin binding to the D2 intracellular domain of CD45 promotes CD45 PTPase activity (13,25,26). To ask if loss of the D1 tyrosine phosphatase domain of CD45 affected fodrin phosphorylation, we determined baseline tyrosine phosphorylation status of fodrin in J45.01 cells expressing either full-length RABC or the D1− CD45 construct. Tyrosine phosphorylation of fodrin was increased approximately two-fold in cells expressing the D1− CD45 construct compared to cells expressing CD45RABC, as shown by examining the ratio of phosphorylated intact fodrin to total intact fodrin in these cells (Fig. 4D). This indicates that the CD45 tyrosine phosphatase domain modulates fodrin phosphorylation status, which may regulate fodrin susceptibility to proteolytic cleavage during galectin-1 cell death.
Enhanced phagocytosis of apoptotic T cells with degraded fodrin
As fodrin degradation is not essential for other events in galectin-1 induced T cell death, such as phosphatidylserine externalization and DNA degradation, we asked if fodrin degradation would enhance phagocyte clearance of dying cells. Clearance of apoptotic cells by phagocytes is important for normal tissue homeostasis, for prevention of inflammation resulting from release of cellular contents, and for removal of intracellular antigens that could be recognized as non-self and thus trigger autoimmunity (1–4,26).
We compared phagocytosis of control or galectin-1 treated Jurkat and J45.01 cells by RAW 264.7 macrophages. T cells were labeled with CFSE to visualize engulfed T cells within macrophages. Labeled T cells were treated with galectin-1 for the indicated period of time and added to a monolayer of RAW 264.7 cells for one hour prior to microscopic examination. After one hour, there was minimal blebbing and disintegration of either Jurkat or J45.01 cells, and similar numbers of Jurkat and J45.01 cells were observed on the RAW 264.7 monolayer. However, after three hours, there was pronounced fragmentation of galectin-1 treated Jurkat cells, and numerous RAW 264.7 cells displayed internalized T cell fragments. In contrast, J45.01 cells remained generally intact three hours after galectin-1 treatment, with minimal blebbing and cell fragmentation, as in Figs. 3 and 4. T cells engulfed by macrophages were clearly visible, although the RAW 264.7 cells contained fewer fragments of J45.01 cells than Jurkat cells.
Phagocytosis of control or galectin-1 treated cells was quantified in two ways. We calculated the phagocytic index (defined in Materials and Methods) (Fig. 5B), as well as the fraction of macrophages containing engulfed T cells or T cell fragments (Fig. 5C). When cells treated with galectin-1 for only one hour were added to RAW 264.7 cells, there was a significant increase in the phagocytic index for both Jurkat and J45.01 cells compared to control treated cells, but no difference in phagocytosis of the two T cell lines compared to each other. However, after three hours of galectin-1 treatment, we observed a significant reduction in the phagocytic index for J45.01 cells compared to Jurkat cells (Fig. 5B), consistent with the reduced fragmentation of J45.01 cells compared to Jurkat cells by this time point (Fig. 5A). In addition, the fraction of macrophages ingesting Jurkat cells treated with galectin-1 for three hours was greater than that observed for Jurkat cells treated for one hour; in contrast, there were fewer macrophages containing J45.01 cells after one or three hours of galectin-1 treatment, compared to Jurkat cells, and increased time of galectin-1 treatment did not appreciably increase the fraction of macrophages with ingested J45.01 cells (Fig. 5C).
Figure 5.

Fodrin cleavage promotes phagocytosis of dying cells. A) Jurkat and J45.01 cells were labeled with CFSE prior to addition of buffer control or galectin-1 for 6 hrs. T cells were added to RAW 264.7 macrophage monolayers for 1 or 3 hr. Phagocytosis of labeled T cells was visualized by confocal microscopy. B) Phagocytotic index of macrophages with Jurkat or J45.01 cells. * at 1 hr, p = 0.39 for Jurkat vs. J45.01 cells treated with galectin-1. ** at 3 hr, p < 0.02 for Jurkat vs. J45.01 cells treated with galectin-1. (C) The fraction of macrophages with ingested cells. * at 1 hr, p = 0.02 for Jurkat vs. J45.01 cells treated with galectin-1. ** at 3 hr, p < 0.01 for Jurkat vs. J45.01 cells treated with galectin-1. For (B) and (C), values are mean +/− SD of triplicate samples from one of three independent experiments.
These data demonstrate that macrophages more efficiently phagocytose dying cells with cleaved fodrin, compared to dying cells in which fodrin proteolysis is prevented. While fodrin proteolysis accompanied membrane blebbing (Figs. 3, 4), membrane blebbing alone may not be sufficient to promote efficient phagocytosis, as inhibition of membrane blebbing during Jurkat cell apoptosis by blocking the Rho kinase ROCK-I did not alter the rate of macrophage ingestion of the cells (27). This suggests that cytoskeletal disassembly, rather than blebbing, contributes to efficient phagocytic clearance of dying cells. Moreover, these experiments identify a novel function for CD45 during galectin-1 T cell death, in promoting fodrin degradation to enhance phagocytic clearance of dying T cells.
Alterations in the intracellular and extracellular domains of CD45 are associated with autoimmune disease in humans and mice (28,29). The requirement for the CD45 D1 tyrosine phosphatase domain for efficient fodrin cleavage (Fig. 4) suggests that mutations in either extracellular or intracellular CD45 domains that affect fodrin cleavage could affect the rate of phagocytic clearance of cells killed by galectin-1 in vivo, and thus contribute to the increased frequency of autoimmunity associated with specific CD45 mutations.
These data describe a novel role for CD45 in regulating fodrin degradation during galectin-1 death. Conversely, is there a role for fodrin degradation in regulating CD45 function? Clustering of CD45 by galectin-1 or spontaneous oligomerization of CD45 both reduce CD45 tyrosine phosphatase activity, although the precise mechanism of this effect has not been elucidated (29,30). Association of the CD45 D2 domain with fodrin enhances tyrosine phosphatase activity of the D1 domain, perhaps by keeping the CD45 cytoplasmic tail in an extended conformation (13,25). It will be of interest to determine if fodrin degradation contributes to reduced CD45 tyrosine phosphatase activity after galectin-1 binding.
Galectin-mediated clustering of cell surface glycoproteins is proposed to regulate a variety of events on cell surface, including TCR segregation to induce anergy in tumor-infiltrating CD8 T cells, restricting TCR signaling during thymocyte selection and peripheral T cell response to antigen, determination of cytokine receptor residency time on mammary tumor cells and glucose transporter 2 residency time on pancreatic beta cells, polarized sorting of glycoproteins in renal epithelial cells, and glycoprotein association with lipid rafts on intestinal epithelial cells (7,31–36). These events may be regulated solely by galectin binding to glycans on the glycoprotein extracellular domains; however, none of these studies have addressed if changes in cytoskeletal tethering via glycoprotein intracellular domains is required for galectin-glycoprotein interactions. Our data clearly demonstrate that the dramatic clustering of CD45 observed after galectin-1 binding requires fodrin degradation, and thus release of the CD45 intracellular domain from cytoskeletal tethering.
These studies demonstrate a novel role for CD45 in cytoskeletal disassembly during a specific cell death pathway, describe a mode of cytoskeletal disassembly that is separable from other cellular events in this death pathway, and support a role for CD45-mediated cytoskeletal disassembly in efficient phagocytic clearance of apoptotic cells. Understanding the processes that regulate apoptotic cell clearance may promote new approaches to halting initiation or progression of autoimmune disease, while identification of novel actions for CD45 may elucidate the roles of this abundant and complex glycoprotein during lymphocyte development and function in the periphery.
MATERIALS AND METHODS
Cell lines and reagents
Jurkat E6-1 cells (gift of Fu-Tong Liu, UC Davis) and J45.01 cells (ATCC, Rockville, MD) were maintained in RPMI 1640 (Invitrogen) with 10% FBS (Hyclone), 10 mM HEPES, 2 mM Glutamax, 2 mM sodium puruvate in 5% CO2 at 37°C. J45.01 cells transfected with CD45 or vector alone were maintained in the above media with 0.6 μg/ml G418. RAW 264.7 cells (ATCC) were maintained in DMEM (Invitroten, 10% FBS, 2 mM Glutamax. Antibodies were from the indicated sources: monoclonal mouse anti-fodrin (Chemicon), polyclonal rabbit CD45 (Abcam), monoclonal mouse phospho-tyrosine (Cell Signaling), monoclonal mouse CD45(LCA) (Dako), monoclonal mouse CD45 Alexa 488 (Biolegend), goat anti-mouse Alexa 594, goat anti-rabbit Alexa 488 (Invitrogen), goat anti-mouse HRP(Biorad). Additional reagents: Annexin V-Alexa 594 (Invitrogen), Prolong Gold Anti-fade mounting media, Annexin V-FITC (Molecular Probes), caspase inhibitor I (z-vad-fmk), calpain inhibitor IV (z-lly-fmk) and calpain inhibitor VI (SJA 6017) (Calbiochem), Apo-Direct kit (BD Biosciences), Enhanced Chemiluminescence (ECL) (GE Healthcare). Galectin-1 was prepared as previously described (9).
T cell death assays
T cell death assays were done as previously described (9–11). Briefly, 1.25 × 106 Jurkat or J45.01 cells in triplicate were pretreated with +/− 20uM calpain inhibitors for 1 h at 37°C in 200ul of the relevant +/− G418 prior to adding 20 μM galectin-1 or buffer control for 6 h at 37°C, 5% CO2. Cells were dissociated with 0.1 M lactose in PBS. Fifty microliters in duplicates were removed for detection of cell death with AnnexinV-FITC and propidium iodide. Data were acquired on a FACscan flow cytometer and analyzed using CellQuest software (BD Biosciences). The remainder of the samples were combined for parallel experiments (immunoblot, immunoprecipitation, immunofluorescence). Terminal dUTP nick end labeling (TUNEL) was performed on cells fixed in 1% paraformaldehyde to manufacturer’s directions (ApoDirect Kit). Data were acquired and analyzed as above.
Immunoblotting
For fodrin analysis, cells were treated with galectin-1 or buffer control as described above. Cells were suspended in 0.1 mM pervanadate for 30 min, 37°C before lysing in 50mM Tris-HCl, pH 7.4, 1% NP-40, 5mM EDTA, 150mM NaCl, 1mM PMSF, 10ug/ml aprotinin, 10ug/ml leupeptin, 10mM sodium orthovanadate for 15 mins on ice (22). Lysates were microfuged for 15 mins at 10,000 rpm. 10 μg of lysates were separated on a 3–8% Tris-acetate gel (Invitrogen NuPAGE Electrophoresis System) and electroblotted onto nitrocellulose (Whatman). The membrane was blocked and probed as previously described (9) and proteins visualized by ECL. For immunoprecipitation, 1 × 107 cells treated with galectin-1 or buffer control were incubated with 3ug fodrin antibody or CD45 antibody with proteinG beads (Pierce) overnight, and washed extensively with lysis buffer prior to processing as above. Samples were denatured in NuPAGE reducing agent and NuPAGE SDS Sample buffer (Invitrogen) prior to loading.
Confocal immunofluorescence microscopy
Cells were treated with galectin-1 or buffer control as above, washed with cold PBS, fixed with 1 ml 4% paraformadehyde in PBS for 30 mins on ice, and quenched with 3 ml 0.2 M glycine in PBS. Pelleted cells were washed with PBS, and blocked overnight with 2% goat serum in PBS at 4°C. Cells not permeabilized were incubated with CD45-Alexa 488 (1:200) and Annexin V-Alexa594 (1:50) for 1 hour, RT. After washing with PBS, cells were refixed briefly with 4% paraformaldehyde in PBS and quenched. Cells were mounted on slides with Prolong Anti-fade Gold mounting media, dried overnight, RT, in the dark and stored at 4°C. Alternatively, cells were permeabilized in 0.1% Triton X-100, 1% BSA, in PBS for 5 mins, washed with PBS, refixed with 4% paraformaldehyde for 1 min, quenched, washed with PBS and blocked with 1ml 10% goat serum in PBS for 1hour, RT. Cells were incubated with mouse anti-human fodrin (1:100) and rabbit anti-CD45 (1:100) in 1% goat serum, PBS overnight at 4°C. Cells were washed with PBS, stained goat anti-mouse Alexa 594 (1:100) and goat anti-rabbit Alexa 488 (1:00) in 1% goat serum, PBS for 1 hour, RT, in the dark. Cells were washed and mounted as above. Slides were visualized on a Fluoview laser scanning confocal microscope(Olympus America, Inc). and images were processing using Fluoview imaging analysis software (version 2.1.39).
Phagocytosis assay
RAW 264.7 macrophages (3×105) were cultured on coverslips in 6-well plates in complete DMEM. Jurkat or J45.01 cells were labeled with 2 μM Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE) (Molecular Probes) for 15 min in PBS, 37°C, in the dark. Unbound CFSE was neutralized with complete medium before resuspending cells in fresh pre-warmed medium and incubating another 30 min. CFSE-labeled T cells were treated with galectin-1 or buffer control as described above. 1×106 labeled T cells were added to the macrophage monolayer for the indicated time at 37 °C. Non-phagocytosed target cells were removed by three washes with PBS. Digital images of randomly selected fields under phase-contrast microscopy were captured by confocal fluorescence microscopy and the number of engulfed cells in more than 300 macrophages was counted. The phagocytic index (37,38) was calculated as follows: phagocytic index = (total number of engulfed T cells/total number of counted macrophages) × (number of macrophages containing engulfed cells/total number of counted macrophages) × 100. The fraction of macrophages containing phagocytosed material was also calculated (number of macrophages containing engulfed cells/total number of counted macrophages × 100).
Acknowledgments
This work was supported in part by NIH R01GM63281 (to LGB) and NIH CA16042 and AI28697 (to the UCLA Jonsson Comprehensive Cancer Center). We thank Karen Pace for valuable advice and insight, and Ken Dorshkind and Benhur Lee for critical reading of the manuscript.
Abbreviations
- PTPase
protein tyrosine phosphatase
- TUNEL
Terminal dUTP nick end labeling
Footnotes
The authors have no competing financial interests.
References
- 1.Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Diff. 2008;15:243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- 2.Hume DA. Bring out your dead. Nat Immunol. 2008;9:12–14. doi: 10.1038/ni0108-12. [DOI] [PubMed] [Google Scholar]
- 3.Nagata S. Autoimmune diseases caused by defects in clearing dead cells and nuclei expelled from erythroid precursors. Immunol Revs. 2007;220:237–250. doi: 10.1111/j.1600-065X.2007.00571.x. [DOI] [PubMed] [Google Scholar]
- 4.Krysko DV, Vandenabeele P. From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Diff. 2008;15:29–38. doi: 10.1038/sj.cdd.4402271. [DOI] [PubMed] [Google Scholar]
- 5.Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 6.Perillo NL, Uittenbogaart C, Nguyen J, Baum LG. Galectin-1, an endogenous lectin produced by thymic epithelial cells, induces apoptosis of human thymocytes. J Exp Med. 1997;185:1851–1858. doi: 10.1084/jem.185.10.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu SD, Whiting CC, Tomassian T, Pang M, Bissel SJ, Baum LG, Mossine VV, Poirier F, Miceli MC. Endogenous galectin-1 enforces class I-restricted TCR functional fate decisions in thymocytes. Blood. 2008 doi: 10.1182/blood-2007-09-114181. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, Rabinovich GA. Differential glycosylation of TH1, TH2 and TH17 effector cells selectively regulates susceptibility to cell death. Nature Immunol. 2007;8:825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 9.Pace KE, Lee C, Stewart PL, Baum LG. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol. 1999;163:3801–11. [PubMed] [Google Scholar]
- 10.Pace KE, Hahn HP, Pang M, Nguyen JT, Baum LG. CD7 delivers a pro-apoptotic signal during galectin-1 induced T cell death. J Immunol. 2000;165:2331–2334. doi: 10.4049/jimmunol.165.5.2331. [DOI] [PubMed] [Google Scholar]
- 11.Hahn HP, Pang M, He J, Hernandez JD, Yang RY, Li LY, Wang X, Liu FT, Baum LG. Nuclear translocation of Endonuclease G in caspase- and cytochrome c-independent galectin-1 induced T cell death. Cell Death Diff. 2004;11:1277–86. doi: 10.1038/sj.cdd.4401485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen JT, Evans DP, Galvan M, Pace KE, Leitenberg D, Bui TN, Baum LG. CD45 modulates galectin-1 induced cell death: regulation by expression of core 2 O-glycans. J Immunol. 2001;167:5697–5707. doi: 10.4049/jimmunol.167.10.5697. [DOI] [PubMed] [Google Scholar]
- 13.Lokeshwar VB, Bourguignon LYW. Tyrosine phosphatase activity of lymphoma CD45 (GP180) is regulated by a direct interaction with the cytoskeleton. J Biol Chem. 1992;267:21551–7. [PubMed] [Google Scholar]
- 14.Iida N, V, Lokeshwar B, Bourguignon LYW. Mapping the fodrin binding domain in CD45, a leukocyte membrane-associated tyrosine phosphatase. J Biol Chem. 1994;269:28576–28583. [PubMed] [Google Scholar]
- 15.Pradhan D, Morrow J. The spectrin-ankyrin skeleton controls CD45 surface display and interleukin-2 production. Immunity. 2002;17:303–315. doi: 10.1016/s1074-7613(02)00396-5. [DOI] [PubMed] [Google Scholar]
- 16.Martin SJ, O’Brien GA, Nishioka WK, McGahon AJ, Mahboubi A, Saido TC, Green DR. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem. 1995;270:6425–6428. doi: 10.1074/jbc.270.12.6425. [DOI] [PubMed] [Google Scholar]
- 17.Brown TL, Patil S, Cianci CD, Morrow JS, Howe PH. Transforming growth factor β induces caspase 3-independent cleavage of αII-spectrin (α-fodrin) coincident with apoptosis. J Biol Chem. 1999;274:23256–23262. doi: 10.1074/jbc.274.33.23256. [DOI] [PubMed] [Google Scholar]
- 18.Wang KKW, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. Simultaneous degradation of αII- and βII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem. 1998;273:22490–22497. doi: 10.1074/jbc.273.35.22490. [DOI] [PubMed] [Google Scholar]
- 19.Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J Biol Chem. 1996;271:31075–31085. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- 20.Zheng TS, Schlosser SF, Dao T, Hingorani R, Crispe IN, Boyer JL, Flavell RA. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc Natl Acad Sci USA. 1998;95:13618–13623. doi: 10.1073/pnas.95.23.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolas G, Fournier CM, Galand C, Malbert-Colas L, Bournier O, Kroviarski Y, Fourgeois M, Camonis JH, Dhermy D, Grandchamp B, Lecomte MC. Tyrosine phosphorylation regulates alpha II spectrin cleavage by calpain. Mol Cell Biol. 2002;22:3527–3536. doi: 10.1128/MCB.22.10.3527-3536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nedrelow JH, Ciani CD, Morrow JS. c-Src binds αII spectrin’s Src homology 3 (SH3) domain and blocks calpain susceptibility by phosphorylating Tyr1176. J Biol Chem. 2003;278:7735–7741. doi: 10.1074/jbc.M210988200. [DOI] [PubMed] [Google Scholar]
- 23.Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, Maki M, Saito TC. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains. J Biol Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- 24.Fajka-Boja R, Szemes M, Ion G, Legradi A, Caron M, Monostori E. Receptor tyrosine phosphatase, CD45, binding galectin-1 but does not mediate its apoptotic signal in T cell lines. Immunol Lett. 2002;82:149–154. doi: 10.1016/s0165-2478(02)00030-5. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Johnson P. Expression of CD45 lacking the catalytic protein tyrosine phosphatase domain modulates Lck phosphorylation and T cell activation. J Biol Chem. 2005;280:14318–14324. doi: 10.1074/jbc.M413265200. [DOI] [PubMed] [Google Scholar]
- 26.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 27.Shiratsuchi A, Mori T, Nakanishi Y. Independence of plasma membrane blebbing from other biochemical and biological characteristics of apoptotic cells. J Biochem. 2002;132:381–386. doi: 10.1093/oxfordjournals.jbchem.a003233. [DOI] [PubMed] [Google Scholar]
- 28.Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T. Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol. 2007;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 29.Hermiston ML, Wu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- 30.Amano M, Galvan M, He J, Baum LG. The ST6Gal I sialyltransferase selectively modifies CD45 and negatively regulates galectin-1 induced CD45 clustering, phosphatase modulation and T cell death. J Biol Chem. 2003;278:7469–7475. doi: 10.1074/jbc.M209595200. [DOI] [PubMed] [Google Scholar]
- 31.Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Leuscher IF, Hivroz C, Nicaise J, Squifflet JL, Mourad M, Godelaine D, Boon T, van der Bruggen P. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity. 2008;28:414–424. doi: 10.1016/j.immuni.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- 33.Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 34.Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 35.Delacour D, Cramm-Behrens CI, Drobecq H, Le Bivic A, Naim HY, Jacob R. Requirement for galectin-3 in apical protein sorting. Curr Biol. 2006;16:408–414. doi: 10.1016/j.cub.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 36.Danielsen EM, Hansen GH. Lipid raft organization and function in brush borders of epithelial cells. Mol Membr Biol. 2006;23:71–79. doi: 10.1080/09687860500445604. [DOI] [PubMed] [Google Scholar]
- 37.Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I, Izui S, Liu FT. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112:389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golpon HA, Fadok VA, Taraseviciene-Stewart, Scerbavicius R, Sauer C, Welte T, Henson PM, Voelkel NF. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. 2004;18:1716–18. doi: 10.1096/fj.04-1853fje. [DOI] [PubMed] [Google Scholar]