

Abstract
Endometriosis is considered as an estrogen-dependent inflammatory disease but its etiology is unclear. To date, a mechanistic role for steroid receptor coactivators (SRCs) in endometriosis progression has not been elucidated. An SRC-1−/− mouse model reveals that the SRC-1 gene plays an essential role in endometriosis progression. Notably, a novel 70-kDa SRC-1 proteolytic isoform is highly elevated both in the endometriotic tissue of mice with surgically induced endometriosis and in endometriotic stromal cells biopsied from endometriosis patients. Tnf−/− and Mmp9−/− mice with surgically induced endometriosis reveal that activation of TNFα-induced MMP9 activity mediates formation of the 70-kDa SRC-1 C-terminal isoform in endometriotic mouse tissue. In contrast to full-length SRC-1, the endometriotic 70-kDa SRC-1 C-terminal fragment prevents TNF-α-mediated apoptosis in human endometrial epithelial cells upon TNF-α treatment and causes the epithelial-mesenchymal transition and invasion of human endometrial cells that are hallmarks of progressive endometriosis. Collectively, the novel TNF-α/MMP9/SRC-1 isoform functional axis promotes pathogenic progression of endometriosis.
The defining feature of endometriosis is that endometrial tissues are deposited and grown onto sites outside of the uterine cavity1. The pathogenesis of endometriosis remains controversial despite extensive research. Among the hypotheses, Sampson's retrograde menstruation theory proposes that endometrial cells are refluxed through the fallopian tubes during menstruation and then are implanted onto the peritoneum or pelvic organs. In addition to retrograde menstruation, however, other factors should be involved in the progression of endometriosis because only 15% of all women of reproductive age have endometriosis even though retrograde menstruation is a common phenomenon.
Estrogen is considered to be one of the key modulators in the pathophysiology and/or pathogenesis of endometriosis because a considerable number of clinical reports have defined endometriosis as an estrogen-dependent disease2,3. The estrogen-dependent growth of endometriotic tissue is enhanced by increased local estrogen levels mediated by aromatase4. Therefore, drugs that suppress estrogen production, such as gonadotropin-releasing hormone analogs and aromatase inhibitors, have been used in endometriosis treatments5–7. In addition to the elevation of local estrogen concentrations, the up-regulation of the Estrogen Receptor(ER)β level in endometriotic tissue is correlated with the progression of endometriosis8. Regulation of ER-β activity by selective estrogen receptor modulators also has been proffered as a potential endometriosis treatment9,10. For example, treatment with ERB-041 (ER-β selective agonist) could regress ectopic lesion growth in mice with endometriosis9. Nevertheless, the role of ER-β in the estrogen-dependent progression of endometriosis has not been clearly elucidated.
Proinflammatory signaling also plays an essential role in the progression of endometriosis because a number of cytokines in the peritoneal fluid of endometriosis patients are actively involved in the pathogenesis and progression of endometriosis11, 12. Interference with the proinflammatory signaling in the peritoneal fluid has been employed to suppress ectopic lesion growth. For example, monoclonal antibody to TNF-α (c5N) treatment in baboons with endometriosis significantly reduces the extent of endometriosis progression13. Nonsteroidal anti-inflammatory drugs, such as ibuprofen, also have been used for endometriosis treatment14.
To advance the endometrial progression program, endometriotic cells express aberrant, elevated levels of matrix metalloproteinases (MMPs) during their migration and implantation into distinct anatomical sites15. Therefore, the inhibition of MMP activity by MMP inhibitor III may prevent the development of endometriosis progression16.
To precisely respond to external hormonal stimuli, steroid receptor coactivators (SRCs) dynamically modulate nuclear receptor (NR)-mediated cellular processes in a tissue-selective manner. Accordingly, altered levels of SRCs and NRs in specific target tissues are frequently associated with human disease progression17. In the case of endometriosis, lowered SRC-1(p160) protein levels have been reported in endometriotic tissue when compared to normal endometrium18, 19. However, the role of SRC-1 in the progression of endometriosis has not been elucidated.
Therefore, the object of this study was to investigate the functional role of the SRC-1 gene in the progression of endometriosis. Based on our results, we suggest that a novel SRC-1 isoform can be a new molecular target for therapy of endometriosis.
Results
Endometriotic tissues contain a 70-kDa SRC-1 isoform
To investigate the role of SRCs in the progression of endometriosis, we employed C57BL/6J mice with surgically induced endometriosis (SIE)20 in parallel with primary human endometriotic stromal cells isolated from individuals who received a diagnosis of endometriosis. In our C57BL/6J mouse model, we successfully implanted ectopic lesions to be progressed in recipient C57BL/6J mice in an estrogen-dependent manner (Supplementary Fig. 1a,b). Next we determined the expression levels of cyclooxygenase-2 (COX-2) to validate the progression of endometriosis; COX-2 has been reported to be elevated in endometriotic endometrium compared to normal endometrium21. We found that endometriotic tissues of mice with SIE had a higher COX-2 expression levels relative to the uterine tissue from sham-treated mice (Supplementary Fig. 1c). To investigate the expression profiles of SRCs between normal and endometriotic tissues, we divided the mice into two groups: “sham-treated”, referring to the uterine tissue from ovariectomized mice treated with estradiol (E2) pellets and surgery but without the implantation of an endometrial fragment, and “endometriosis,” referring to the ectopic and eutopic endometrium of ovariectomized mice with SIE in the presence of E2 pellets (Fig. 1a). We found that the uterine tissue of the sham-treated mice had a 160-kDa intact SRC-1 protein (Fig. 1a). However, both ectopic and eutopic endometrium of mice with SIE had lower level of intact full-length SRC-1 but higher level of a 70-kDa truncated form versus the sham-treated uterine tissue (Fig. 1 a,b). To evaluate the nature of the 70-kDa fragment, we isolated uteri from ovariectomized SRC-1−/− mice treated with E2 pellets. The uterine tissue from SRC-1−/− mice did not have intact SRC-1 nor the 70-kDa fragment (Fig. 1 c). Therefore, the 70-kDa fragment was substantiated as an isoform of SRC-1. An immunostaining analysis revealed that SRC-1 isoform had similar endometrial compartment-specific (such as luminal epithelium and stroma) expression patterns as compared to those of full-length SRC-1 in normal endometrium (Fig. 1d). However, we found that sham-treated uterine tissue primarily had an intact SRC-1 in the nuclei, but endometriotic tissue had the SRC-1 isoform in both the cytoplasm and the nuclei (Fig. 1d).
Figure 1.

The endometriotic 70-kDa SRC-1 isoform. (a) Western blot analysis of SRC-1 expression in uterus of sham-treated mice and in the ectopic and eutopic endometrium of mice with SIE. Tubulin acts as a protein loading control. (b) The ratio of the 70-kDa fragment to intact SRC-1 in each type of endometrium (n=6/group). (c) Western blot analysis of SRC-1 expression pattern in the uterus of sham-treated mice, the eutopic endometrium of mice with SIE and the uterus of sham-treated SRC-1−/− mice. Tubulin acts as a protein loading control (d) Immunohistochemical analysis of SRC-1 expression pattern in uterus of SRC-1−/− mice, uterus of sham-treated mice and in the ectopic and eutopic endometrium of mice with SIE. (e) Western blot analysis of expression patterns of SRC-1, the 70-kDa SRC-1 isoform, ER-β and ER-α in cultured primary HESCs lines isolated from women without endometriosis (n=1) and from the ectopic lesions of women with endometriosis (n=6; lanes 1 to 6). Tubulin acts as a protein loading control. (f–h) The ratios of the SRC-1 isoform to tubulin (f), ER-β to tubulin (g) and ER-α to tubulin (h) in cultured primary HESC lines isolated from women without endometriosis (n=3) and from the ectopic and eutopic endometrium of endometriosis patients (n=10). * P<0.05, ** P<0.01 by Student's t test. Scale bars, 50 μm.
Next we examined whether human endometriotic tissue also contained the 70-kDa SRC-1 isoform. We determined an SRC-1 expression profile in primary human endometrial stromal cells (HESCs) isolated from women without endometriosis (n=3) and from women with endometriosis (n=10). We found that eight of ten primary HESCs from women with endometriosis had higher levels of this 70-kDa truncated SRC-1 form compared to the primary HESCs of women without endometriosis (Fig. 1e,f and Supplementary Fig. 2a). Consistent with SRC-1−/− mice, SRC-1 knockdown in the HESCs of women with endometriosis validated that the 70-kDa fragment is also an SRC-1 isoform in humans (Supplementary Fig. 2b). Next we examined whether external hormone changed the 70-kDa SRC-1 isoform status in human endometriotic stromal cells. We found that estrogen, progesterone or their combination did not change the status of the SRC-1 isoform in primary human endometriotic stromal cells during cell culture (Supplementary Fig. 2d–h). Primary human endometriotic stromal cells had variable ratios of the SRC-1 isoform to full-length SRC-1 between them (Fig. 1e and Supplementary Fig. 2a). However, examination of the subject information for these cells (Supplementary Fig. 2c) revealed that the differential ratio of the SRC-1 isoform to full-length SRC-1 was significantly correlated with the site of biopsy. The SRC-1 isoform was predominantly detected in HESCs isolated from ectopic lesions of women with endometriosis (Fig. 1e). However, a low ratio of the SRC-1 isoform to full-length SRC-1 was detected in the HESCs of the eutopic endometrium (Supplementary Fig. 2a). Therefore, ectopic lesions had active SRC-1 processing as compared to the eutopic endometrium. The menstrual cycle did not impact the ratio of the SRC-1 isoform to full-length SRC-1. In addition to uterus, endometriosis impacted the SRC-1 milieus of other organs of the abdominal cavity because intestine and liver of mice with SIE had higher levels of endometriotic SRC-1 isoform versus those of sham treated mice (Supplementary Fig. 2i). In the case of estrogen receptor (ER) expression, we found that the primary HESCs of women with endometriosis had higher ER-β expression levels versus the normal primary HESCs, as described previously (Fig. 1e,g and Supplementary Fig. 2a)22,23. In contrast to ER-β, however, we found that ER-α expression levels were not changed in the HESCs of women with endometriosis when compared to the HESCs of women without endometriosis (Fig. 1e,h and Supplementary Fig. 2a).
In addition to SRC-1, we found that endometriotic mouse tissue also had some different altered forms of SRC-2 and SRC-3 (Supplementary Fig. 3a,b). However, only the ectopic lesions, but not eutopic endometrium, had truncated forms of SRC-2 (110 kDa) and SRC-3 (85 kDa) in mice with SIE. In contrast to the SRC-1 isoform, we found that HESCs isolated from the ectopic lesions of women with endometriosis did not have altered forms of SRC-2 and SRC-3 as compared to normal HESCs (Supplementary Fig. 3c,d). Collectively, these data revealed that mouse and human endometriotic tissues had the SRC-1 70 kDa isoform when compared to SRC-2 and SRC-3.
Notably, we found that SRC2−/− and SRC-3−/− ectopic lesions had lower ectopic lesion volume compared to wild-type ectopic lesions in mice with SIE (Supplementary Fig. 3e). Therefore, SRC-2 and SRC-3 genes might play some permissive or cooperative role with SRC-1 isoform to modulate the growth of ectopic lesions in mice with SIE.
SRC-1 gene modulates progression of ectopic lesion
The observation of the endometriotic SRC-1 isoform raised the question of whether it is required for endometriosis. To address this question, we surgically induced endometriosis with SRC-1−/− mice. We transplanted the endometrial tissue fragments generated from the uteri of SRC-1−/− mice or congenic C57BL/6J mice into ovariectomized C57BL/6J recipient mice treated with E2 pellets to induce endometriosis. We found that the endometrial tissue fragments from C57BL/6J donors successfully developed into ectopic lesions (Fig. 2a). However, the deletion of SRC-1−/− gene reduced volumes of ectopic lesions by over 500% versus the wild-type ectopic lesions (Fig. 2a,b). Therefore, the SRC-1 gene is an essential factor for the growth of ectopic lesion. To further validate the role of the SRC-1 gene in ectopic lesion growth in mice with SIE, we employed GFP transgenic mice [C57BL/6-Tg(CAG-GFP)10sb/J] that ubiquitously express GFP in all tissue types. To obtain SRC-1−/− uterine tissue that expressed GFP, we generated bigenic SRC-1−/−:GFP mice by crossing SRC-1−/− mice with GFP transgenic mice. We isolated the wild-type and SRC-1−/− uterine fragments expressing GFP and then implanted them into ovariectomized C57BL/6J recipient mice treated with E2 pellets to induce endometriosis. After the progression of endometriosis, we detected ectopic lesions expressing GFP in the mesenteric membrane of recipient mice under a fluorescence dissecting microscope (Fig. 2c). The SRC-1−/− ectopic lesions have lower GFP density versus wild-type ectopic lesions (Fig. 2d).
Figure 2.
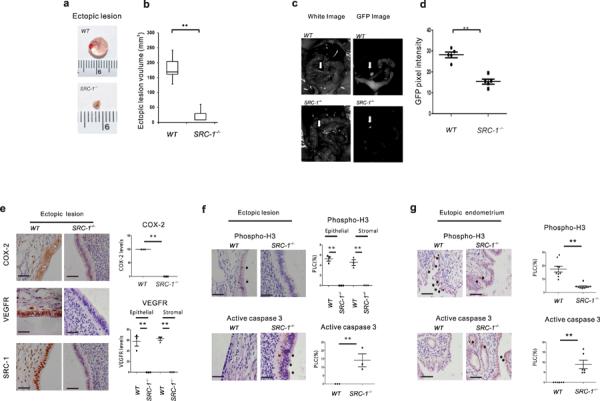
The SRC-1 gene had an essential role in the progression of endometriosis. (a) Wild-type ectopic lesions and SRC-1−/− ectopic lesions isolated from C57BL/6J recipient mice after the induction of endometriosis. (b) The volume analysis of wild-type and SRC-1−/− ectopic lesions (n=4/group). (c) The GFP images analysis of a wild-type and an SRC-1−/− ectopic lesion expressing GFP in mice with SIE. (d) The GFP pixel density of wild-type and SRC-1−/− ectopic lesions expressing GFP in mice with SIE. (e) Immunohistochemical analysis of COX-2, VEGFR and SRC-1 expression levels in wild-type ectopic lesions and SRC-1−/− ectopic lesions and quantification of COX-2 and VEGFR expression levels in each type of ectopic lesion (n=3/group). (f,g) Immunohistochemical analysis of expression levels of phospho-histone (H) 3 and activated caspase 3 in wild-type and SRC-1−/− ectopic lesions (f) or in wild-type and SRC-1−/− eutopic endometrium (g) and quantification of their expression levels in each type of ectopic lesion (n=3/group) and eutopic endometrium (n=9/group). PLC, percentage of labeled cells, **P<0.01 by Student's t test. Scale bars, 50 μm.
To further analyze the growth defects in the ectopic lesions stemming from SRC-1−/− endometrial fragments, we determined the expression levels of COX-2 and the vascular endothelial growth factor receptor (VEGFR) because endometriotic tissues had higher COX-2 and VEGFR expression relative to normal tissues24,25. We found that SRC-1−/− ectopic lesions had significantly lower expression levels of both endometriosis marker genes versus wild-type ectopic lesions (Fig. 2e). In addition, a phospho-histone(H) 3 level analysis revealed that the SRC-1−/− ectopic lesions also had lower proliferative activity relative to wild-type ectopic lesions (Fig. 2f). In contrast to proliferation, however, we found that SRC-1−/− ectopic lesions had higher active caspase 3 levels compared to wild-type ectopic lesions (Fig. 2f). In addition to ectopic lesions, we found that the proliferative activity and apoptosis signaling also were decreased and increased, respectively, in the SRC-1−/− eutopic endometrium when compared to the wild-type eutopic endometrium of mice with SIE (Fig. 2g). Collectively, these results suggest that the SRC-1 gene has a similar effect on both the ectopic and eutopic endometrium of mice with SIE.
The 70-kDa of SRC-1 is a proteolytic SRC-1 C-terminal isoform
We next attempted to discern the exact origin of the SRC-1 isoform in endometriotic tissue. A Northern blot analysis with a probe against full-length mouse SRC-1 revealed that the eutopic endometrium of mice with SIE did not have alternative forms of the SRC-1 RNA compared to sham-treated uterine tissue (Supplementary Fig. 4). This result implied that the SRC-1 isoform is not an alternative RNA splicing variant but may be a posttranslational and site-specific protease-mediated proteolytic cleavage product.
To identify the SRC-1 isoform, we employed a heterologous mouse model of endometriosis with severe combined immune deficiency (SCID) mutant mice and immortalized human endometrial cells as previously described26. We utilized immortalized human endometrial epithelial cells (IHEECs) obtained by stably transfecting the catalytic subunit (hTERT) of the human telomerase complex27 and immortalized human endometrial stromal cell lines (IHESCs) generated by the transfection of hTERT using a retroviral system28. To determine whether the endometriotic SRC-1 isoform contained the N-terminus or C-terminus, we modified the human SRC-1 cDNA with a Flag tag on the N-terminal region and a MYC tag on the C-terminal region (named F-SRC-1-M). Using this lentiviral vector containing the F-SRC-1-M gene, we modified IHEECs and IHESCs to stably express the exogenous F-SRC-1-M gene. To efficiently induce endometriosis in the SCID mouse model, we mixed and cultured recombinant IHESCs expressing F-SRC-1M with recombinant IHEECs expressing F-SRC-1-M on Matrigel because paracrine communication between the stromal and epithelial compartments is a critical factor for modulating endometrial function in vivo29. We then transplanted the mixed-cultured recombinant human endometrial cells into the pelvic cavities of ovariectomized SCID mice treated with E2 pellets (Fig. 3a). To precisely analyze the exogenous F-SRC-1-M processing pattern in ectopic lesions using antibodies to the Flag and MYC tags, we produced another type of ectopic lesion through the transplantation of mixed-cultured parental IHESCs and IHEECs into SCID mice because ectopic lesions developed from the parental human endometrial cells do not express FSRC-1-M (Fig. 3a).
Figure 3.

The endometriotic SRC-1 isoform was the SRC-1 C-terminal fragment. (a) Experimental scheme of the heterologous mouse model of endometriosis. (b) Western blot analysis of expression levels of ER-β, COX-2, VEGFR2 and Tubulin in mixed-cultured recombinant human endometrial cells and the ectopic lesions that developed from these cells. (c–e) Western blot analysis of SRC-1processing pattern in mixed-cultured recombinant human endometrial cells expressing F-SRC-1-M (Mixed-cultured:F-SRC-1-M), ectopic lesions developed with recombinant human endometrial cells expressing F-SRC-1-M (Ectopic:W/ FSRC-1-M), and parental ectopic lesions developed from parental human endometrial cells that did not express F-SRC-1-M (Ectopic: W/O F-SRC-1-M) using a Flag-specific antibody (c), a MYC-specific antibody (d) and an SRC-1-specific antibody (e). Tubulin acts as a protein loading control. WB; Western Blot analysis. *; nonspecific bands.
We found that the ectopic lesions were well developed in ~80% of the SCID mice transplanted with mixed-cultured human endometrial cells (Supplementary Fig. 5a–c). Immunostaining with antibodies to vimentin (an endometrial stromal cell biomarker) and cytokeratin 18 (an endometrial epithelial cell biomarker) revealed that ectopic lesions successfully developed from mixed-cultures of IHEECS and IHESCs (Supplementary Fig. 5d–f). In addition, we found that human ectopic lesions developed in SCID mice had higher levels of several endometriosis marker genes (such as ER-β, VEGFR2 and COX-2) versus the mixed-cultured human endometrial cells (Fig. 3b). Therefore, the mixed-cultured IHEECs and IHESCs successfully developed into typical ectopic lesions in SCID mice.
For the identification of the SRC-1 isoform, we conducted Western blot analysis with antibodies to the Flag or MYC peptides. We found that the antibody to Flag did not detect the 70-kDa SRC-1 isoform and SRC-1 fragments of other sizes in recombinant ectopic lesions expressing F-SRC-1-M (Ectopic:W/F-SRC-1-M) (Fig. 3c). In contrast to the antibody to Flag, however, the antibody to MYC recognized the 70-kDa protein in recombinant ectopic lesions expressing F-SRC-1-M (Ectopic:W/F-SRC-1-M) but not in the other control samples (Fig. 3d). We found that this 70-kDa fragment containing the MYC tag in recombinant ectopic lesions also was recognized by the antibody to SRC-1 that was used for the detection of the 70-kDa SRC-1 isoform in endometriotic mouse tissue (Fig. 3e). Therefore, the 70-kDa SRC-1 isoform in endometriotic tissues is a posttranslational proteolytic C-terminal isoform of SRC-1.
The TNF-α/MMP-9 axis generates endometriotic SRC-1 isoform
A protease cleavage site prediction program (http://pops.csse.monash.edu.au/pops-cgi/index.php) indicated that MMP9 can generate the 70-kDa SRC-1 C-terminal isoform from intact SRC-1 because SRC-1 has a potential MMP9 cleavage site at P790-M791. Gelatin zymography analysis revealed that eutopic endometrium of mice with SIE and primary human endometriotic stromal cells of women with endometriosis had higher MMP9 activity versus endometrium of sham-treat mice and normal HESCs, respectively (Fig. 4a,b). In contrast to MMP9, however, both mouse and human endometriotic cells had very limited MMP2 activity (Fig. 4a,b). The inhibition of MMP9 activity by treatment with MMP2/MMP9 inhibitor III (MMP2/9 In) significantly reduced the volume of the ectopic lesions in mice with SIE, and did not produce the endometriotic 70-kDa SRC-1 C-terminal isoform in the eutopic endometrium when compared to vehicle treatment (Fig. 4c–e). In addition to MMP2/9 In treatment, the MMP9 gene also had an essential role in ectopic lesion growth because Mmp9−/− ectopic lesions in Mmp9−/− mice with SIE had a lower ectopic lesion volume versus the wild-type ectopic lesions of congenic FVB/NJ mice (Supplementary Fig. 6a). In addition to the regression of ectopic lesion growth, we found that the eutopic endometrium of Mmp9−/− mice with SIE did not have the endometriotic SRC-1 isoform when compared to the congenic FVB/NJ mice (Supplementary Fig. 6b). Collectively, these results suggested that MMP9 plays an essential role in both ectopic lesion growth and in the generation of the SRC-1 C-terminal isoform in the endometriotic tissue of mice with SIE.
Figure 4.

The TNF-α/MMP9 functional axis had an essential role in the progression of endometriosis and endometriotic SRC-1 isoform formation in endometriotic tissue. (a) Gelatin zymography analysis of MMP2 and MMP9 activity in uterus of sham-treated mice and eutopic endometrium of mice with SIE and ratio of MMP9 activity to tubulin in each type of endometrium (n=4/group). (b) Gelatin zymography analysis of MMP2 and MMP9 activity in primary HESCs isolated from women without endometriosis (W/O endometriosis) and from women with endometriosis (W/ endometriosis) and ratio of MMP9 activity to tubulin in each type of cells (n=3/group). (c) Gelatin zymography analysis of the MMP9 activity in the eutopic endometrium of C57BL/6J mice with SIE treated with vehicle or 2.5 mg kg−1 of MMP2/MMP9 inhibitor III (MMP2/9 In) and ratio of MMP9 activity to tubulin in each eutopic endometrium (n=4/group). (d) Ectopic lesions isolated from mice with SIE treated with vehicle or 2.5 mg kg−1 of MMP2/9 In, and volume analysis of each isolated ectopic lesion (n=4/group). (e) Western blot analysis of SRC-1 expression patterns in the eutopic endometrium of mice with SIE treated with vehicle or MMP2/9 In. and the ratio of the 70-kDa SRC-1 isoform to intact SRC-1 in each type of endometrium. (f) Gelatin zymography analysis of MMP9 activity in the eutopic endometrium of Tnf−/− mice and congenic wild-type mice with SIE and the ratio of MMP9 activity to tubulin in each type of eutopic endometrium (n=3/group) (g) Ectopic lesions isolated from congenic wild-type mice and Tnf−/− mice with SIE, and volume analysis of each isolated ectopic lesion (n=4/group). (h) Western blot analysis of expression patterns of SRC-1 in the eutopic endometrium of Tnf−/− mice and congenic wild-type mice with SIE and ratio of the 70-kDa SRC-1 isoform to intact SRC-1 in each type of eutopic endometrium (n=3/group). ** P<0.01, *** P<0.001 by Student's t test.
How is MMP9 activity regulated in endometriotic tissues during the progression of endometriosis? Previous studies revealed that pro-inflammatory signaling is closely associated with tissue-remodeling pathways in inflammatory disease progression. For example, TNF-α induces the expression of invasion mediators, such as MMPs, to promote cell mobility30. In our mouse model, we also found that the eutopic and ectopic endometrium of mice with SIE had a higher TNF-α level as compared to normal endometrium (Supplementary Fig. 6c). Notably, gelatin zymography revealed that the eutopic endometrium of Tnf−/− mice with SIE had lower MMP9 activity versus congenic wild-type mice with SIE (Fig. 4f). In addition to the reduction of MMP9 activity, we found that Tnf−/− mice with SIE had lower volumes of ectopic lesion versus congenic wild-type mice with SIE (Fig. 4g). The eutopic endometrium of Tnf−/− mice with SIE did not have SRC-1isoform as compared to endometrium of wild-type mice with SIE (Fig. 4h). Collectively, these results suggested that the induction of TNF-α signaling activates MMP9 activity to promote ectopic lesion growth and to generate the SRC-1 C-terminal isoform in endometriotic tissue.
The above observation raised the question of whether SRC-1 is a direct substrate for MMP9 in endometriotic tissue. To address this question, we conducted in vitro MMP9 processing assays with the purified double-tagged F-SRC-1-M protein and activated recombinant MMP9 protein. The antibody to Flag revealed that recombinant MMP9 generated a 60-kDa N-terminal SRC-1 fragment in vitro (Fig. 5a). However, the human ectopic lesions developed in SCID mice did not have this N-terminal SRC-1 fragment in vivo (Fig. 3c). In contrast to the antibody to Flag, MYC specific antibody detected the 70-kDa SRC-1 C-terminal isoform (Fig. 5b). We found that this 70-kDa SRC-1 C-terminal fragment also was recognized by an SRC-1 specific antibody that has been used for SRC-1 isoform detection in mouse endometriotic tissue (Fig. 5c). Therefore, MMP9 directly recognized SRC-1 and processed it to the 70-kDa SRC-1 C-terminal isoform in vitro. In contrast to MMP9, however, MMP2 did not produce the 70 kDa SRC-1 C-terminal isoform in vitro (Supplementary Fig. 6d)
Figure 5.

MMP9 directly cleaved human SRC-1 at P790-M791 in vitro and in vivo. (a–c) Western blot analysis of the resulting processed forms of SRC-1 by MMP9 with antibodies to Flag (a), MYC (b) and SRC-1 (c). (d–f). Western blot analysis of the resulting processed forms of the SRC-1(P790A) mutant by MMP9 with antibodies to Flag (d), MYC (e) and SRC-1 (f). (g) Experimental scheme for the transplantation of recombinant IHEECs stably expressing wild-type F-SRC-1-M or the F-SRC-1-M(P790A) into SCID mice to induce endometriosis. (h) Western blot analysis of SRC-1 expression pattern in ectopic lesions expressing F-SRC-1-M or the FSRC-1-M(P790A) with a MYC-specific antibody and the ratio of the 70-kDa SRC-1 isoform to intact SRC-1 in each ectopic lesion (n=3/group). *; Non-specific bands. (i) The processing pattern of SRC-1 to the endometriotic SRC-1 C-terminal isoform [791-1441 amino acids(aa)] by MMP9 in endometriotic tissues. bHLH, basic helix-loop-helix. RID, receptor interacting domain. AD, activation domain. Q, proline-rich domain. aa, amino acid. *** P<0.001 by Student's t test.
A protease prediction program revealed that the MMP9 can recognize P790-M791 dipeptide in SRC-1 to process the 70-kDa SRC-1 C-terminal isoform. To confirm this prediction, we substituted Pro790 with Ala to generate an MMP9 cleavage-defective SRC-1 (P790A) mutant that had a Flag tag in the N-terminal region and a MYC tag in the C-terminal region. An in vitro MMP9 processing assay with the purified SRC-1(P790A) mutant revealed that MMP9 also generated a 60-kDa N-terminal SRC-1 fragment from the SRC-1 (P790A) mutant as was found in wild-type SRC-1 (Fig. 5d). However, Western blot analysis with the MYC-specific antibody revealed that MMP9 did not generate the 70-kDa SRC-1 C-terminal isoform from the SRC-1 (P790A) mutant in vitro (Fig. 5e). In addition to the MYC-specific antibody, the antibody to SRC-1 also did not recognize the 70-kDa fragment from the SRC-1 (P790A) mutant after MMP9 incubation (Fig. 5f). These findings indicated that MMP9 cleaved SRC-1 at the P790-M791 site to produce the 70-kDa SRC-1 C-terminal isoform in vitro.
The above observation raised the question of whether MMP9 also cleaves the P790-M791 site of SRC-1to produce the SRC-1 C-terminal isoform in endometriotic tissue in vivo. To address this question, we transplanted mixed-cultures of recombinant IHEECs stably expressing the F-SRC-1 (P790A)-M mutant and IHESCs into SCID mice to induce ectopic lesions (Fig. 5g). Western blot analysis with the MYC-specific antibody revealed that ectopic lesions expressing F-SRC-1-M (P790A) mutant did not generate the 70-kDa SRC-1 C-terminal isoform when compared to wild-type F-SRC-1-M (Fig. 5h). Taken together, these results suggested that MMP9 cleaves at the P790-M791 site of SRC-1 to generate the 70-kDa SRC-1 C-terminal isoform in endometriotic tissue (Fig. 5i).
The role of SRC-1 isoform in the progression of endometriosis
What is the differential role of the SRC-1 C-terminal isoform compared to full-length SRC-1 in endometriotic tissue? To address this question, we generated IHEECs that stably express the SRC-1 C-terminal isoform [791-1441 amino acids(aa)] using a lentivirus-mediated gene transfer protocol (Supplementary Fig. 7a). Immunostaining analysis revealed that SRC-1 C-terminal isoform was located in both the nuclear and cytoplasmic compartments (Supplementary Fig. 7b).
In NR mediated signaling, the SRC-1 C-terminal isoform interacted with ER-α, ER-β and Progesterone Receptor(PR) (Supplementary Fig. 8a–c) and activated ER-α-, ER-β- and PR-mediated gene transcription, as does full-length SRC-1 (Supplementary Fig. 8d–g).
The endometriosis-associated proinflammatory pelvic environment normally causes cell death, such that menstrual cell masses are cleared. Therefore, the evasion of TNF-α-induced cell death is an essential step for the development of stable ectopic lesions during the progression of endometriosis. Therefore, we investigated the role of the SRC-1 C-terminal isoform in the evasion of TNF-α-induced cell death. The parental and two different recombinant IHEECs expressing full-length SRC-1 or the SRC-1 isoform grew well under vehicle treatment relative to their growth before treatment (Fig. 6a). However, TNF-α treatment prevented the growth of parental (Fig. 6a,b) and recombinant IHEECs expressing full-length SRC-1 (Fig. 6a,c) when compared to the vehicle treatment. In contrast, however, IHEECs expressing the SRC-1 C-terminal isoform did not have significant TNF-α-induced growth inhibition compared to the parental IHEECs and those expressing full-length SRC-1 (Fig. 6a,d). Therefore, we concluded that the endometriotic SRC-1 C-terminal isoform prevented the TNF-α-induced growth inhibition of IHEECs.
Figure 6.

Endometriotic SRC-1 prevented TNF-α-induced cell death and increased EMT to improve the invasive capacity of human endometrial epithelial cells. (a) The crystal violet cell proliferation assay of parental IHEECS and recombinant IHEECS stably expressing full-length SRC-1 or the SRC-1 C-terminal isoform in the absence or presence of 10 ng ml−1 TNF-α for 2 d. (b–d) The viable cell numbers of the parental IHEECs (b) and the recombinant IHEECs expressing full-length SRC-1 (c) or the SRC-1 isoform (d) per field at the indicated time points. (e) Western blot analysis of expression levels of procaspase 8 (57 kDa) and its cleaved form (10 kDa) in parental IHEECS and recombinant IHEECS stably expressing full-length SRC-1 or the SRC-1 C-terminal isoform upon 10 ng ml−1 TNF-α treatment at the indicated time points and the ratio of the 10kDa isoform to the 50 kDa isoform in each type of cell at the indicated time points. (f) Western blot analysis of expression levels of apoptosis markers (tBid and activated caspase 3) in parental IHEECS and recombinant IHEECS stably expressing full-length SRC-1 or the SRC-1 C-terminal isoform upon 10 ng ml−1 TNF-α treatment at the indicated time points. (g) Immunoprecipitation (IP) of caspase 8 with parental IHEECs or IHEECs stably expressing full-length SRC-1 (SRC-1) or the SRC-1 C-terminal isoform (SRC-1 Iso) treated with vehicle or 10 ng ml−1 of TNF-α for 8 h. Western blot analysis of levels of the Flag-SRC-1 C-terminal isoform, SRC-1, Flag-SRC-1 and caspase 8 in immunoprecipitated pellets. *; Non-specific bands. (h) Transwell invasion assays of parental HEECS and recombinant IHEECS stably expressing full-length SRC-1 or the SRC-1 C-terminal isoform for 2 d and the average number of invasive cells per field at the indicated time points. (i) Western blot analysis of expression patterns of EMT markers (such as N-cadherin, Vimentin, Slug, Snail, β-catenin, and TCF8) in parental IHEECS and recombinant IHEECS stably expressing full-length SRC-1 or the SRC-1 C-terminal isoform upon 10 ng ml−1 TNF-α treatment at the indicated time points. (j) TNF-α/MMP9/SRC-1 isoform functional axis in human endometriotic epithelial cells. *** P<0.001 by Student's t test. Scale bars, 50 μm.
The activation of the caspase 8/caspase 3/tBid-mediated apoptosis pathway has a key role in TNF-α-induced growth inhibition31. Therefore, we examined whether the expression of the endometriotic SRC-1 isoform inhibited TNF-α-induced apoptosis signaling in IHEECs. Consistent with previous observations, we found that TNF-α treatment increased the levels of cleaved caspase 8, tBid and activated caspase 3 in parental IHEECs versus vehicle (Fig. 6e,f). Notably, upon TNF-α treatment, the recombinant IHEECs expressing full-length SRC-1 had higher expression levels of these apoptosis markers relative to the parental IHEECs (Fig. 6e,f). In contrast, however, recombinant IHEECs expressing the SRC-1 C-terminal isoform did not elevate the levels of cleaved caspase 8, tBid and activated caspase 3 upon TNF-α treatment relative to the parental and recombinant IHEECs expressing full-length SRC-1 (Fig. 6e,f). To determine how the SRC-1 isoform inhibits TNF-α-induced apoptosis in IHEECs, we performed a co-immunoprecipitation assay. We found that the SRC-1 C-terminal isoform, but not the full-length SRC-1 (F-SRC-1-M or endogenous SRC-1), interacted with procaspase 8 in IHEECs in the absence or presence of TNF-α (Fig. 6g). Therefore, we concluded that the SRC-1 C-terminal isoform protected IHEECs from TNF-α-induced apoptosis by interacting with procaspase 8 to prevent its activational processing.
In addition to evasion of TNF-α-induced cell death, the extensive invasion of ectopic endometrium into target tissues (such as the ovary, uterus, intestine and bladder) is another essential step for the progression of endometriosis. We found that the recombinant IHEECs expressing full-length SRC-1 had higher invasive capacity versus the parental IHEECs (Fig. 6h). However, we found that the invasive capacity of the IHEECs was more significantly enhanced by the overexpression of the endometriotic SRC-1 C-terminal isoform compared to full-length SRC-1 (Fig. 6h).
The Epithelial-Mesenchymal Transition (EMT) plays a key role in metastasis and is characterized by the conversion of epithelial cancer cells to a more motile phenotype that facilitates invasion. Therefore, we investigated whether the endometriotic SRC-1 C-terminal isoform induced EMT in IHEECs to enhance the invasive capacity of these cells. We found that the recombinant IHEECs that overexpressed the endometriotic SRC-1 C-terminal isoform had higher expression levels of many of the acquired markers of EMT (N-cadherin, Vimentin, Slug, Snail, β-catenin and TCF8) when compared to parental IHEECs and to those overexpressing full-length SRC-1 in the absence or presence of TNF-α treatment (Fig. 6i). Therefore, the SRC-1 isoform enhanced EMT to facilitate invasion by the ectopic endometrium during the progression of endometriosis.
In summary, to efficiently promote endometriosis, the activated TNF-α/MMP9 axis processes SRC-1 to its C-terminal isoform to protect the ectopic endometrium from a proinflammatory cytokine-mediated cell death, which then is accompanied by EMT and enhancement of the invasive capacity of the ectopic endometrium (Fig. 6j).
Discussion
Despite their well-known and essential role in estrogen- and progesterone-mediated cellular processes, the roles of SRCs in the progression of endometriosis have not been well studied. In this paper, we propose a new TNF-α/MMP9/SRC-1 isoform functional axis as a novel pathogenic pathway for endometriosis. The SRC-1 isoform promotes endometriosis progression by causing EMT and invasion of endometrial epithelial cells. An accompanying alteration of proinflammatory signaling is closely associated with inflammatory diseases and cancer progression because proinflammatory signaling changes the molecular properties of target cells32,33. For example, TNF-α treatment changes steroid hormone responsiveness in uterine smooth muscle cells by suppressing the expression of SRC-1 and SRC-234. In the present study, we found that TNF-α also changes the molecular properties of SRC-1 by proteolytic processing to a 70kDa form that protects ectopic lesions from immune surveillance. Collectively, these results suggested that TNF-α signaling has an essential role in the initiation of endometriotic cellular processes. In addition to uterus, the TNF-α induced inflammatory response to endometriosis can impact the SRC-1 milieus of other organs of the abdominal cavity, such as intestine and liver, In this way, endometriosis may be accompanied by serious harmful systemic effects secondary to a widespread inflammatory response.
We found that MMP9, induced by TNF-α, is responsible for processing SRC-1 to the 70 kDa isoform. It preciously was suggested that MMPs are secreted and then activated to remodel the mesothelial lining of the peritoneum and to allow tissue invasion during progression of endometriosis35. However, we found that both pro-MMP9 and processed MMP9 activities were detected in the eutopic endometrium when compared to the sham-treated uterus. These data imply that the intracellular activation of MMP9 can occur within endometriotic tissue. In support of our conclusion, the co-localization of trypsin-2 and MMP-9 has been reported in “intracellular pro-MMP-9 processing” that causes fully or partially activated MMP-9 in the intracellular vesicles of carcinoma cells36. Therefore, it is likely that intracellular MMP9 activation in endometriosis is performed by a specific TNF-α-induced protease to process SRC-1 for the progression of endometriosis. In addition to the proteolytic activity of MMP9, the non-proteolytic activity of MMP-9 may be involved in the progression of endometriosis because non-proteolytic MMP9 activity is associated with cell migration37.
Proinflammatory signaling is frequently associated with tissue-remodeling pathways in other inflammatory diseases and cancer. For example, TNF-α-induced MMP9 activation is associated with the progression of various cancers38. Therefore, the TNF-α/MMP9 functional axis may play an essential role not only in endometriosis but also in other types of inflammatory diseases and cancer. Consequently, this functional axis should be considered a new molecular therapeutic target for the treatment of these diseases.
What is the pathobiological function of SRC-1 C-terminal isoform-mediated anti-apoptotic processing in human endometrial epithelial cells during the progression of endometriosis? The communication between the epithelial and stromal compartments plays an essential role in endometrial function. Therefore, paracrine factors produced from stromal cells regulate cell survival signaling in epithelial cells in the normal endometrium. In endometriosis, paracrine factors are also released from stromal cells to modulate cell survival signaling in epithelial cells in the endometriotic eutopic endometrium39. In contrast to the eutopic endometrium, however, ectopic stromal cells lose the ability to regulate cell survival signaling in ectopic epithelial cells39. Therefore, epithelial cells in ectopic lesions must acquire a new cell survival system. In this context, epithelial cells in ectopic lesions activate the TNF-α/MMP9/SRC-1 isoform pathway in response to TNF-α during the progression of endometriosis. The activation of this pathway processes SRC-1 to the SRC-1 C-terminal isoform, protecting epithelial cells in ectopic lesions from TNF-α-induced cell death and, thus, promoting their survival. In this way, the activation of the TNF-α/MMP9/SRC-1 isoform pathway in ectopic lesions can compensate for the loss of paracrine survival signaling generated from ectopic stromal cells.
Endometriosis has been known as an estrogen-dependent inflammatory disease. What is the role of the SRC-1 C-terminal isoform in the estrogen-dependent nature of endometriosis? Ectopic lesions require estrogen-dependent cellular processes for growth. However, ectopic lesions cannot use SRC-1 for estrogen-dependent gene regulation because full-length SRC-1 increased TNF-α-mediated cell death in IHEECs. For survival, SRC-1 must be processed to the SRC-1 C-terminal isoform in the epithelial cells of ectopic lesions to modulate estrogen-dependent cellular processes during activated proinflammatory signaling. In this way, the endometriosis-induced microenvironment actively changes the molecular properties of SRC-1 by proteolytic processing to prevent apoptosis but can still allow an estrogen-dependent progression of endometriosis.
We propose that this novel TNF-α/MMP9/SRC-1 C-terminal isoform functional axis is a new pathogenic pathway for the progression of endometriosis that has potential as a therapeutic target for endometriosis therapy.
Methods
Mouse information
We purchased five-week-old normal mice (C57BL/6J), Tnf−/− mice (B6;129S-Tnftm1Gkl/J), Mmp9−/− mice (FVB.Cg-Mmp9tm1Tvu/J), GFP-expressing mice (C57BL/6-Tg(CAG-GFP)10sb/J) and SCID mice (NOD.Cg-PrkfcscidB2mtm1UncIl2rgtm1Wjl/Szj) from Jackson Laboratory. We generated the bigenic SRC-1−/−:GFP-expressing mice by crossing SRC-1−/− mice40 with GFP-expressing mice. We maintained the animals under a standard 12-h photoperiod and fed a normal chow diet. All procedures were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine (AN-0544 and AN-5284).
Surgically induced endometriosis
We performed the surgical treatments of mice under aseptic conditions with anesthesia. We surgically induced endometriosis in mice with a modification of a previously described method41. A) Autotransplantation: We ovariectomized 6-wk-old C57BL/6 mice, Tnf−/− mice, and MMP9 −/− mice. After two weeks, we implanted sterile 60-day release pellets containing 0.36 mg of 17-β estradiol (Innovative Research of America) into ovariectomized mice. Two days later, we isolated one uterine horn of each mouse under anesthesia. In a petri dish containing warmed Dulbecco's modified Eagle medium (DMEM)/F-12 (Invitrogen) supplemented with 100 U ml−1 penicillin and 100 μg ml−1 streptomycin, we longitudinally cut the uterine horns with a pair of scissors. We then isolated tissue samples using a 2-mm dermal biopsy punch (Miltex) and subsequently sutured them to the mesenteric membrane in the same mouse through a midline incision (7–0 braided silk suture; Ethicon). We closed the abdominal incision with a 5–0 braided silk suture (Ethicon) in a continuous fashion. On day 21 after the endometriosis challenge, we sacrificed mice and carefully isolated the ectopic lesions and eutopic endometrium from the surrounding tissue. B) Heterotransplantation with mouse tissue: We ovariectomized the donor mice (SRC-1−/− mice, GFP-expressing mice, and bigenic SRC-1−/−:GFP expressing mice) and recipient mice (C57BL/6J mice), and then implanted sterile 60-day release pellets containing 0.36 mg of 17-β estradiol. After two days, we isolated one uterine horn of the donor mouse under anesthesia. We isolated tissue samples using a 2-mm dermal biopsy punch and subsequently sutured them to the mesenteric membrane in the C57BL/6J recipient mouse through a midline incision. After that, we closed the abdominal incision with a 5–0 braided silk suture in a continuous fashion. On day 21 after the endometriosis challenge, we sacrificed mice and carefully isolated the ectopic lesions and eutopic endometrium from the surrounding tissue. C) Transplantation of mixed-cultured IHEECs and IHESCs into SCID mice: We cultured IHEECs and IHESCs in DMEM/F12 (Sigma, St. Louis, MO) containing 10% fetal bovine serum (Hyclone) and penicillin (100 U mL−1), streptomycin (100 mg ml−1), and amphotericin-B (2.5 mg ml−1) (Invitrogen) in humidified 5% CO2 and 95% air at 37°C. No special steroid treatment was employed. We changed the medium every three days. The IHEECs and IHESCs were trypsinized with 0.1% trypsin-ethylenediaminetetraacetic acid (Invitrogen Inc.). We mixed all cell lines (2×105 cells) together in 0.5 ml DMEM/F12. The Matrigel (BD Biosciences) was diluted in a 1:1 ratio with ice-cold DMEM/F12, and then 250 μl of the Matrigel:medium mixture was added into the wells of a 24-well plate and incubated for another 30 min. Next, we gently dropped 0.5 ml of mixed cells onto the top of the solidified gel. We cultured the cells for 1 week, and changed media at every 2–3 d. The spheroid formation and projection growth were monitored daily under light microscopy. We implanted sterile 60-day release pellets containing 0.36 mg of 17-β estradiol into ovariectomized SCID mice. Two days later, we anesthetized the mice and opened the abdomen. We carefully removed the Matrigel containing the mixed-cultured cells from the well and transplanted into the pelvic cavity. We implanted the culture from one well of a 24-well plate into the pelvic cavity of one SCID mouse. We closed the abdominal incision with a 5–0 braided silk suture in a continuous fashion. On day 21 after the endometriosis challenge, we sacrificed mice and carefully isolated the endometriotic lesions from the surrounding tissue. According to the formula [Volume (mm3) = 0.52 × width × length], we calculated the volumes of the endometriotic lesions.
Western Blot Analysis
The following primary antibodies were used: mouse SRC-1 (Santa Cruz Biotechnologies), human SRC-1 (Abcam), MYC (Abcam), Flag (Sigma), tubulin (Santa Cruz Biotechnologies), ER-α (Santa Cruz Biotechnologies), ER-β (Santa Cruz Biotechnologies), PR (Santa Cruz Biotechnologies), COX-2 (Abcam), VEGFR (Abcam), active caspase 3 (Cell Signaling), Bid (Santa Cruz Biotechnologies) and an EMT-specific antibody sample kit (Cell Signaling). Subsequently, we incubated the membranes with secondary HRP-tagged antibodies (Sigma), and visualized the signals with ECL plus (Amersham).
Immunohistochemistry
We performed the immunostaining in 10% neutral-buffered, formalin-fixed and paraffin-embedded sections of mouse tissue as previously described42. Antibodies to mouse SRC-1 (Santa Cruz Biotechnologies), COX-2 (Santa Cruz Biotechnologies), VEGFR2 (Santa Cruz Biotechnologies), active caspase 3 (Cell Signaling) and phospho-histone 3 (Cell Signaling) were used. We visualized specific antigens using the DAB substrate kit (Vector). We quantified the immunostaining intensity using ImageJ program (NIH).
Primary HESCs
All tissues used for this study were collected with Baylor College of Medicine, Institutional Review Board (H-21138 and H26382) approval. We isolated and cultured HESCs as described previously43.
Cell Culture, Transfection and Lentiviral Infections
We cultured IHEECs and IHESCs in DMEM/F12 supplemented with 10% FCS and penicillin/streptomycin. We transfected cells with 25 nM siRNAs and Dharmafect 1(Dharmacon) according to the manufacturer's recommendations for transfection. We conducted the lentiviral transfection according to the manufacturer's instructions (System Biology).
In vitro MMP9 cleavage assay
We purified double-tagged SRC-1 (F-SRC-1-M) and an MMP9 cleavage-defective mutant [F-SRC-1-M(p790A)] from recombinant IHEECs that stably expressed each type of SRC-1 using an anti-Flag M2 affinity gel (Sigma-Aldrich) according to the manufacturer's protocol. To activate recombinant human (rh) MMP9 (R&D), 200 ng/ul of rhMMP9 was incubated at 37°C for 2 h in 1 mM p-aminophenylmercuric acetate (APMA). After activation, 10 and 5 ng of APMA-activated MMP-9 were incubated with 200 ng of purified wild-type and mutant SRC-1 at 30 °C for 2 h in 50 mM Tris buffer (5 mM CaCl2 and 150 mM NaCl). Upon the completion of the incubation, we heated the samples for 5 min at 95 °C. We analyzed the processed forms of the wild-type or mutant SRC-1 by MMP9 using Western blot analysis.
Gelatin zymography
We generated cell lysates from mouse endometrium or HESCs and applied them to an 8% polyacrylamide gel copolymerized with 2 mg ml−1 gelatin (15 μg per lane). After electrophoresis, we washed the gels three times for 20 min with 2.5% Triton X-100 to remove sodium dodecyl sulfate. We then rinsed the gels with incubation buffer (50 mM Tris-Cl (pH 7.6), 5 mM CaCl2, and 150 mM NaCl) and incubated in the same buffer at 37 °C for 12 h. We stained the gels with 2% Coomassie Brilliant Blue G-250, 25% methanol and 10% acetic acid for 2 h and then destained for 1 h in 2% methanol/4% acetic acid.
Co-immunoprecipitation and Western blotting
For the co-immunoprecipitation assays, we generated lysates from the eutopic endometrium of mice with SIE. We incubated the lysates with the indicated antibodies with constant rotation. Protein G beads were added 2 h later and incubated for an additional 2 h followed by extensive washing (20 mM HEPES [pH 7.6], 150 mM KCl, 1 mM DTT, 0.1% NP-40 and 8% glycerol supplemented with protease inhibitors and phosphatase inhibitors). We directly boiled the Protein G beads in 1× Laemmli buffer, and the precipitated proteins were separated by SDS-PAGE. We transferred the samples onto nitrocellulose membranes and used them for analysis by Western blotting. We performed the Western blotting and detected signals as previously described44.
Modification of the human SRC-1 gene
To generate the double-tagged human SRC-1 gene, we introduced the Flag peptide (DYKDDDDK) and the MYC peptide (EQKLISEEDL) by PCR using a Flag-tagging primer (5'-GCGGCCGCAATCTAGAGCCACCATGGATTACAAGGATGACGACGATAAGATGAGTGGCCTCGGGGACAGTT -3'), a MYC-tagging primer (5'-GCGGCCGCAATCTAGAGCCACCATGGATTACAAGGATGACGACGATAAGATGAGTGGCCTCGGGGACAGTT-3') and the human (h) SRC-1 cDNA. We transferred this double-tagged hSRC-1 (F-SRC-1-M) gene to the pCDH-CMV-MCS-EF-1-Puro lentivector (System Bioscience). We generated the endometriotic SRC-1 C-terminal isoform (amino acids 791-1441 of SRC-1) using the primers 5'-ATCACCACTTTGTCTGTCGAGCCT-3' and 5'-TTATTCAGTCAGTAGCTGCTGAAGGAG-3' and then inserted into the pCDH-CMV-MCS-EF-1-Puro lentivector. We generated the MMP9 cleavage-defective double-tagged SRC-1 (P790A) with the primers 5'-CATGTAATACAAACCCAACCGCAATGACCAAACCCACTCCTG-3' and 5'-CAGGAGTGGGTTTGGTCATTGCGGTTGGGTTTGTATTACATG-3', and the pCDH-CMV-MCS-EF-1-Puro vector containing F-SRC-1-M gene using the QuikChange® Lightning site-directed mutagenesis kit (Stratagene). The mutation site was confirmed by sequencing.
Two-chamber transwell cell invasion assay
We analyzed the cell invasion using a cell invasion assay (Millipore) according to the manufacturer's instructions. We detached parental IHEECs and recombinant IHEECs expressing full-length F-SRC-1M or the F-SRC-1 C-terminal isoform with trypsin-EDTA and washed them once with serum-free medium. We suspended cells in serum-free medium. We added a total of 0.5 ml of complete culture medium to the bottom of each well, and 0.5×106 cells to each transwell insert. We pre-coated the inserts with growth factor-reduced Matrigel, and allowed the cells to migrate for 48 h in a 37 °C cell incubator. We removed the cells in the upper surface of the transwell using cotton swabs. We fixed the migrated cells attached to the undersurface with 4% paraformaldehyde for 10 min and stained them with crystal violet solution (0.5% in water) for 10 min. We counted the cells under a microscope.
Statistical Analysis
Data were expressed as the means ± s.d. The statistical significance was assessed by independent two-tailed Student's t test. P< 0.05 was considered statistically significant.
Supplementary Material
Acknowledgement
We thank C. J. Lockwood (Department of Obstetrics, Gynecology and Reproductive Science and Department of Genetics, Yale University School of Medicine) and T. Klonisch (Department of Human Anatomy and Cell Science, University of Manitoba) for providing immortalized human endometrial stromal and epithelial cells for this work. We thank MJ. Park for her technical support in animal experiments. We also thank the US National Institutes of Health-designated Diabetes and Endocrinology Research Center and the Proteomics Core in Dan L. Duncan Cancer Center at Baylor College of Medicine for supporting this work.
This work was supported by grants from US National Institute of Diabetes and Digestive and Kidney Diseases (U54HD0077495 and 5K12HD050128 to S.H, U54HD007495 to F.D., and R01 HD08188 to B.O., U54HD007495 pilot grant to SJ. H.), a grant from US Nation Cancer Institute (R01 CA077030 to J.L.)
Footnotes
Author contribution SJ. H. led the project and designed and performed most of experiments. K.B., S.Y.J., and J.Q. provided technical expertise. S.M.H. and E.K. provided primary normal HESCs and primary human endometriotic stromal cells. J.P.L. and F.J.D. provided intellectual inputs. B.W.O. supervised the entire project. SJ.H. and B.W.O wrote the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Bulun SE. Endometriosis. N Engl J Med. 2009;360:268–279. doi: 10.1056/NEJMra0804690. [DOI] [PubMed] [Google Scholar]
- 2.Missmer SA, Cramer DW. The epidemiology of endometriosis. Obstet Gynecol Clin North Am. 2003;30:1–19. vii. doi: 10.1016/s0889-8545(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 3.Meldrum DR, et al. “Medical oophorectomy” using a long-acting GNRH agonist--a possible new approach to the treatment of endometriosis. J Clin Endocrinol Metab. 1982;54:1081–1083. doi: 10.1210/jcem-54-5-1081. [DOI] [PubMed] [Google Scholar]
- 4.Noble LS, Simpson ER, Johns A, Bulun SE. Aromatase expression in endometriosis. J Clin Endocrinol Metab. 1996;81:174–179. doi: 10.1210/jcem.81.1.8550748. [DOI] [PubMed] [Google Scholar]
- 5.Fedele L, Bianchi S, Zanconato G, Tozzi L, Raffaelli R. Gonadotropin-releasing hormone agonist treatment for endometriosis of the rectovaginal septum. Am J Obstet Gynecol. 2000;183:1462–1467. doi: 10.1067/mob.2000.108021. [DOI] [PubMed] [Google Scholar]
- 6.Imai A, Takagi A, Tamaya T. Gonadotropin-releasing hormone analog repairs reduced endometrial cell apoptosis in endometriosis in vitro. Am J Obstet Gynecol. 2000;182:1142–1146. doi: 10.1067/mob.2000.104804. [DOI] [PubMed] [Google Scholar]
- 7.Simsa P, et al. Selective estrogen-receptor modulators and aromatase inhibitors: promising new medical therapies for endometriosis? Womens Health (Lond Engl) 2007;3:617–628. doi: 10.2217/17455057.3.5.617. [DOI] [PubMed] [Google Scholar]
- 8.Fujimoto J, Hirose R, Sakaguchi H, Tamaya T. Expression of oestrogen receptor-alpha and -beta in ovarian endometriomata. Mol Hum Reprod. 1999;5:742–747. doi: 10.1093/molehr/5.8.742. [DOI] [PubMed] [Google Scholar]
- 9.Harris HA, Bruner-Tran KL, Zhang X, Osteen KG, Lyttle CR. A selective estrogen receptor-beta agonist causes lesion regression in an experimentally induced model of endometriosis. Hum Reprod. 2005;20:936–941. doi: 10.1093/humrep/deh711. [DOI] [PubMed] [Google Scholar]
- 10.Xiu-li W, Wen-jun C, Hui-hua D, Su-ping H, Shi-long F. ERB-041, a selective ER beta agonist, inhibits iNOS production in LPS-activated peritoneal macrophages of endometriosis via suppression of NF-kappaB activation. Mol Immunol. 2009;46:2413–2418. doi: 10.1016/j.molimm.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 11.Bedaiwy MA, et al. Prediction of endometriosis with serum and peritoneal fluid markers: a prospective controlled trial. Human Reproduction. 2002;17:426–431. doi: 10.1093/humrep/17.2.426. [DOI] [PubMed] [Google Scholar]
- 12.Berkkanoglu M, Arici A. Immunology and endometriosis. Am J Reprod Immunol. 2003;50:48–59. doi: 10.1034/j.1600-0897.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 13.Falconer H, et al. Treatment with anti-TNF monoclonal antibody (c5N) reduces the extent of induced endometriosis in the baboon. Hum Reprod. 2006;21:1856–1862. doi: 10.1093/humrep/del044. [DOI] [PubMed] [Google Scholar]
- 14.Efstathiou JA, et al. Nonsteroidal antiinflammatory drugs differentially suppress endometriosis in a murine model. Fertil Steril. 2005;83:171–181. doi: 10.1016/j.fertnstert.2004.06.058. [DOI] [PubMed] [Google Scholar]
- 15.Osteen KG, Yeaman GR, Bruner-Tran KL. Matrix metalloproteinases and endometriosis. Semin Reprod Med. 2003;21:155–164. doi: 10.1055/s-2003-41322. [DOI] [PubMed] [Google Scholar]
- 16.Nap AW, Dunselman GAJ, de Goeij AFPM, Evers JLH, Groothuis PG. Inhibiting MMP activity prevents the development of endometriosis in the chicken chorioallantoic membrane model. Human Reproduction. 2004;19:2180–2187. doi: 10.1093/humrep/deh408. [DOI] [PubMed] [Google Scholar]
- 17.Lonard DM, Lanz RB, O'Malley BW. Nuclear Receptor Coregulators and Human Disease. Endocrine Reviews. 2007;28:575–587. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki A, et al. Immunohistochemical detection of steroid receptor cofactors in ovarian endometriosis: involvement of down-regulated SRC-1 expression in the limited growth activity of the endometriotic epithelium. Virchows Arch. 2010;456:433–441. doi: 10.1007/s00428-010-0884-x. [DOI] [PubMed] [Google Scholar]
- 19.Kumagami A, Ito A, Yoshida-Komiya H, Fujimori K, Sato A. Expression patterns of the steroid receptor coactivator family in human ovarian endometriosis. J Obstet Gynaecol Res. 2011;37:1269–1276. doi: 10.1111/j.1447-0756.2010.01509.x. [DOI] [PubMed] [Google Scholar]
- 20.Fang Z, et al. Intact progesterone receptors are essential to counteract the proliferative effect of estradiol in a genetically engineered mouse model of endometriosis. Fertil Steril. 2004;82:673–678. doi: 10.1016/j.fertnstert.2004.01.048. [DOI] [PubMed] [Google Scholar]
- 21.Hirata T, et al. Interleukin-17F increases the secretion of interleukin-8 and the expression of cyclooxygenase 2 in endometriosis. Fertil Steril. 2011;96:113–117. doi: 10.1016/j.fertnstert.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 22.Matsuzaki S, et al. Expression of estrogen receptor alpha and beta in peritoneal and ovarian endometriosis. Fertility and sterility. 2001;75:1198–1205. doi: 10.1016/s0015-0282(01)01783-6. [DOI] [PubMed] [Google Scholar]
- 23.Bergqvist A, Bruse C, Carlberg M, Carlstrom K. Interleukin 1beta, interleukin-6, and tumor necrosis factor-alpha in endometriotic tissue and in endometrium. Fertility and sterility. 2001;75:489–495. doi: 10.1016/s0015-0282(00)01752-0. [DOI] [PubMed] [Google Scholar]
- 24.Chishima F, et al. Increased expression of cyclooxygenase-2 in local lesions of endometriosis patients. Am J Reprod Immunol. 2002;48:50–56. doi: 10.1034/j.1600-0897.2002.01101.x. [DOI] [PubMed] [Google Scholar]
- 25.Wang HB, et al. [Expression of vascular endothelial growth factor receptors in the ectopic and eutopic endometrium of women with endometriosis] Zhonghua Yi Xue Za Zhi. 2005;85:1555–1559. [PubMed] [Google Scholar]
- 26.Banu SK, Starzinski-Powitz A, Speights VO, Burghardt RC, Arosh JA. Induction of peritoneal endometriosis in nude mice with use of human immortalized endometriosis epithelial and stromal cells: a potential experimental tool to study molecular pathogenesis of endometriosis in humans. Fertil Steril. 2009;91:2199–2209. doi: 10.1016/j.fertnstert.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 27.Hombach-Klonisch S, et al. Regulation of functional steroid receptors and ligand-induced responses in telomerase-immortalized human endometrial epithelial cells. Journal of Molecular Endocrinology. 2005;34:517–534. doi: 10.1677/jme.1.01550. [DOI] [PubMed] [Google Scholar]
- 28.Krikun G, et al. A Novel Immortalized Human Endometrial Stromal Cell Line with Normal Progestational Response. Endocrinology. 2004;145:2291–2296. doi: 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
- 29.Rubel CA, Jeong JW, Tsai SY, Lydon JP, Demayo FJ. Epithelial-stromal interaction and progesterone receptors in the mouse uterus. Semin Reprod Med. 2010;28:27–35. doi: 10.1055/s-0029-1242990. [DOI] [PubMed] [Google Scholar]
- 30.Grund EM, et al. Tumor necrosis factor-alpha regulates inflammatory and mesenchymal responses via mitogen-activated protein kinase kinase, p38, and nuclear factor kappaB in human endometriotic epithelial cells. Mol Pharmacol. 2008;73:1394–1404. doi: 10.1124/mol.107.042176. [DOI] [PubMed] [Google Scholar]
- 31.Sanz AB, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol. 2008;19:1634–1642. doi: 10.1681/ASN.2007121336. [DOI] [PubMed] [Google Scholar]
- 32.Overton C, Fernandez-Shaw S, Hicks B, Barlow D, Starkey P. Peritoneal fluid cytokines and the relationship with endometriosis and pain. Hum Reprod. 1996;11:380–386. doi: 10.1093/humrep/11.2.380. [DOI] [PubMed] [Google Scholar]
- 33.Shakiba K, Falcone T. Tumour necrosis factor-alpha blockers: potential limitations in the management of advanced endometriosis? A case report. Hum Reprod. 2006;21:2417–2420. doi: 10.1093/humrep/del179. [DOI] [PubMed] [Google Scholar]
- 34.Leite RS, Brown AG, Strauss Iii JF. Tumor necrosis factor-α suppresses the expression of steroid receptor coactivator-1 and -2: A possible mechanism contributing to changes in steroid hormone responsiveness. The FASEB Journal. 2004 doi: 10.1096/fj.04-1684fje. [DOI] [PubMed] [Google Scholar]
- 35.Pino M, et al. Association between MMP1 and MMP9 activities and ICAM1 cleavage induced by tumor necrosis factor in stromal cell cultures from eutopic endometria of women with endometriosis. Reproduction. 2009;138:837–847. doi: 10.1530/REP-09-0196. [DOI] [PubMed] [Google Scholar]
- 36.Vilen S-T, et al. Intracellular co-localization of trypsin-2 and matrix metalloprotease-9: Possible proteolytic cascade of trypsin-2, MMP-9 and enterokinase in carcinoma. Experimental Cell Research. 2008;314:914–926. doi: 10.1016/j.yexcr.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 37.Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J. Role of matrix metalloproteinase-9 (MMP-9) dimers in cell migration: design of inhibitory peptides. J. Biol. Chem. 2010 doi: 10.1074/jbc.M109.091769. M109.091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott KA, et al. TNF-[alpha] regulates epithelial expression of MMP-9 and integrin [alpha]v[beta]6 during tumour promotion. A role for TNF-[alpha] in keratinocyte migration? Oncogene. 2004;23:6954–6966. doi: 10.1038/sj.onc.1207915. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, et al. Endometriotic stromal cells lose the ability to regulate cell-survival signaling in endometrial epithelial cells in vitro. Molecular Human Reproduction. 2009;15:653–663. doi: 10.1093/molehr/gap069. [DOI] [PubMed] [Google Scholar]
- 40.Xu J, et al. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science. 1998;279:1922–1925. doi: 10.1126/science.279.5358.1922. [DOI] [PubMed] [Google Scholar]
- 41.Cummings AM, Metcalf JL. Induction of endometriosis in mice: a new model sensitive to estrogen. Reprod Toxicol. 1995;9:233–238. doi: 10.1016/0890-6238(95)00004-t. [DOI] [PubMed] [Google Scholar]
- 42.Han SJ, et al. Dynamic Cell Type Specificity of SRC-1 Coactivator in Modulating Uterine Progesterone Receptor Function in Mice. Mol. Cell. Biol. 2005;25:8150–8165. doi: 10.1128/MCB.25.18.8150-8165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hawkins SM, et al. Functional MicroRNA Involved in Endometriosis. Molecular Endocrinology. 2011;25:821–832. doi: 10.1210/me.2010-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu RC, et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol. 2002;22:3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.