

Abstract
The molecular reaction mechanism of the GTPase-activating protein (GAP)-catalyzed GTP hydrolysis by Ras was investigated by time resolved Fourier transform infrared (FTIR) difference spectroscopy using caged GTP (P3-1-(2-nitro)phenylethyl guanosine 5′-O-triphosphate) as photolabile trigger. This approach provides the complete GTPase reaction pathway with time resolution of milliseconds at the atomic level. Up to now, one structural model of the GAP⋅Ras⋅GDP⋅AlFx transition state analog is known, which represents a “snap shot” along the reaction-pathway. As now revealed, binding of GAP to Ras⋅GTP shifts negative charge from the γ to β phosphate. Such a shift was already identified by FTIR in GTP because of Ras binding and is now shown to be enhanced by GAP binding. Because the charge distribution of the GAP⋅Ras⋅GTP complex thus resembles a more dissociative-like transition state and is more like that in GDP, the activation free energy is reduced. An intermediate is observed on the reaction pathway that appears when the bond between β and γ phosphate is cleaved. In the intermediate, the released Pi is strongly bound to the protein and surprisingly shows bands typical of those seen for phosphorylated enzyme intermediates. All these results provide a mechanistic picture that is different from the intrinsic GTPase reaction of Ras. FTIR analysis reveals the release of Pi from the protein complex as the rate-limiting step for the GAP-catalyzed reaction. The approach presented allows the study not only of single proteins but of protein–protein interactions without intrinsic chromophores, in the non-crystalline state, in real time at the atomic level.
The GTP-binding protein Ras is a major regulator of many signal transduction processes that lead to cell growth and differentiation (1). Like other GTP-binding proteins, it acts as a molecular switch, cycling between an inactive GDP-bound and an active GTP-bound conformation. The GTP β–γ bond is cleaved by a nucleophilic attack of a water molecule at the γ phosphate (2). In the active GTP-bound state Ras interacts with effectors and downstream targets in the signaling cascade. The intrinsic GTPase reaction of Ras is very slow, but GTPase-activating proteins (GAPs) can accelerate it maximally ca. 105 times (3). Many oncogenic mutants of Ras escape regulation by RasGAPs. This misfunction leads to a high concentration of Ras⋅GTP, which is a key event in malignant transformation of cells. Therefore, investigations have focused on understanding the molecular mechanism by which GAPs catalyze the hydrolysis reaction (4). Structural models of the Ras⋅GDP⋅AlF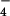 ⋅GAP complex, a useful transition state analog (5, 6), have revealed as a key element for catalysis a Gln residue on Ras and a highly conserved arginine on GAP interacting with AlF3 in the transition state analog. The additional positively charged “Arg-finger” is proposed to stabilize the developing negative charges on γ-GTP in the transition state and thereby catalyze the reaction.
⋅GAP complex, a useful transition state analog (5, 6), have revealed as a key element for catalysis a Gln residue on Ras and a highly conserved arginine on GAP interacting with AlF3 in the transition state analog. The additional positively charged “Arg-finger” is proposed to stabilize the developing negative charges on γ-GTP in the transition state and thereby catalyze the reaction.
Although the transition state structural model provided a milestone in the understanding of the mechanism, central questions still need to be discussed: The exact positioning of the catalytic arginine deviates slightly in structural models of two different GTP binding proteins (4) with interactions occurring with the previous β-γ-bridging oxygen and/or AlFx (5, 7, 8). The latter mimics the oxygens of the γ phosphate. A charge shift toward the β phosphate has been interpreted as favoring a more dissociative mechanism whereas a charge on the terminal γ phosphate would favor a more associative mechanism (9). Positive residues contacting γ phosphate oxygens in the transition state would favor a more associative mechanism.
For the understanding of the molecular mechanism, the determination of the charge shifts within GTP and GDP because of GAP binding is crucial. The initial GAP⋅Ras⋅GTP and final GAP⋅Ras⋅GDP three-dimensional structures have not been determined. All this makes it important to gather additional information in real time at atomic resolution of the complete GTPase reaction. Thereby, the Ras⋅GDP⋅AlF3⋅GAP complex (5), which represents a reasonable “snapshot” close to the transition state, can be understood in the context of the complete reaction and compared with a presumed ground state complex with Cdc42 and RhoGAP (10).
Time-resolved Fourier transform infrared (FTIR) difference spectroscopy has been successfully applied to monitor the molecular reaction mechanisms of proteins (11). Performing difference spectra between a ground state and an activated state selects the absorbance changes of the few residues involved in the reaction from the quiescent background absorption of the whole protein (12). The intrinsic GTPase reaction of Ras has already been successfully studied by time-resolved FTIR difference spectroscopy using caged GTP (P3-1-(2-nitro)phenylethyl guanosine 5′-O-triphosphate) as the photo labile trigger compound (13). The FTIR experiments reveal that binding to Ras forces the flexible GTP into a specific configuration and draws negative charge to the non-bridging β oxygens (13). This charge shift induces in GTP a charge distribution that is closer to the one in GDP and mimics more the charge distribution of a dissociative transition state, where negative charge is accumulated on the β phosphate. Thereby, the activation energy for the β–γ bond cleavage is reduced. Recently, this first study was refined by improving some band assignments, which support this proposal (14, 15). In addition to the IR studies, Raman spectroscopy has also been successfully applied, providing complementary band assignments and insight into the GTPase mechanism (16).
In this work, FTIR spectroscopy is used to investigate the GAP-catalyzed GTPase reaction. The accumulation of an intermediate can be monitored, providing insights into the molecular mechanism by which GAP catalyzes the GTPase reaction. An approach is established that is able to monitor at the atomic level in real time a protein–protein interaction leading to an enzymatic reaction.
Materials and Methods
Materials.
Wild-type full-length H-Ras were prepared from Escherichia coli (17). Wild-type NF1–333 was isolated from E. coli CK600 (18). A detailed description and more detailed literature for 18O labeling of GTP and GDP is given in ref. 15. For the FTIR measurements, the sample solution was prepared as described in detail (13, 15). The sample solutions for the measurements with NF1–333 contained 6.0 mM Ras⋅caged nucleotide, 6.6 mM wild-type NF1–333, 20 mM MgCl2, 20 mM DTT, 200 mM Mes (pH 6.0), and 12% ethylene glycol. NF1–333 was in slight excess over Ras⋅caged nucleotide to investigate the NF1–333-catalyzed GTPase reaction under single turnover conditions. DTT was used to scavenge the reactive photolysis byproduct 2-nitrosoacetophenone. For the measurements in H218O, the sample solution was picked up three times with an excess of H218O.
Methods.
The FTIR measurements were performed as described by using the fast scan technique (12), and the same conditions as detailed in (15) were used. The photolysis of caged GTP was performed at 308 nm by an LPX 240 XeCl excimer laser (Lambda Physics, Göttingen, Germany). Thirty flashes were applied to achieve 60 to 70% degree of conversion of caged GTP to GTP in 90 ms. The chosen number of flashes was a compromise between the time of photolysis and the degree of conversion. The temperature of 260 K was chosen to slow down the hydrolysis reaction rate for the application of the fast scan technique but to avoid freezing of the sample. The photolysis measurements with Ras⋅caged GTP were also performed at 260 K.
The data of the time-resolved fast scan measurements were analyzed between 1800 and 950 cm−1 with a global fit method (19). In global fit analysis, the absorbance changes ΔA in the infrared are analyzed with sums of nr exponentials with apparent rate constants kl and amplitudes al.
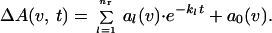 |
In the analysis, the weighted sum of squared differences f between the fit with nr rate constants kl and data points at nw measured wavelengths νi and nt time-points tj is minimized (19).
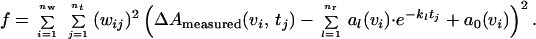 |
Results
The GTPase reaction of the GAP⋅Ras⋅caged GTP complex was initiated by UV laser flashes, which release GTP from caged GTP. To achieve single turnover conditions, a 1:1 stochiometry of GAP:Ras was used. As GAP, the catalytic domain NF1–333 of neurofibromin (NF1) (18) was used. The IR absorbance changes of the GAP-catalyzed reaction and, for comparison, the absorbance changes of the intrinsic reaction are shown in Fig. 1. The absorbance changes reflect only the reactions of those groups that are involved in the GTPase reaction. The curves relax to zero because the last measured spectrum is taken as reference. The kinetic analysis by the global method used here particularly improves the determination of small absorbance changes by analyzing them simultaneously with large absorbance changes between 1800 and 950 cm−1 (19). Increase of the reaction from 2 h 50 min to 80 s by GAP (Fig. 1) is well resolved, and an intermediate accumulates that is observed only in the GAP-catalyzed reaction. This difference can be seen, for example, at 1143 cm−1. The intrinsic reaction of Ras can be described by a single exponential function with the rate constant k = (5.1 ± 0.5) × 10−4 s−1 at 310 K. The GAP-catalyzed reaction has to be described by at least three exponential functions. This result implies at least a three step mechanism:
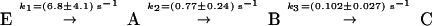 |
To determine the molecular reactions described by the apparent rate constant kl, the corresponding amplitude spectra al(ν) are analyzed. The amplitude spectra al(ν) reflect the absorbance changes in the transition described by kl. Because this manuscript focuses on the influence of GAP on the phosphate moiety of Ras⋅GTP and Ras⋅GDP during the GTPase reaction, only the phosphate region between 1350 and 950 cm−1 will be shown and discussed.
Figure 1.

The absorbance changes in the infrared between 1800 and 950 cm−1 during the intrinsic GTPase reaction of Ras and during the GAP-catalyzed reaction are shown. The absorbance changes of the intrinsic reaction can be described by a single exponential function as seen for example at 1143 cm−1 for GTP and 1104 cm−1 for GDP. In the GAP-catalyzed reaction, an intermediate accumulates as seen for example at 1143 cm−1. The fitted curves of the global multiexponential kinetic analysis are shown (note the difference in log time scale). Absorbance changes at single wave numbers as function of time are shown in Fig. 4.
The E to A Transition.
The amplitude spectrum of k1 (data not shown) agrees in general with the photolysis amplitude spectrum of the intrinsic reaction presented in ref. 15. This agreement implies that, in the E to A reaction, the photolysis of the GAP⋅Ras⋅caged GTP complex to a GAP⋅Ras⋅GTP ground state complex occurs. The complex formation between GAP⋅Ras⋅caged GTP was confirmed by gel filtration (results not shown). Because also the rate constants for the formation of Ras⋅GTP from Ras⋅caged GTP in the presence or absence of GAP are the same, GAP binding seems to have almost no influence on the photolysis reaction.
The A to B Transition.
In Fig. 2aA, the amplitude spectrum of k2 is presented (black line). The amplitude spectra follow the convention that negative bands are associated with disappearing molecular groups or interactions and positive bands with the formation of molecular groups or interactions. In general, only those bands that have been assigned to specific groups and are measured reproducibly are labeled. The disappearing bands at 1259, 1216, 1160, and 1141 cm−1 agree nicely with the α-, β-, and γ-GTP bands of the intrinsic reaction (13–16). The positive bands at 1186 cm−1 and 1113 cm−1 of the intermediate B are not observed in the intrinsic reaction because this intermediate has not been detected previously.
Figure 2.

(a) The amplitude spectra of k2, which describe the A to B transition are shown (black lines). Negative bands belong to the GTP state and positive bands to the intermediate. In addition, the amplitude spectra of the labeled compounds are shown (colored lines). The 18O-labeled positions in GTP are indicated by colored oxygens. (b) Differences between the amplitude spectra of labeled and unlabeled GTP as shown in a are presented to visualize the frequency shifts. The shifts of labeled bands cause difference bands, illustrated by shaded areas. The original frequency is given in black and the shifted position in color. This is available enlarged as Figs. 7 and 8, which are published as supplemental data on the PNAS web site, www.pnas.org.
To assign the bands to specific residues, different site-specific 18O-labeled caged GTP were used. Isotopic labeling shifts the IR bands of the labeled group to lower frequencies. Because the phosphate vibrations become localized by Ras binding, the bands can be assigned to vibrations of single groups (13–16). Because of γ-18O labeling (Fig. 2aA, red line), the γ-GTP bands at 1160 and 1141 cm−1 are shifted to 1129 and 1118 cm−1. To visualize the frequency shifts, double difference spectra are used. In principle, in the double difference spectra, only those bands should appear that are shifted because of labeling. However, depending on the signal to noise ratio, some band shifts are clearly identified whereas other band shifts are just above the noise. For bands with lower intensity, we rely on the band shifts seen in the difference spectra and use the double difference spectra only for illustration of the identified band shifts. In the double difference spectrum (Fig. 2bA), the GTP band shifted because of γ-18O labeling appears at its unlabeled position, as positive bands at 1157 and 1145 cm−1 and, at its labeled positions, as negative bands at 1129 and 1116 cm−1. Because of these frequency shifts, the bands at 1160 and 1141 cm−1 in Fig. 2aA are assigned to the γ-PO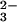 phosphate vibration as for the intrinsic reaction (13–16). For the γ-PO
phosphate vibration as for the intrinsic reaction (13–16). For the γ-PO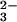 group, a degenerate vibration with only one band is generally expected. The distinct splitting into two vibrations at 1160 and 1141 cm−1 can be explained by two configurations of γ phosphates in slightly different environments (15). In agreement, NMR experiments propose two states of GTP (20). The GAP binding greatly increases the intensity of the band at 1160 cm−1, as compared with the intrinsic reaction (14, 15), and thereby the population of the second conformation.
group, a degenerate vibration with only one band is generally expected. The distinct splitting into two vibrations at 1160 and 1141 cm−1 can be explained by two configurations of γ phosphates in slightly different environments (15). In agreement, NMR experiments propose two states of GTP (20). The GAP binding greatly increases the intensity of the band at 1160 cm−1, as compared with the intrinsic reaction (14, 15), and thereby the population of the second conformation.
The positive intermediate bands at 1186 cm−1 and 1113 cm−1 are downshifted to 1170 cm−1 and 1093 cm−1, respectively (Fig. 2aA). These shifts appear as difference bands at 1191/1169 cm−1 and 1113/1091 cm−1 in the double difference spectrum (Fig. 2bA). Because the bands at 1186 and 1113 cm−1 are shifted, they represent vibrations of either the γ phosphate group or, if cleavage of the β–γ bond has already taken place in the intermediate, the released Pi.
The isotopic shifts because of the α/γ 18O-labeling are not shown, but data are available on request. The shifts already identified in A because of γ-labeling are also present. The intensity is reduced because only one P–O bond is labeled, but the same frequency shifts are seen because of γ labeling. In addition, a shift from 1260 cm−1 to 1236 cm−1 is observed caused by the α-PO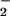 vibration. The assignment agrees well with the α vibration of the intrinsic GTPase reaction (14, 15).
vibration. The assignment agrees well with the α vibration of the intrinsic GTPase reaction (14, 15).
To determine whether attack of water on the γ phosphate group has taken place in the intermediate, experiments were performed in H218O. The nucleophilic attack of labeled water led to an 18O-labeled phosphate group and its bands should shift. This band shift of 18O-labeled Pi is shown for the intrinsic reaction (15). Only small but reproducible shifts are seen from 1186 cm−1 to 1170 cm−1, at 1156/1141 cm−1, and from 1113 cm−1 to 1091 cm−1 (Fig. 2aB). Even if these changes are small, they are clearly above the noise level. The shifts of the positive intermediate bands are visualized in the double difference spectrum at 1191/1169 and 1118/1091 cm−1. They agree with the shifts observed because of γ labeling and α/γ labeling as indicated in Fig. 2b by straight lines. The intensity of the downshifted bands in Fig. 2bB at 1186 and 1113 cm−1 is much smaller than in 2bA because only one P–O bond is labeled instead of four. However, it is similarly frequency shifted and has comparable intensity to that of the α/γ labeling also with only one P–O γ bond labeled. Further independent evidence for the shifts of the intermediate bands at 1186 and 1113 cm−1 in H218O is provided below from the amplitude spectra of k3. In summary, because of the band shifts in H218O, it must be concluded that attack of a water molecule has taken place in the intermediate.
The amplitude spectra also indicate a downshift of the negative GTP-γ phosphate bands at 1156 cm−1 and 1141 cm−1 visualized in the double difference spectra at 1157/1145 cm−1 (Fig. 2 a and b). This downshift is not expected after bond cleavage. However, a fast equilibrium between GTP and GDP⋅Pi as observed for ATP/ADP⋅Pi in myosin would lead to the incorporation of labeled 18O into bound GTP (14, 15). The shift of the γ band at 1156/1143 cm−1 might indicate such an equilibrium reaction.
In Fig. 2aC, the shifts because of (SP)-[β-18O]GTP are shown. The β-PO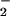 -GTP vibration of the intrinsic reaction has been assigned to 1219 cm−1 (14, 15). Beside the expected shift of the band at 1216 cm−1 down to 1186 cm−1, an additional unexpected shift appears from 1141 cm−1 to 1108 cm−1. These shifts are visualized in the double difference at 1217/1190 cm−1 and 1140/1117 cm−1. Interestingly, similar shifts at 1140 cm−1 are observed for the γ vibration in Fig 2 aA and aB. In principle, vibrational coupling between β and γ vibrations could cause the downshift of the band at 1140 cm−1. In such a case, the γ labeling should reciprocally influence the β vibration at 1217 cm−1. But no such shift is observed at 1217 cm−1 in Fig. 2 aA, aB, or aC. Therefore, the band at 1140 cm−1 represents a not coupled (Sp)β P-O vibration just at the same frequency as the γ-PO
-GTP vibration of the intrinsic reaction has been assigned to 1219 cm−1 (14, 15). Beside the expected shift of the band at 1216 cm−1 down to 1186 cm−1, an additional unexpected shift appears from 1141 cm−1 to 1108 cm−1. These shifts are visualized in the double difference at 1217/1190 cm−1 and 1140/1117 cm−1. Interestingly, similar shifts at 1140 cm−1 are observed for the γ vibration in Fig 2 aA and aB. In principle, vibrational coupling between β and γ vibrations could cause the downshift of the band at 1140 cm−1. In such a case, the γ labeling should reciprocally influence the β vibration at 1217 cm−1. But no such shift is observed at 1217 cm−1 in Fig. 2 aA, aB, or aC. Therefore, the band at 1140 cm−1 represents a not coupled (Sp)β P-O vibration just at the same frequency as the γ-PO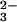 vibration. The band at 1140 cm−1 represents a remarkable downshift of the (Sp)β vibration as compared with the intrinsic reaction.
vibration. The band at 1140 cm−1 represents a remarkable downshift of the (Sp)β vibration as compared with the intrinsic reaction.
There is no shift of a positive band remaining, which could be assigned to a β-GDP intermediate vibration. The β-GDP band might be masked by overlap with other bands or its intensity is largely reduced similar to the α-GDP intermediate bands, and therefore the GDP bands of the intermediate could not be resolved.
Similar to the intrinsic reaction, the intensity of the (Rp)β band at 1216 cm−1 is reduced in Fig. 2aD as compared with the (SP)-β band. This lower intensity is also seen by comparing the difference bands 1217/1190 cm−1 in the double difference spectra (Fig. 2 bC and bD). The band at 1216 cm−1 contains contributions of (Sp) and (Rp)β PO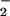 vibrations. In contrast, the clear downshift from 1140 cm−1 to 1117 cm−1 because of (Sp)18O labeling is not observed with (Rp) labeling. There is a reduced intensity around 1141 cm−1, but no clear band shift to 1117 cm−1 is seen (Fig. 2aD). The observed deviations at 1140 cm−1 could be caused by base line drifts. This observation is also illustrated by the double difference in Fig. 2bD, where no clear difference band is identified at 1140/1117 cm−1 as compared with Fig. 2bC. Within our experimental error there is no evidence for a large contribution of the Rp P-O vibration to the band at 1141 cm−1 in contrast to the Sp P-O vibration. Therefore, we conclude that the band at 1140 cm−1 mainly represents the (Sp)P-O− vibration. Two (Sp)-PO− vibrations at 1216 cm−1 and 1140 cm−1 can be explained by two different conformations of the (Sp)-PO− group leading to different interactions with the protein similar to that discussed for γ-GTP above.
vibrations. In contrast, the clear downshift from 1140 cm−1 to 1117 cm−1 because of (Sp)18O labeling is not observed with (Rp) labeling. There is a reduced intensity around 1141 cm−1, but no clear band shift to 1117 cm−1 is seen (Fig. 2aD). The observed deviations at 1140 cm−1 could be caused by base line drifts. This observation is also illustrated by the double difference in Fig. 2bD, where no clear difference band is identified at 1140/1117 cm−1 as compared with Fig. 2bC. Within our experimental error there is no evidence for a large contribution of the Rp P-O vibration to the band at 1141 cm−1 in contrast to the Sp P-O vibration. Therefore, we conclude that the band at 1140 cm−1 mainly represents the (Sp)P-O− vibration. Two (Sp)-PO− vibrations at 1216 cm−1 and 1140 cm−1 can be explained by two different conformations of the (Sp)-PO− group leading to different interactions with the protein similar to that discussed for γ-GTP above.
Characterization of the B to C Transition.
In Fig. 3aA the amplitude spectrum of k3 is shown. The former positive bands at 1186 cm−1 and 1114 cm−1 of the intermediate now become negative because it decays with k3 to the final state. The downshifts of the negative bands at 1186 cm−1 and 1114 cm−1 because of γ labeling are seen again, as reflected by difference bands at 1189/1172 cm−1 and 1115/1098 cm−1 in Fig. 3bA. Shifts of the final state positive bands are seen from 1137 cm−1 down to around 1114 cm−1, and 1081 cm−1 to 1046 cm−1 (Fig. 3aA). The band from 992 cm−1 also shifts down. However, the position of the downshifted band is not clearly resolved. The shifts of positive bands are reflected in the double difference spectrum by difference bands at 1138/1115, 1078/1046 cm−1, and at 992 cm−1 (Fig. 3bA). The shift at 1138 cm−1 is due to the formerly labeled bridging oxygen between β and γ phosphate, which is labeled and remains at the β phosphate group after the bond splitting. The band at 1138 cm−1 has therefore to be assigned to a νa(β-PO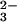 ) vibration of GDP. The band absorbs at the same frequency as the νa(γ-PO
) vibration of GDP. The band absorbs at the same frequency as the νa(γ-PO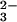 ) group in the GTP state, but now reflects the vibration of the β-GDP PO
) group in the GTP state, but now reflects the vibration of the β-GDP PO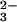 group.
group.
Figure 3.

(a) The amplitude spectrum of k3 (black lines), which describes the transition from the intermediate to the final GDP state. Negative bands now belong to the intermediate and positive bands to the GDP state. In addition, the amplitude spectra of the labeled compounds are shown (colored lines) as in Fig. 2a. (b) Differences between the amplitude spectra of labeled and unlabeled GTP as shown in a, presented as in Fig. 2a. This is available enlarged as Figs. 9 and 10, which are published as supplemental data.
The positive bands at 1081 cm−1 and 992 cm−1 which are shifted because of γ labeling can be assigned to Pi vibrations (Fig. 3aA). The released Pi of the intrinsic GTPase reaction also absorbs at 1078 cm−1 and 992 cm−1, and the bands are downshifted by γ labeling (14, 15). The frequencies indicate formation of HPO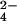 .
.
The same shifts as observed for 4-fold 18O γ labeling are also detected for the α/γ labeling. The band shifts are seen at 1189/1176, 1115/1098, 1078/1046, and 992 cm−1 (see supplemental Figs. 9 and 10). Again, the intensity in the spectrum of the single α/γ labeled GTP is reduced in comparison to the 4-fold γ-labeled GTP, but the same frequency shifts are seen.
The bands at 1186 and 1114 cm−1 are downshifted because of H218O (Fig. 3aB) as in the k2 amplitude spectrum and appear in the double difference spectrum (Fig. 3bB) as difference bands at 1189/1172 cm−1 and 1115/1098 cm−1. This result confirms the former assignments in the k2 amplitude spectrum above. Also the Pi vibrations at 1078 cm−1 and 992 cm−1 are downshifted by H218O similarly to the intrinsic reaction (15). The intensities of these Pi bands are similar to those of the Pi bands in the intermediate at 1186 cm−1 and 1113 cm−1.
In Fig. 3 aC and bC, the isotopic shifts because of (SP)-β-18O are presented. There is no indication of a shift of a negative band, which might indicate the β-GDP vibration in the intermediate. The positive bands at 1137 cm−1 and 1098 cm−1 are downshifted, which is visualized in the double difference spectra by difference bands at 1138/1117 cm−1 and 1104/1084 cm−1. Therefore, the bands can be assigned to β-PO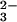 GDP vibrations. Interestingly, the β-PO
GDP vibrations. Interestingly, the β-PO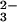 vibrations of GDP appear in the hydrolysis spectrum of the intrinsic GTPase reaction at the same positions (14, 15) and are also unexpectedly split in two bands. The two bands may be caused either by a very asymmetric environment or by two different GDP conformations (14, 15). The two state explanations are more likely, because splitting is already observed for the GTP vibrations.
vibrations of GDP appear in the hydrolysis spectrum of the intrinsic GTPase reaction at the same positions (14, 15) and are also unexpectedly split in two bands. The two bands may be caused either by a very asymmetric environment or by two different GDP conformations (14, 15). The two state explanations are more likely, because splitting is already observed for the GTP vibrations.
Frequency shifts because of (RP)-β-18O labeling in Fig. 3 aD and bD are not observed above the noise level. The deviations reflect baseline drifts. This observation is illustrated in the difference of the amplitude spectra, which show no difference band (Fig. 3b).
In summary, the B to C transition describes the appearance of the Ras-bound GDP and the release of Pi. Pi has the same frequencies as in the intrinsic reaction. This agreement shows that GAP binding has almost no influence on the final GDP and Pi state.
Time Course of the Reaction.
Based on these band assignments, the reactions of the phosphate groups can now be seen by following the absorbance changes of the respective phosphate groups against time. Absorbance changes of the intrinsic reaction are already shown in Fig. 1 and more detailed in ref. 15. They can be described by a single exponential function. In Fig. 4, the absorbance changes of the GAP-catalyzed GTPase reaction are presented. The absorbance charges relax to zero because the final GDP state is used as reference. At 1143 cm−1 the γ phosphate group appears with k1. The absorbance change reflects the photolysis of caged GTP to GTP in the E to A transition. GTP disappears almost completely with k2, reflecting the cleavage of the γ phosphate. Simultaneously, the Pi vibration at 1113 cm−1 rises with k2, indicating the appearance of the protein bound Pi in the intermediate. The intermediate decays with k3, and Pi is released with k3 from the Ras⋅GAP complex as monitored at 1078 cm−1. Simultaneously, the GDP vibration of the final Ras⋅GDP state appears at 1104 cm−1. The small deviations at 1098 cm−1 and 1104 cm−1 appear in the time course, because the bands overlap. Within the experimental error there is nice agreement.
Figure 4.

Typical absorbance changes of the GAP-catalyzed reaction are shown. Besides the data (crosses), the fitted curves (solid line) and the individual contributions (ki) of the multi-exponential fit analysis are shown. In contrast to the intrinsic Ras-catalyzed reaction, which can be described with one exponential function and the rate constant k = (5.1 ± 0.5) × 10−4 s−1 at 310 K, the absorbance changes have to be described with at least three rate constants k1 = (6.8 ± 4.1) s−1, k2 = (0.77 ± 0.24) s−1, and k3 = (0.102 ± 0.027) s−1 at 260 K. k1 represents the photolysis of caged GTP and the appearance of released γ phosphate as indicated at 1143 cm−1. k2 shows the cleavage of γ phosphate again seen at 1143 cm−1 and the appearance of protein-bound Pi at 1113 cm−1. k3 shows the decay of the protein bound Pi at 1113 cm−1 and its release from the protein–protein complex into the bulk medium at 1078 cm−1. k3 represents the rate-limiting step. It is clearly seen that GAP catalyses the bond cleavage reaction.
Discussion
All band assignments performed in the manuscript are summarized in Fig. 5 A, B, and C. To illustrate the frequency shifts of GTP/GDP vibrations because of GAP binding, the shifts because of Mg and Ras binding are also listed (13–16, 21–23). The implications of Mg- and Ras-induced shifts have already been discussed in detail (15). Which additional band shifts because of GAP binding are observed and what can be learned from them? Comparing the phosphate frequencies of Ras⋅Mg⋅GTP/GDP and GAP⋅Ras⋅Mg⋅GTP/GDP in Fig. 5, only the (SP)-β-GTP vibration is shifted significantly because of GAP binding to 1140 cm−1, whereas the other vibrations are not shifted at all. Also a coupled (SP)/(RP)-β-PO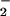 -GTP vibration at 1217 cm−1 is observed, which is not shifted by GAP as compared with Ras. These two IR bands of the (SP)-β-P-O group can be explained by two conformations in the GAP bound state. Two conformations of GTP are also proposed by NMR studies (20).
-GTP vibration at 1217 cm−1 is observed, which is not shifted by GAP as compared with Ras. These two IR bands of the (SP)-β-P-O group can be explained by two conformations in the GAP bound state. Two conformations of GTP are also proposed by NMR studies (20).
Figure 5.

The frequency shifts of GTP, GDP, and Pi vibrations induced by different binding partners are listed (12–16, 22).
The β-PO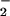 -GTP vibration at 1217 cm−1 in Ras is downshifted as compared with Mg by 28 cm−1. It has been argued that in Ras the phosphate vibrations are almost not coupled and to a first approximation they mainly indicate changes of the force constant of the respective vibration. Changes of the force constant are related to bond order changes (24). The frequency downshift of the β-PO
-GTP vibration at 1217 cm−1 in Ras is downshifted as compared with Mg by 28 cm−1. It has been argued that in Ras the phosphate vibrations are almost not coupled and to a first approximation they mainly indicate changes of the force constant of the respective vibration. Changes of the force constant are related to bond order changes (24). The frequency downshift of the β-PO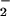 vibration because of Ras binding indicates a bond order decrease and thereby an increase of negative charge at both non-bridging β oxygens (13, 14, 15). At the γ oxygens, the negative charge decreases and the overall charge movements in GTP result in a charge distribution that resembles that expected for a more dissociative transition state (9, 13, 14, 15). The similarity in the charge distribution between educt and transition state has been proposed to reduce the activation free energy and contribute to catalysis by Ras (13, 15). In addition, it supports the charge transfer from γ-PO
vibration because of Ras binding indicates a bond order decrease and thereby an increase of negative charge at both non-bridging β oxygens (13, 14, 15). At the γ oxygens, the negative charge decreases and the overall charge movements in GTP result in a charge distribution that resembles that expected for a more dissociative transition state (9, 13, 14, 15). The similarity in the charge distribution between educt and transition state has been proposed to reduce the activation free energy and contribute to catalysis by Ras (13, 15). In addition, it supports the charge transfer from γ-PO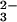 in GTP to β-PO
in GTP to β-PO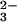 in GDP during hydrolysis. This charge transfer contributes also to catalysis as proposed by theoretical analysis (14, 15, 25). Binding of GAP induces an even larger downshift of 79 cm−1 of the (Sp)-β-P-O vibration, thereby creating a further accumulation of negative charge at the (Sp)-β oxygen. This shift is the only change in the charge distribution of GTP because of GAP binding and conclusively this must be a decisive factor in GAP-mediated catalysis. In Ras Lys-16 and the NH groups of the P-loop shifting charges towards β-phosphate (13, 15, 32, 33).
in GDP during hydrolysis. This charge transfer contributes also to catalysis as proposed by theoretical analysis (14, 15, 25). Binding of GAP induces an even larger downshift of 79 cm−1 of the (Sp)-β-P-O vibration, thereby creating a further accumulation of negative charge at the (Sp)-β oxygen. This shift is the only change in the charge distribution of GTP because of GAP binding and conclusively this must be a decisive factor in GAP-mediated catalysis. In Ras Lys-16 and the NH groups of the P-loop shifting charges towards β-phosphate (13, 15, 32, 33).
The question arises, which additional positively charged group of GAP draws more negative charge onto the (SP)-β oxygen? Arg-789 (residue from p120GAP) in the transition state model is close enough to the β phosphate to provide this interaction under the reasonable assumption that the mimic of the transition state structure is similar to that of the GAP⋅Ras⋅GTP complex observed in our FTIR experiments (5). However, this assumption would require Arg-789 to move from AlF3 and the β,γ-bridging oxygen in the transition state analog to the (SP)-β oxygen. The catalytic Arg is a likely candidate for such a move even if no interaction of Arg-GAP residues with (SP)-β oxygens has been observed in the presumed ground-state structure of the Cdc42⋅GppNHp⋅p50rhoGAP complex (10). This disagreement could, however, mean that the Cdc42⋅GppNHp complex does not resemble the GTP state of the real educt state or is different from the ground state complex between Ras and RasGAP. As an alternative to the catalytic Arg, the binding of GAP to Ras might shift the invariant positively charged Lys-16 from the P-loop toward the (SP)-β oxygen (Fig. 6). A shift of the P-loop lysine as an important element of catalysis has previously been proposed for adenylate kinase (26).
Figure 6.

The cartoon summarizes the results: the shift of negative charges from γ-GTP to β oxygens because of Ras binding and the additional larger shift mostly to the (SP)-β oxygen because of GAP binding by Arg-789 and/or Lys-16 and backbone NH groups of the highly conserved P-loop are illustrated. Mg2+, Lys, and Arg are considered to be the key charged residues holding like “molecular tweezers” charges on β oxygens. This charge shift reduces the free activation energy for β–γ bond cleavage in the A to B transition with k2 (compare Fig. 4, 1143 cm−1). The Pi is strongly bound, most likely to GAP, in the intermediate B. Pi release is rate limiting, with k3 in the B to C transition (compare Fig. 4, 1113, 1078, 1104 cm−1). The structural arrangement is based on structural models of refs. 5, 32, and 33.
The largest influence of GAP on the phosphate vibrations is observed in the intermediate. Two prominent Pi bands at 1191 cm−1 and 1116 cm−1 appear with the intermediate (Fig. 5). Their frequencies are unusually upshifted as compared with the typical Pi vibrations at 1078 cm−1 and 992 cm−1 in the final GDP state (Fig. 5). How can these unusual frequency up shifts of Pi be explained? It is unlikely that the bands represent metaphosphate of the transition state. Such a reactive species would not be expected to survive for the long periods seen in this study. In addition, there is a disagreement in the frequencies observed here and the frequencies of model compounds for metaphosphate (e.g., BaOP2O5).
Pi frequencies at 1191 and 1116 cm−1 are typical for phosphorylated enzymes. A Mg-coordinated phosphorylated aspartyl in the E2P state in the Ca-ATPase shows exactly these two bands at 1191 and 1116 cm−1 (27, 28). Phosphorylated serine and threonine absorb similarly, at 1194 and 1085 cm−1 (29). Therefore, a phosphorylation-like Pi strongly bound to the enzyme could explain the unusual frequencies. Although a mechanism with a phosphorylated intermediate does not appear likely on the basis of present knowledge, it cannot be dismissed out of hand.
In principle, noncovalent binding of Pi to a positively charged group could also upshift the frequencies. But the binding of Pi to positively charged guanidinium in solution failed to upshift the Pi vibrations (M. Blessenohl, unpublished data). It cannot completely be ruled out that in solution water molecules may weaken the electrostatic interaction and that, within a hydrophobic protein environment, the Pi vibrations might somehow upshift. It would, nevertheless, be surprising to find exactly the same bands as for a phosphorylated-like state described above.
Both Pi bands are shifted when the reaction is performed in H218O, which would not immediately be expected for a phosphorylated intermediate formed by an amino acid reacting with the γ phosphate. A water molecule should attack the phosphorylated enzyme in the next step, whereas our results show that the phosphate group in the intermediate has already incorporated 18O. A possible explanation would be that the hydrolysis of the phosphorylated intermediate is easily reversible, allowing rephosphorylation by the freshly produced inorganic phosphate. This equilibrium is actually observed in the actin-stimulated ATPase reaction of myosin (30), and there might be a similar reaction mechanism in both systems. Interestingly, in myosin, no catalytic Arg finger is observed, but it does contain a bona fide P-loop.
The results of this work can be summarized as follows (Fig. 6): In the A to B reaction, the γ phosphate is cleaved, and Pi is formed. This reaction is largely catalyzed by drawing more negative charge onto the (SP)-β oxygen by additional positive charges provided by GAP binding. Likely positive charged residues near the (SP)-β oxygen are Lys-16 of the P-loop and/or Arg-789 of GAP. Pi with phosphorylation-like IR absorption is strongly bound to the protein in the intermediate B. The rate-limiting step for the GAP-catalyzed reaction is the Pi release into the bulk medium during the B to C transition. This result is in harmony with two transition state structural models (5, 7), which show that GAP bound to Ras covers the AlF3 and might therefore block Pi release (Fig. 6). This conclusion could also be inferred from the Pi release measurements reported by Nixon et al. (31).
This work is an example of the investigation of a protein–protein interaction by FTIR in a system that does not contain an intrinsic chromophore. The approach presented here can be applied to other GTP-binding proteins and phosphoryl transfer reactions to yield insight into their mechanism and into protein–protein interactions without requiring the protein complex to be crystallized (x-ray) or solubilized (NMR).
Supplementary Material
Acknowledgments
We especially thank Dr. R. S. Goody for very critically reading the manuscript and useful suggestions, M. Blessenohl and Ken Holmes for helpful discussions, Kate Dalton for help with the English style, and Christian Kandt for producing Fig. 6. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 394-B1).
Abbreviations
- caged GTP
P3-1-(2-nitro)phenylethyl guanosine 5′-O-triphosphate
- FTIR
Fourier transform infrared
- GAP
GTPase-activating protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Barbacid M. Annu Rev Biochem. 1987;56:799–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Feuerstein J, Goody R S, Webb M R. J Biol Chem. 1989;264:6188–6190. [PubMed] [Google Scholar]
- 3.Wittinghofer A, Scheffzek K, Ahmadian M R. FEBS Lett. 1997;410:63–67. doi: 10.1016/s0014-5793(97)00321-9. [DOI] [PubMed] [Google Scholar]
- 4.Sprang S R. Science. 1997;277:329–330. doi: 10.1126/science.277.5324.329. [DOI] [PubMed] [Google Scholar]
- 5.Scheffzek K, Ahmadian M R, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 6.Scheffzek K, Ahmadian M R, Wittinghofer A. Trends Biochem Sci. 1998;27:257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- 7.Rittinger K, Walker P A, Eccleston J F, Smerdon S J, Gamblin S J. Nature (London) 1997;389:758–762. doi: 10.1038/39651. [DOI] [PubMed] [Google Scholar]
- 8.Nassar N, Hoffman G R, Manor D, Clardy J C, Cerione R A. Nat Struct Biol. 1998;5:1047–1052. doi: 10.1038/4156. [DOI] [PubMed] [Google Scholar]
- 9.Maegley K A, Admiraal S J, Herschlag D. Proc Natl Acad Sci USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rittinger K, Walker P A, Eccleston J F, Nurmahomed K, Owen D, Laue E, Gamblin S J, Smerdon S J. Nature (London) 1997;388:693–697. doi: 10.1038/41805. [DOI] [PubMed] [Google Scholar]
- 11.Gerwert K. Curr Opin Struct Biol. 1993;3:769–773. [Google Scholar]
- 12.Gerwert K, Souvignier G, Hess B. Proc Natl Acad Sci USA. 1990;87:9774–9778. doi: 10.1073/pnas.87.24.9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cepus V, Goody R S, Gerwert K. Biochemistry. 1998;37:10263–10271. doi: 10.1021/bi973183j. [DOI] [PubMed] [Google Scholar]
- 14.Du X, Frei H, Kim S. J Biol Chem. 2000;275:8492–8500. doi: 10.1074/jbc.275.12.8492. [DOI] [PubMed] [Google Scholar]
- 15.Allin C, Gerwert K. Biochemistry. 2001;40:3037–3046. doi: 10.1021/bi0017024. [DOI] [PubMed] [Google Scholar]
- 16.Wang J H, Xiao D G, Deng H, Webb M R, Callender R. Biochemistry. 1998;37:11106–11116. doi: 10.1021/bi980471m. [DOI] [PubMed] [Google Scholar]
- 17.Tucker J, Sczakiel G, Feuerstein J, John J, Goody R S, Wittinghofer A. EMBO J. 1986;5:1351–1358. doi: 10.1002/j.1460-2075.1986.tb04366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmadian M R, Stege P, Scheffzek K, Wittinghofer A. Nat Struct Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 19.Hessling B, Souvignier G, Gerwert K. Biophys J. 1993;65:1929–1941. doi: 10.1016/S0006-3495(93)81264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geyer M, Schweins T, Herrmann C, Prisner T, Wittinghofer A, Kalbitzer H R. Biochemistry. 1996;35:10308–10320. doi: 10.1021/bi952858k. [DOI] [PubMed] [Google Scholar]
- 21.Cepus V, Ulbrich C, Allin C, Troullier A, Gerwert K. Methods Enzymol. 1998;291:223–245. doi: 10.1016/s0076-6879(98)91015-1. [DOI] [PubMed] [Google Scholar]
- 22.Wang J H, Xiao D G, Deng H, Callender R, Webb M R. Biospectroscopy. 1998;4:219–227. doi: 10.1002/(SICI)1520-6343(1998)4:4%3C219::AID-BSPY1%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 23.Chapman A C, Thirlwell L E. Spectrochim Acta. 1964;20:937–947. [Google Scholar]
- 24.Pohle W, Bohl M, Böhling H. J Mol Struct. 1990;242:33–342. [Google Scholar]
- 25.Glennon T M, Villà J, Warshel A. Biochemistry. 2000;39:9641–9651. doi: 10.1021/bi000640e. [DOI] [PubMed] [Google Scholar]
- 26.Reinstein J, Schlichting J, Wittinghofer A. Biochemistry. 1990;29:7451–7459. doi: 10.1021/bi00484a014. [DOI] [PubMed] [Google Scholar]
- 27.Barth A. J Biol Chem. 1999;274:22170–22175. doi: 10.1074/jbc.274.32.22170. [DOI] [PubMed] [Google Scholar]
- 28.Ulbrich C. Ph.D. thesis. Bochum, Germany: Ruhr-Universität; 2000. [Google Scholar]
- 29.Sanchez-Ruiz J M, Martinez-Carrion M. Biochemistry. 1988;27:3338–3342. doi: 10.1021/bi00409a033. [DOI] [PubMed] [Google Scholar]
- 30.Bowater R, Zimmerman R W, Webb M R. J Biol Chem. 1990;265:171–176. [PubMed] [Google Scholar]
- 31.Nixon A E, Brune M, Lowe P N, Webb M R. Biochemistry. 1995;34:15592–15598. doi: 10.1021/bi00047a026. [DOI] [PubMed] [Google Scholar]
- 32.Pai E F, Krengel U, Petsko G A, Goody R S, Kabsch W, Wittinghofer A. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheidigk A J, Burmester C, Goody R S. Structure Fold Des. 1999;7:1311–1324. doi: 10.1016/s0969-2126(00)80021-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}