

Abstract
The vast majority of studies addressing the induction of autophagy have focused upon cytoplasmic aspects of its regulation. Recently, we have started to expand our knowledge regarding the nuclear events of autophagic induction. Many autophagy-related genes are transcriptionally upregulated upon induction of autophagy, but only in a limited number of cases do we know the pathways leading to this upregulation. Few transcription factors have been implicated in controlling autophagy genes in yeast. However, many of the ATG genes show some level of transcriptional induction upon starvation. Now, we show that transcription of ATG8 is repressed under growing conditions by the Ume6-Sin3-Rpd3 complex.
Keywords: autophagosome, lysosome, phagophore, stress, vacuole
One of the ATG genes that shows the strongest induction is ATG8, a core component of the autophagic machinery, whose mRNA levels increase more than 10-fold upon starvation. This increase occurs quickly, with mRNA levels peaking 30 min after starvation. Atg8 is conjugated to the lipid phosphatidylethanolamine (PE) and lines the inner and outer membrane of the expanding phagophore, where it is involved in the recognition of specific cargos during selective autophagy. When Atg8 levels during starvation are artificially reduced by the use of alternate promoters, smaller autophagosomes are formed, indicating that the amount of this protein regulates autophagosome size. Therefore, control of ATG8 mRNA levels is an important aspect of autophagic regulation.
In mammalian cells, the FOXO family of transcription factors upregulates the expression of the ATG8 homologs LC3 and GABARAPL1, along with ATG12. Similarly, the transcription factor E2F1 was recently shown to directly upregulate LC3 and ULK1. In both cases this induction leads to increased autophagy. However, there is not yet a complete picture of how LC3 transcription is controlled; moreover, until now little was known about what factors regulate the expression of ATG8 in yeast, the model organism where control of autophagic induction is probably best understood. Therefore, we decided to explore the mechanism controlling ATG8 transcription, and we discovered that the Ume6-Sin3-Rpd3 complex acts in an inhibitory capacity.
When UME6 is deleted, Atg8 protein levels increase dramatically, even when nutrients are plentiful. The same effect is seen when RPD3 or SIN3 are deleted, suggesting that these three proteins work together to suppress ATG8 transcription during nutrient-rich conditions. The ATG8 promoter contains a consensus binding site for Ume6, and chromatin immunoprecipitation analysis using a TAP-tagged Ume6 shows that it does indeed bind to this region. Consistent with these findings, a β-galactosidase reporter under the control of the ATG8 promoter shows increased transcription levels in a ume6∆ strain.
In agreement with the idea that Atg8 levels control autophagosome size and thereby total autophagic activity, a ume6∆ strain shows higher levels of autophagy than the isogenic wild type as measured by the quantitative Pho8∆60 assay. To estimate the size of the autophagosomes formed in the absence of Ume6, we used electron microscopy to measure the autophagic bodies that accumulated in pep4∆ vps4∆ and pep4∆ vps4∆ ume6∆ strains after starvation. The autophagic bodies were significantly larger in the absence of Ume6, accounting for the higher autophagy activity seen in the ume6∆ strain. We also observed a small increase in the number of autophagic body cross-sections in the ume6∆ strain; however, we think this is a secondary effect of the larger size of the bodies, as larger bodies are more likely to be caught by a random section and thus will be sampled more often.
Ume6 is phosphorylated under starvation conditions in a manner dependent on Rim15 kinase. Deletion of RIM15 leads to lower Atg8 protein levels, whereas the rim15∆ ume6∆ double-deletion strain has higher than normal Atg8 levels, similar to the ume6∆ single deletion. This suggests that phosphorylation of Ume6 may be one way that Rim15 exerts its autophagy-inducing effects. We have previously shown that Rim15 acts downstream of protein kinase A (PKA) and Sch9. Therefore, we propose a model in which upstream sensors such as PKA inactivate Rim15 during growing conditions, allowing Ume6 to inhibit ATG8 transcription. The low level of constitutive Atg8 is adequate for the cytoplasm-to-vacuole targeting pathway and basal autophagy. In the absence of nutrients, PKA is inactivated, and Rim15 now phosphorylates and inhibits Ume6, resulting in the upregulation of ATG8 (Fig. 1).
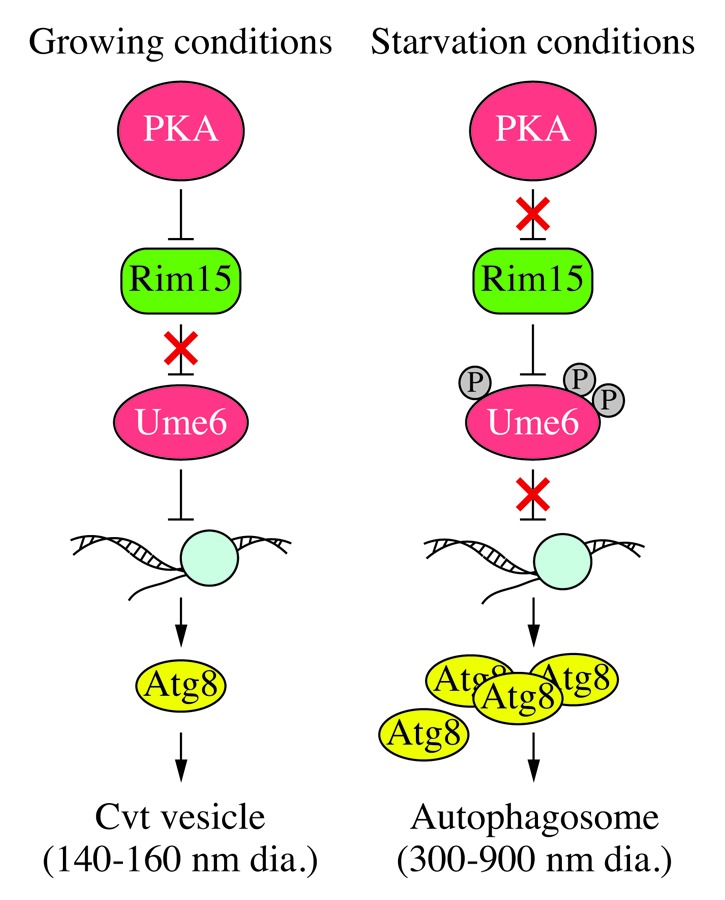
Figure 1. Schematic model for Ume6-dependent regulation of ATG8 transcription. In growing conditions, upstream kinase sensors such as PKA inhibit the Rim15 kinase; active Ume6 keeps ATG8 transcription at a low basal level, which is adequate for the production of enough Atg8 to make relatively small Cvt vesicles. Under starvation conditions, PKA activity is inhibited and the now active Rim15 phosphorylates and inhibits Ume6, resulting in a higher level of ATG8 transcription; the subsequently increased amount of Atg8 allows the generation of large autophagosomes.
UME6 has no clear homolog in mammals, but there are homologs for the corepressor SIN3 and the deacetylase RPD3, suggesting that elements of this pathway might be conserved. Indeed, double knockdown of SIN3A and SIN3B with shRNA leads to an increase in LC3 levels in three different human cell lines. This effect is not mediated by the autophagy regulatory protein MTOR, whose activity is unchanged, suggesting that it might be a direct effect of SIN3 on LC3 transcription.
The involvement of the deacetylase Rpd3 in controlling ATG8 transcription is interesting, as this joins other recent studies in showing a role for histone acetylases and deacetylases in controlling autophagy, both in yeast and mammals. Some of these effects appear to be cytoplasmic, via direct action of these enzymes on components of the autophagic machinery, but others appear to be nuclear, as in the case of Ume6-Sin3-Rpd3.
Although we have now established a clear link between the transcriptional regulation of ATG8 (and hence autophagy activity) by Ume6, many details of this regulation remain to be worked out. For example, during meiosis, Ume6 is phosphorylated and degraded, releasing transcriptional inhibition. Under starvation conditions, Ume6 is likewise phosphorylated, by Rim15, but does not seem to leave the ATG8 promoter, raising the question of how ATG8 transcription is promoted upon induction of autophagy. It may be that in this case Ume6 phosphorylation disrupts its interaction with Sin3 and Rpd3, or inactivates the complex without causing it to dissociate. Interestingly, the induction of ATG8 expression is only temporary, and it appears to be turned off again after a few hours of starvation. Could it be that Ume6 remains at the ATG8 promoter in order to help subsequently repress ATG8 expression, and thus moderate the autophagic response?
Finally, although the role of SIN3 is conserved in mammalian cells, there is presumably another transcription factor that plays a role equivalent to Ume6 in binding the LC3 promoter and recruiting the corepressor complex to inhibit LC3 transcription and autophagy during growing conditions. The identity of this factor, and how it interplays with the already known positive factors FOXO3 and E2F1, remains to be discovered.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21845