

Abstract
Huntington disease (HD) results from CAG repeats that encode expanded polyglutamine tracts in the HTT/huntingtin protein. HD belongs to a large category of inherited and sporadic neurodegenerative disorders in which production of a misfolded protein initiates the pathogenic cascade. Previous studies have shown that misfolded proteins become resistant to cellular protein turnover pathways by eluding and disabling the ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway. Based upon earlier work implicating impaired PPARGC1A function in HD, we derived inducible PPARGC1A mice and crossed them with HD mice. We found that PPARGC1A overexpression can rescue HD neurological phenotypes and neurodegeneration. An unexpected outcome of the rescue was the virtual elimination of huntingtin aggregates, and we found that PPARGC1A-mediated aggregate elimination required the autophagy pathway. Moreover, we observed decreased expression of transcription factor EB (TFEB), a master regulator of the autophagy-lysosome pathway, in HD cells and mice, and documented PPARGC1A co-activation of TFEB in these model systems, noting that PPARGC1A is upstream of TFEB in promoting proteostasis. These findings underscore the importance of bioenergetics and autophagy in neurodegeneration, and indicate that PPARGC1A promotes mitochondrial quality control to support high-energy production states in cells, such as neurons. As impaired energy production and altered protein–organelle quality control appear inextricably linked in disorders such as HD, Parkinson disease, and Alzheimer disease, efforts directed at enhancing PPARGC1A and TFEB action may represent viable strategies for therapy development in neurodegeneration.
Keywords: PGC-1-alpha, TFEB, autophagy, Huntington disease, mitochondria, neurodegeneration, oxidative stress, transcription, aggregate
Huntington disease results from a CAG repeat expansion in the HTT gene, yielding HTT protein with an expanded polyglutamine (polyQ) tract. To understand the molecular basis of polyQ neurotoxicity, investigators generated numerous in vitro and in vivo models, and demonstrated that polyQ-expanded proteins misfold and become sequestered into “aggregates.” The discovery of such protein inclusions linked HD and the polyQ diseases with virtually all neurodegenerative disorders. Previous work on HD has shown that autophagy pathway dysfunction is a prominent feature of disease pathogenesis, suggesting that modulation of the autophagy pathway might hold promise as a potential treatment for HD.
In 2006, my group and the laboratory of Dimitri Krainc independently discovered that impaired function of a transcriptional co-activator, known as peroxisome proliferator-activated receptor gamma, coactivator 1 α (PPARGC1A), contributes to HD neurodegeneration, by linking PPARGC1A dysfunction to thermoregulatory defects in HD mice, mitochondrial abnormalities in HD cell models, and coordinate downregulation of PPARGC1A target genes in HD patient brain samples. To determine if increased PPARGC1A function can ameliorate HD, my research group established an inducible PPARGC1A mouse model and crossed these mice with HD transgenic mice. We found that PPARGC1A overexpression rescues neurological phenotypes in HD mice, confirming the importance of PPARGC1A dysfunction in HD. Moreover, and rather unexpectedly, we noted that PPARGC1A overexpression also leads to a virtual eradication of HTT protein aggregates in the brains of these mice, accompanied by rescue of HD neuropathology. When we analyzed the basis for this, we determined that increased PPARGC1A function promoted mitochondrial ATP production and diminished reactive oxygen species (ROS). However, PPARGC1A-mediated HTT aggregate reduction requires a fully functional autophagy pathway, and when we examined HD mouse brain and cultured cells, we noted significant reductions in TFEB expression, which can be rescued by PPARGC1A overexpression. We chose to study TFEB, as recent work from Andrea Ballabio’s lab has established TFEB as a master regulator of the autophagy-lysosome pathway.
To determine the mechanistic basis of PPARGC1A regulation of TFEB, we performed chromatin immunoprecipitation and determined that PPARGC1A occupies the TFEB promoter. We used this information to create a TFEB promoter-reporter, and then found that PPARGC1A expression can robustly induce TFEB promoter-reporter activity. We also observed a marked reduction in TFEB promoter-reporter activity in striatal-like neurons from homozygous HD knockin mice, and found that PPARGC1A can rescue the impaired TFEB induction. When we assayed TFEB target genes in the brains of HD transgenic mice, we detected significant reductions, and observed marked increases in TFEB target gene expression in HD mice induced to overexpress PPARGC1A. To determine if PPARGC1A-mediated rescue of HTT protein aggregation is TFEB-dependent, we monitored the extent of HTT protein aggregation in response to PPARGC1A or TFEB co-expression, respectively knocking down TFEB or PPARGC1A. Although PPARGC1A knockdown does not blunt TFEB-mediated reductions in HTT protein aggregation, TFEB knockdown in the presence of PPARGC1A overexpression prevents any appreciable reduction in HTT protein aggregation. These findings indicate that PPARGC1A’s ability to promote clearance of HTT protein aggregates and rescue HTT neurotoxicity stems from its induction of TFEB.
When one studies the molecular basis of neurodegenerative diseases, such as HD, one must consider the features of neurons that make these cells so unique. In addition to being postmitotic cells that must survive throughout one’s entire lifetime, neurons face special challenges as they are under constant demand for substantial amounts of energy, and they must ceaselessly maintain quality control for proteins and organelles. These features of neurons highlight bioenergetics and autophagy as key pathways likely to be involved in neurodegenerative disease. In our current paper, we show that PPARGC1A overexpression can rescue HD mitochondrial dysfunction and impaired proteostasis, underscoring the linkage between bioenergetics and protein–organelle quality control in neurons.
In addition to implicating bioenergetics and protein–organelle quality control in neuronal dysfunction, and highlighting how these pathways are inextricably linked, our study provides new insights into the biology of PPARGC1A. The principal action of PPARGC1A is to promote mitochondrial biogenesis and biochemical processes that yield high levels of ATP. An unavoidable byproduct of high-energy generation is the creation of dangerous ROS; hence, PPARGC1A induces the expression of genes whose protein products break down or remove ROS. Another consequence of high-energy production is that mitochondria become damaged and must be removed, a feat that is accomplished by the autophagy-lysosome pathway. We now report that PPARGC1A promotes mitochondrial quality control by positively regulating TFEB expression (Fig. 1). The realization that PPARGC1A co-regulates the expression of genes to counter both ROS production and mitochondrial damage further extends our understanding of how PPARGC1A insures that cells, such as neurons, can maintain a high-energy production state.
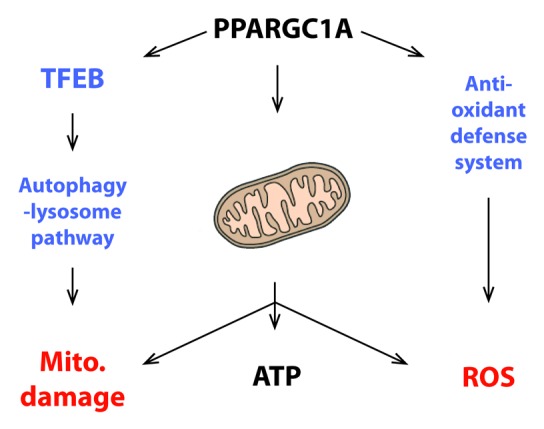
Figure 1. PPARGC1A induces TFEB to maintain mitochondrial quality control during high-energy production states. PPARGC1A drives mitochondrial biogenesis and co-activates the expression of target genes required for metabolic pathways that yield high levels of ATP. Maximal operation of the oxidative phosphorylation pathway creates ROS as an unavoidable byproduct of heavy utilization of this metabolic process; hence, as previously shown by the Spiegelman lab, PPARGC1A counters ROS production by co-activating the expression of genes that comprise an anti-oxidant defense system. An additional consequence of elevated mitochondrial function is an increase in the number of damaged mitochondria. Turnover of damaged mitochondria is accomplished by the autophagy-lysosome pathway through the process of mitophagy. In the current work, we report that PPARGC1A co-activates the expression of TFEB, a master regulator of the autophagy-lysosome pathway, providing another arm to PPARGC1A’s regulation of cellular homeostasis in the face of elevated mitochondrial energy production.
One final implication of our study is that TFEB does appear to be a key regulator of cellular protein and organelle homeostasis, suggesting that further investigation into its biology and regulation is warranted. If TFEB is as central to protein and organelle quality control as PPARGC1A is pivotal for mitochondrial biogenesis and ATP production, then TFEB also deserves careful consideration as a target for therapy development for disorders involving altered protein and organelle quality control. Delineating pathways of PPARGC1A and TFEB regulation may yield plausible strategies for modulating processes of energy production and protein–organelle quality control, especially if we can determine how these pathways become impaired in the course of disease.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21862