

Abstract
One of the first methods to encapsulate drugs within polymer nanospheres was developed by Fessi and coworkers in 1989 and consisted of one-step nanoprecipitation based on solvent displacement. However, proteins are poorly encapsulated within polymer nanoparticles using this method because of their limited solubility in organic solvents. To overcome this limitation, we developed a two-step nanoprecipitation method and encapsulated various proteins with high efficiency into poly(lactic-co-glycolic)acid (PLGA) nanospheres (NP). In this method, a protein nanoprecipitation step is used first followed by a second polymer nanoprecipitation step. Two model enzymes, lysozyme and α-chymotrypsin, were used for the optimization of the method. We obtained encapsulation efficiencies of >70%, an amount of buffer-insoluble protein aggregates of typically <2%, and a high residual activity of typically >90%. The optimum conditions identified for lysozyme were used to successfully encapsulate cytochrome c(Cyt-c), an apoptosis-initiating basic protein of similar size, to verify reproducibility of the encapsulation procedure. The size of the Cyt-c loaded-PLGA nanospheres was around 300–400 nm indicating the potential of the delivery system to passively target tumors. Cell viability studies, using a human cervical cancer cell line (HeLa), demonstrate excellent biocompatibility of the PLGA nanoparticles. PLGA nanoparticles carrying encapsulated Cyt-c were not efficient in causing apoptosis presumably because PLGA nanoparticles are not efficiently taken up by the cells. Future systems will have to be optimized to ascertain efficient cellular uptake of the nanoparticles by, e.g., surface modification with receptor ligands.
Keywords: Nanoprecipitation, Nanoparticle, Pharmaceutical protein, Protein stability, Sustained release
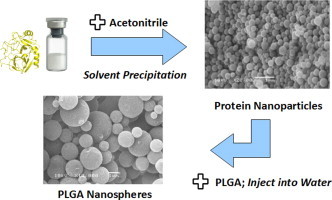
1. Introduction
Nanoparticles can be used to design or even comprise excellent drug delivery systems [1,2]. For example, due to the enhanced permeability and retention (EPR) effect, nanoparticles can passively target tumors and accumulate in them [1,3]. Nanoparticles can increase the stability of drugs including proteins in blood, are secreted less readily by the kidney, which often results in increased therapeutic efficacy and can reduce side effects of other therapies. [1,3–8]. In this work, we focus on nanoparticles comprised of poly(lactic-co-glycolic acid) (PLGA) because the polymer is an intensely studied material in the field of sustained release, has received FDA approval in various invasive applications including drug delivery, and is biocompatible, biodegradable, and non-toxic [2,9–11].
A promising method to obtain PLGA nanoparticles is by nanoprecipitation, a procedure that was developed by Fessi and coworkers and enables production of particles in the 100–300 nm range [12]. Advantages of this method include that it is a single step not requiring extended shearing/stirring rates, sonification, or high temperatures. The method is characterized by the absence of an oil–aqueous interface which is detrimental to protein structure and function [13,14]. However, the nanoprecipitation method, as developed, is mostly suitable for hydrophobic compounds that are soluble in ethanol or acetone, but display limited solubility in water. For example, Barichello et al. obtained encapsulation efficiencies close to 100% with the lipophilic drugs cindomethacin and cyclosporine A, but less than 15% for the hydrophilic drugs vacomycin and phenobarbital [15].
In order to overcome these limitations, the original nanoprecipitation method was modified by Bilati et al. using a wide range of water-miscible organic solvents [13]. This work provided evidence that nanoprecipitation could occur with solvents other than acetone or ethanol, and that an accurate solvent and non-solvent selection could be extended to enable nanoprecipitation of more hydrophilic drugs. It remains difficult to identify two suitable solvents, because one of them must be able to dissolve both drug and polymer (solvent or diffusing phase), while the polymer should be insoluble in the second solvent (non-solvent or dispersing phase). In a second study, they selected the water-miscible organic solvent DMSO as the diffusing phase and tested the encapsulation using the model proteins lysozyme and insulin [16]. The authors were able to load nanospheres efficiently with lysozyme, but not with insulin. Note that the study by Bilati et al. [13] did not include protein stability experiments. This is troublesome because DMSO is reported to irreversible unfold most proteins [17,18] and it is therefore unlikely that the developed method is generally applicable.
We set out to overcome the aforementioned problems by developing a new nanoencapsulation procedure. Overcoming the obstacles in protein encapsulation by one-step nanoprecipitation is challenging. First, it is difficult to find a common solvent for the quite hydrophobic PLGA and the hydrophilic protein. Second, the organic solvent can induce deleterious protein structural and functional loss. We therefore designed a novel two-step nanoprecipitation method (Fig. 1) and tested its capability to encapsulate two model proteins, lysozyme and α-chymotrypsin, into PLGA nanospheres. The two model proteins were chosen because we have employed them frequently in the past to follow encapsulation procedures [14]. While lysozyme is quite stable, α-chymotrypsin easily denatures and is an excellent sensor for the potential impact of the procedure on protein structure and function [14]. The first step in this new method consists in solvent-induced nanoprecipitation of the protein. Then, encapsulation was accomplished by a subsequent polymer nanoprecipitation step. In contrast to Bilati et al. who used DMSO to dissolve the proteins [16], we suspended the dehydrated protein nanoparticles obtained by solvent precipitation in organic solvents incapable of dissolving proteins, but capable of dissolving PLGA. Results from solid-state protein formulations show that in the absence of water, protein conformational mobility is reduced so that the stability of proteins in contact with the organic solvent is enhanced [14,19,20]. Results from non-aqueous enzymology support this assumption [14,21–23]. By determining protein aggregation and function after encapsulation, we tested whether our assumptions with respect to the advantages of reduced protein structural mobility were correct or not.
Fig. 1.
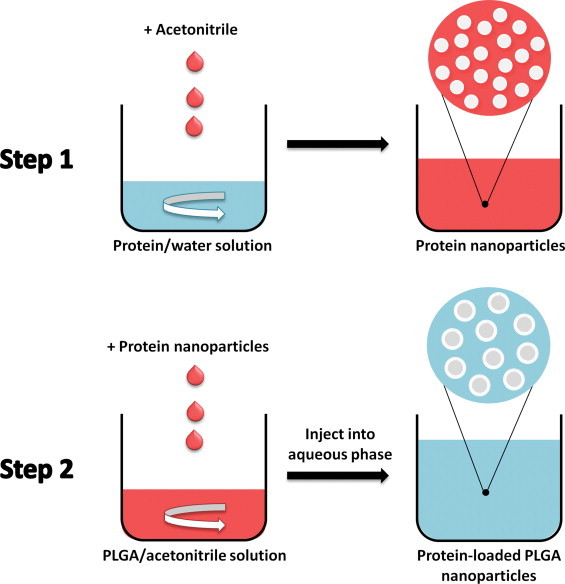
Scheme of the encapsulation of proteins into PLGA nanospheres by two-step nanoprecipitation.
After optimizing the methodology, we employed the processing parameters established for lysozyme to encapsulate an unrelated basic protein of similar size, horse heart cytochrome c(Cyt-c), in PLGA nanospheres to test the potential of the drug delivery system for applications in cancer treatment [24]. Cyt-c is an important mediator of apoptosis when it is released from the mitochondria to the cytoplasm. This process normally takes place in response to DNA damage, but in many cancer cells it is inhibited. The targeted delivery of Cyt c directly to the cytoplasm of cancer cell could selectively initiate apoptosis.
2. Materials and methods
2.1. Chemicals
Poly(d,l-lactic-co-glycolic)acid (PLGA) with a co-polymer ratio of 50:50 and 65:35 [lactide-to-glycolide] and a MW of 10,000 (not endcapped), was from Lakeshore Biomaterials (Birmingham, AL). The MW is an average value determined by the supplier. Bovine pancreatic α-chymotrypsin, hen egg-white lysozyme, equine heart cytochrome c(Cyt c), micrococcus cells, and poly(vinyl)alcohol (PVA, 87%–89% hydrolyzed with a MW of 13,000–23,000) were from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN, HPLC grade) was from Fisher Scientific (Pittsburgh, PA). Succinyl-Ala-Ala-Pro-Phe-p-nitroanilide was from Bachem Laboratories (Torrens, CA).
2.2. Protein precipitation and encapsulation
Protein nanoparticles were obtained using a similar method as described by Weber et al. [25]. Briefly, lysozyme and α-chymotrypsin were solvent-precipitated from 0.8 and 1 ml of aqueous solutions at concentrations of 25 and 15 mg/ml, respectively, by adding the water-miscible solvent acetonitrile at a 1:4 volume ratio. The resulting protein suspension was stirred for 5 min with a magnetic stir bar. PLGA was dissolved in acetonitrile at 190 and 28.5 mg/ml and 2 and 10 ml added to the lysozyme and α-chymotrypsin suspensions, respectively. The resulting mixtures (6 and 14 ml) were added directly through a syringe needle into 240 and 560 ml of a 10% PVA solution under stirring (60 ml/min) with a magnetic stir bar (5.08 cm length). The volume ratio of dispersing phase to diffusing phase was 1:40. Polymer nanoprecipitation was immediately visible upon injection of the protein suspensions. The PLGA nanoparticles formed were immediately centrifuged for 10 min at 8000 rpm, the supernatant discarded, and the pellet re-suspended in distilled water. This washing step was thrice repeated and the samples subsequently freeze-dried by first rapidly freezing them in liquid nitrogen followed by lyophilization at a condenser temperature of −45 °C and a pressure of <60 μm of Hg [26]. Cyt-c encapsulation was performed using the same optimum conditions established by us for lysozyme since it has a similar size and net charge.
2.3. Determination of the precipitation yield
After protein nanoprecipitation, the resulting protein suspension was centrifuged at 5000 rpm for 10 min. The supernatant was discarded and the pellet vacuum dried for 30 min. Protein concentration and protein aggregates in the pellet were determined as described by us in detail [26–29]. In brief, the protein pellet was suspended in 2 ml of potassium phosphate buffer for 2 h to dissolve the buffer-soluble fraction. The samples were then subjected to centrifugation at 5000 rpm for 5 min and the supernatant used to determine the concentration of soluble protein. Next, 1 ml of 6 M urea was added to the pellet to dissolve the buffer-insoluble protein fraction and used to determine the concentration of aggregated protein by measuring the UV absorbance at 280 nm. The precipitation yield was calculated from the actual and theoretical quantity of protein recovered after nanoprecipitation and rehydration. The experiments were performed in triplicate, the results averaged, and the standard deviations calculated.
2.4. Dynamic light scattering
The size of protein nanoparticles and PLGA nanospheres was determined by dynamic light scattering using a DynaPro Titan with MicroSampler from Wyatt Technology Corporation (Santa Barbara, CA) as described by us in detail [20]. Protein particles were measured as a suspension in acetonitrile and the PLGA nanospheres as a suspension in water at 100% power intensity. Data analysis was performed using the Dynamic 6.7.6 software supplied with the instrument. The instrument was periodically calibrated using BSA as a standard. In the past, we found that scanning electron microscopy images and size data from dynamic light scattering were consistent [20].
2.5. Determination of actual protein loading and encapsulation efficiency
The actual protein loading of nanospheres was determined following a methodology developed in our laboratory [27]. In brief, 20 mg of PLGA nanospheres were dissolved in 2 ml of ethyl acetate and stirred for 2 h followed by centrifugation at 9000 rpm for 10 min. The supernatant was discarded and the pellet vacuum dried for 30 min. This pellet, consisting mostly of protein, was suspended in 2 ml of potassium phosphate buffer for 2 h to dissolve the buffer-soluble protein fraction. The samples were then subjected to centrifugation at 9000 rpm for 10 min and the supernatant used to determine the concentration of soluble protein. Next, 1 ml of 6 M urea was added to the pellet to dissolve the water-insoluble protein fraction. In all cases, a clear solution without noticeable light scattering was obtained and used to determine the concentration of aggregated protein by measuring the UV absorbance at 280 nm and by BCA assay at 562 nm. The encapsulation efficiency was calculated from the actual and theoretical loading of protein in the nanospheres. The experiments were performed in triplicate, the results averaged, and the standard deviations calculated to highlight the reproducibility of the experiments.
2.6. Determination of enzyme activity
To determine the enzyme activity after encapsulation, ethyl acetate was used to dissolve PLGA because it does not cause enzyme inactivation in the process [27]. Activity of α-chymotrypsin was determined using succinyl-Ala-Ala-Pro-Phe-p-nitroanilide as the substrate [28]. The reaction was carried out in 1 ml of 0.1 M Tris-HCl buffer containing 0.05 mg/ml enzyme, 0.35 mM substrate, and 10 mM CaCl2 at pH 7.8 and our data for α-chymotrypsin as purchased are comparable to those reported [28]. The activity of 0.01 mg/ml lysozyme was determined by measuring the decrease in turbidity at 450 nm of a 0.015% (w/v) suspension of Micrococcus lysodeikticus cells in 1 ml of 66 mM potassium phosphate buffer at pH 6.2 and 25 °C as described by us [29]. The peroxidase-like activity of Cyt-c which is not a natural enzyme was obtained as described [30]. Briefly, the reaction was followed at 415 nm using 0.25 ml of 0.01 mg/ml Cyt-c, 0.2 ml of 300 mM H2O2, and 0.55 ml of 0.05 mM 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) in 20 mM potassium phosphate buffer at pH 7. The data obtained by us for commercial Cyt-c are comparable to those reported in the literature [30].
The activity was obtained by plotting the time-dependent absorbance changes vs. time. The linear portions of the graphs at less than 10% substrate conversion were used to obtain the initial velocities (V0). In all cases the specific activity (mM of substrate converted into product per min and per mg of protein) was calculated. The experiments were performed in triplicate and the results averaged and the standard deviations calculated.
2.7. In vitro release studies
In vitro release studies were conducted as described by us in the past [26–29]. In brief, nanospheres (30 mg) were placed in 1 ml of 10 mM phosphate buffered saline (PBS) at pH 7.3 and incubated at 37 °C. At pre-determined times (typically every 24 h) the supernatant was removed after a short centrifugation. The concentration of the released protein in the supernatant was determined by absorbance measurement at 280 nm (the absorbance was corrected by the very small absorbance produced by degrading empty PLGA nanospheres). The concentration of the released protein was used to construct cumulative release profiles. Release experiments were performed at least in triplicate, the results averaged, and the standard deviations calculated.
2.8. Cell culture
Human HeLa epithelial adenocarcinoma cells (American Type Culture Collection (ATCC), Manassas, VA, catalog number CCL-2) were cultured in Eagle's Minimum Essential Medium (Invitrogen Corp.) supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen Corp.), penicillin (100 U/ml), and 1% glutamine as described by the ATCC. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
2.9. In vitro cell studies – non-radioactive cell poliferation assay
The non-radioactive cell proliferation assay was performed according to the manufacturer instructions (Promega). HeLa cells were seeded into 96-well plates at 7.5×104 cells/well and incubated at 37 °C for 24 h. Then, cells were subjected to medium replacement containing 1% FBS and incubated overnight. Various concentrations of Cyt-c-PLGA NPs dispersed in the cell culture medium were added to cells followed by further incubation at 37 °C for 24, 48, 72, or 96 h. The tested concentrations of Cyt-c-PLGA NPs are equivalent to Cyt-c concentrations of 0.61, 1.21, 3.10, 6.19 and 12.38 μg/ml. Control experiments were performed using blank PLGA NPs. At the day of the experiment, the cells were washed once with PBS and 100 μl fresh medium was added. Background values were recorded at 492 nm using a microplate reader. Then, cells were treated with 20 μL of MTS (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoleum; 5 μg/μl) for 1 h and the absorbance was measured at 492 nm. The results were based on at least three independent experiments and the data averaged.
2.10. Statistical analysis
All experiments were performed at least in triplicate, the data averaged, and the standard deviations calculated. The standard deviations are included in all tables as ± values. To establish statistical significance when comparing multiple groups we used one-way multiple Tukey comparison post-test ANOVA. A P value of <0.05 was considered statistically significant.
3. Results and discussion
3.1. Development and optimization of a two-step nanoprecipitation method
3.1.1. Protein nanoprecipitation
First we explored the effects of different desolvating agents, different excipients, and the protein concentration on the protein nanoprecipitation process using lysozyme and α-chymotrypsin as model proteins. In order to optimize the processing conditions, the precipitation yield, protein particle size, and residual enzyme activity were determined. While protein precipitates were obtained with acetonitrile and acetone, propanol and ethanol were inefficient for both enzymes (data not shown, for conditions see footnotes in Table 1). We also tested the addition of common stabilizing excipients on the process outcome (poly(ethylene glycol) with a MW of 8000, methyl-β-cyclodextrin, and trehalose at a 1:1 excipient-to-protein weight ratio). It was found that excipients did not improve the process outcome in the case of lysozyme and hindered precipitation in the case of α-chymotrypsin. We therefore did not employ excipients subsequently to avoid such complications—.
Table 1.
Properties of the protein precipitates using acetonitrile (ACN) and acetone as desolvating agent.*
| Protein/solvent | Precipitation yield (%) | Insoluble aggregates (%) | Residual activity (%) |
|---|---|---|---|
| Lysozyme | |||
| ACN | 79 ± 4 | 0 ± 0 | 96 ± 8 |
| Acetone | 54 ± 28 | 6 ± 3 | 81 ± 3 |
| α-Chymotrypsin | |||
| ACN | 80 ± 5 | 3 ± 2 | 73 ± 1 |
| Acetone | 82 ± 3 | 1 ± 2 | 75 ± 8 |
Protein concentration: 10 mg/ml; volume ratio of water-to-organic solvent: 1:4.
Focusing on the best solvents identified, ACN and acetone, the outcome of the nanoprecipitation process was characterized in detail (Table 1). While the precipitation efficiency was comparable for both solvents, acetonitrile caused less enzyme aggregation and inactivation in the case of lysozyme. This solvent was therefore chosen for all subsequent work.
In order to further optimize the precipitation conditions, we varied the volume ratio of acetonitrile-to-water. Similar to Weber et al. [25] who used ethanol as desolvating agent, we found that a 1:4 water-to-ACN volume ratio was sufficient to precipitate both proteins (data not shown).
Next, we tested the effect of the protein concentration on precipitation results (Table 2). The precipitation yield and particle size increased at increasing protein concentration under otherwise constant precipitation conditions. While for both proteins, no significant amounts of buffer-insoluble aggregates were formed regardless of the protein concentration, the residual activity increased at increasing protein concentration. We interpret this as an indication, that protein molecules close to the solvent-interface are more prone to denaturation than molecules buried in the interior of the precipitates. Such observations have been made before in solid-in-oil-in-water encapsulation procedures [28]. It is apparent that protein concentrations of 20–30 mg/ml give optimum results. For α-chymotrypsin concentrations higher than 40 mg/ml, unstable suspensions of the precipitated protein resulted and thus did not allow for the subsequent encapsulation process.
Table 2.
Characterization of the nano-precipitation results at various protein concentrations at a 1:4 volume ratio of water-to-acetonitrile.
| Protein | Concentration (mg/ml) | Precipitation yield (%) | Insoluble aggregates (%) | Residual activity (%) | Diameter (nm) |
|---|---|---|---|---|---|
| Lysozyme | 10 | 70 ± 11 | 2 ± 1 | 87 ± 5 | 86 ± 16 |
| 20 | 95 ± 4 | 1 ± 0 | 83 ± 1 | 92 ± 15 | |
| 30 | 99 ± 1 | 1 ± 1 | 96 ± 2 | 168 ± 14 | |
| α-Chymotrypsin | 10 | 77 ± 5 | 0 ± 0 | 78 ± 0 | 66 ± 16 |
| 20 | 83 ± 3 | 0 ± 0 | 100 ± 1 | 174 ± 16 | |
| 30 | 83 ± 4 | 0 ± 0 | 100 ± 2 | 200 ± 12 |
We can surmise from the above that similar to findings by Giteau et al. [19], a variety of precipitation conditions was identified by us leading to nano-particulate enzyme precipitates without causing activity loss or formation of buffer-insoluble aggregates.
3.1.2. Protein nanoparticle encapsulation
After optimizing the protein precipitation conditions, we proceeded to encapsulate the model proteins into PLGA nanospheres. Previously, Giteau et al. precipitated proteins to ensure their stability upon subsequent encapsulation within PLGA microspheres using a solid-in-oil-in-water (s/o/w) technique [19]. After protein precipitation with glycofurol, proteins were centrifuged and the pellet suspended in acetonitrile (ACN) containing the polymer and encapsulated within PLGA microspheres. Our method used the same desolvating agent (ACN) to precipitate the protein and to dissolve the polymer. Additionally, several steps in the encapsulation procedure were changed systematically to assure obtaining nanosized PLGA spheres with high protein loading while aiming at avoiding enzyme inactivation and aggregation. Initially, we selected PLGA with a co-polymer ratio of 65% lactic acid and 35% glycolic acid, a theoretical loading of 2% (w/w), and ACN as the diffusing phase. We tested two commonly used emulsifying agents, namely, poly(vinyl alcohol) (PVA) and poly(ethylene glycol) (PEG, MW = 8000) using a set of defined conditions (Table 3) [28]. It was found that the highest encapsulation efficiency of ca. 70% was achieved using 10% of PVA without compromising protein stability.
Table 3.
Effect of the emulsifier on selected properties of lysozyme-loaded PLGA nanospheres.*
| Dispersing phase | Encapsulation efficiency (%) | Protein aggregates (%) | Residual activity (%) |
|---|---|---|---|
| Water | 48 ± 23 | 0 ± 0 | 100 ± 12 |
| 10% PEG | <10 | n.d. | n.d. |
| 5% PVA | 58 ± 28 | 3 ± 5 | 88 ± 21 |
| 10% PVA | 71 ± 15 | 0 ± 5 | 90 ± 3 |
Lysozyme concentration: 25 mg/ml; volume ratio of water to ACN: 1:4; concentration of PLGA 65:35 in acetonitrile: 28.5 mg/ml; total volume of the diffusing phase: 12 ml, and for the dispersing phase: 150 ml; theoretical loading: 2% (w/w).
We tried to increase the protein loading to 5%, but surprisingly the encapsulation failed when the protein nanoparticles suspended in PLGA solution were added to the PVA solution. However, using PLGA with a co-polymer ratio of 50:50 resulted in nanoencapsulation, but the encapsulation efficiency needed improvement. When we increased the volume of the diffusing phase to accomplish faster particle hardening, the encapsulation efficiency increased substantially to >80% at a 1:40 volume ratio of dispersing-to-diffusing phase (Table 4). We also tested the polymer concentration in this context. It has been shown that a higher polymer concentration leads to higher encapsulation efficiency and larger size of the nanoparticles [31,32]. At a high PLGA concentration, the viscosity of the diffusing phase increases which should result in improved encapsulation by reduction of lysozyme nanoparticles leaking into the dispersing phase. Indeed, we found increasing lysozyme encapsulation efficiency at increasing polymer concentration as expected (Table 5). In a similar fashion encapsulation efficiency was improved for α-chymotrypsin. Changing the polymer concentration proved only somewhat successful in this case, possibly because at increased PLGA concentrations the polymer shell thickness also increased [33]. The encapsulation efficiency remained with a maximum of 30% too low for practical purposes (Table 5). Reducing the particle size of α-chymotrypsin by employing a lower protein concentration of 15 mg/ml (Table 2) resulted in an improved encapsulation efficiency of 74% (Table 6).
Table 4.
Effect of the ratio of dipersing-to-diffusing phase on the encapsulation efficiency of lysozyme in PLGA nanoparticles.*
| Ratio of dispersing phase to diffusing phase | Encapsulation efficiency (%) |
|---|---|
| 1:10 | 9 ± 3 |
| 1:20 | 49 ± 9 |
| 1:30 | 71 ± 7 |
| 1:40 | 84 ± 8 |
Protein concentration: 25 mg/ml; volume ratio between water and ACN: 1:4; PLGA 50:50 concentration in ACN: 90 mg/ml; theoretical loading: 5% (w/w).
Table 5.
Effect of different polymer concentrations on the protein encapsulation efficiency in PLGA nanopheres.*
| Batch | Concentration of PLGA 50:50 in ACN (mg/ml) | Encapsulation efficiency (%) |
|
|---|---|---|---|
| Lysozyme | α-Chymotrypsin | ||
| 1 | 38 | 26 ± 6 | 11 ± 4 |
| 2 | 63 | 45 ± 12 | 24 ± 4 |
| 3 | 95 | 68 ± 7 | 30 ± 1 |
| 4 | 190 | 94 ± 5 | 23 ± 3 |
Protein concentration: 25 mg/ml; volume ratio of water to organic solvent: 1:4; polymer mass: 380 mg; volume ratio of dispersing-to-diffusing phase: 1:40; theoretical protein loading: 5%.
Table 6.
Effect of the polymer concentration on α-chymotrypsin encapsulation efficiency in PLGA nanospheres.*
| Batch | Concentration of PLGA 50:50 in ACN (mg/ml) | Encapsulation efficiency (%) |
|---|---|---|
| 1 | 28.5 | 74 ± 4 |
| 2 | 47.5 | 49 ± 9 |
| 3 | 71.25 | 48 ± 4 |
| 4 | 142.5 | 38 ± 18 |
Protein concentration: 15 mg/ml; volume ratio between water and organic solvent: 1:4; polymer mass: 285 mg; volume ratio of dispersing-to-diffusing phase: 1:40 ml; theoretical protein loading: 5%.
The data show how sensitive the results respond to encapsulation conditions in this method highlighting the fact that encapsulation likely has to be optimized in a similar fashion as described here for other proteins. However, there are only a few processing parameters requiring adjustment and the process is straight forward and reproducible as demonstrated by the small standard deviations obtained for encapsulation parameters under optimized conditions.
The optimum conditions to encapsulate lysozyme and α-chymotrypsin in PLGA nanoparticles are summarized in Table 7. The size of the protein loaded PLGA particles obtained by dynamic light scattering was ca. 300–400 nm in diameter (Table 7). However, while lysozyme encapsulation afforded a highly active enzyme, substantial enzyme inactivation and formation of buffer-insoluble aggregates were observed for α-chymotrypsin. The formation of buffer-insoluble aggregates and loss in specific activity found for α-chymotrypsin is similar to results obtained before upon α-chymotrypsin encapsulation in PLGA microspheres using a s/o/w technique [27,28,34–36]. The use of stabilizing additives (e.g., methyl-β-cyclodextrin or poly(ethylene glycol)) was necessary in the latter case to preserve protein integrity. Such strategies have to be developed for the new two-step nanoprecipitation procedure as well.
Table 7.
Properties of lysozyme and α-chymotrypsin-loaded PLGA nanospheres produced by two-step nanoprecipitation.*
| Protein | Encapsulation efficiency (%) | Insoluble aggregates (%) | Residual activity (%) | Diameter (nm) |
|---|---|---|---|---|
| Lysozyme | ||||
| 94 ± 5 | 0 ± 0 | 100 ± 8 | 336 ± 40 | |
| α-Chymotrypsin | ||||
| 74 ± 4 | 14 ± 17 | 49 ± 2 | 440 ± 16 | |
3.1.3. Cytochrome c-loaded PLGA nanoparticles
Having accomplished protein loaded nano-sized PLGA particles, we tested the development of the sustained release nanoparticles into an application platform. We selected Cyt-c as model protein because it has been employed in experiments geared towards better cancer treatment options [24]. The size of our particles makes them potentially useful in passive and also active targeting of cancer tissues [37,38]. For example, Santra et al. [24] demonstrated recently the therapeutic potential of Cyt-c in nanoparticles by their capability to induce apoptosis in lung carcinoma cells after uptake by the cells by endocytosis. However, their vehicle consisted of a water-soluble hyperbranched polyhydroxyl polymer not approved in medical applications. In contrast, our nanoparticles employ an already FDA approved and commercially available polymer (PLGA) and a straight forward encapsulation method.
We hypothesized that encapsulation of Cyt-c via the two step nanoprecipitation method should work using the optimum conditions identified for lysozyme (Table 7) because both proteins have a similar molecular weight (12 and 14 kDa, respectively) and are basic [39]. The encapsulation efficiency for Cyt-c was with 72% is similar to that obtained for lysozyme under identical conditions (Table 8). The peroxidase activity of Cyt-c was comparable to values prior to precipitation and encapsulation and only few aggregates were formed indicating good preservation of structural integrity during the process. The size of the particles obtained was 340 nm and thus potentially useful to enable passive delivery to cancer tissues based on the EPR effect [37,38].
Table 8.
Properties of Cyt-c after precipitation and encapsulation in PLGA nanospheres.*
| Precipitation | Encapsulation | |
|---|---|---|
| Precipitation efficiency (%) | 81 ± 1 | N/A |
| Encapsulation efficiency (%) | N/A | 72 ± 2 |
| Insoluble aggregates (%) | 0 ± 0 | 5 ± 3 |
| Residual activity (%) | 96 ± 6 | 98 ± 3 |
| Particle size (nm) | 80 ± 17 | 342 ± 62 |
For conditions see batch 4, Table 5.
In vitro release of Cyt-c from the PLGA nanoparticles showed an initial “burst” release within 24 h that was reasonably small with ca. 20% (Fig. 2). Burst release values of >20% are frequently found for such systems, in particular when nanosized systems are being used [40]. During a 100-day incubation period, Cyt c was released completely from the nanospheres. Since the release was slow, the amount of protein released per day was small and the residual activity during release could not be measured with accuracy. Future experiments using cell cultures and animal models will shed light into the bioactivity of the developed system. However, since 100% of the protein was released, we can exclude the formation of buffer-insoluble Cyt-c during the release period.
Fig. 2.
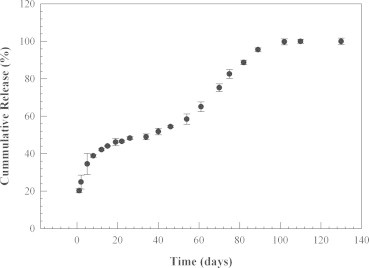
In vitro release profile of Cyt-c from PLGA nanospheres prepared by two step nanoprecipitation.
3.2. Cyt-c-PLGA nanoparticle cytotoxic effects in cancer cells
Since there are some reports that PLGA nanoparticles could be internalized by cells, we investigated whether the Cyt-c-PLGA NPs would be toxic to cancer cells. We selected a human cervical cancer cell line (HeLa) as a model system and incubated the cells for 24, 48, 72, and 96 h at 37 °C under 5% CO2 with various concentrations of drug-loaded and empty PLGA nanoparticles and determined the cell viability (Fig. 3). It was observed that after 72 h of incubation, the HeLa cells treated with Cyt-c-PLGA NPs had a significantly reduced viability for the highest Cyt-c concentration used (12.38 mg/ml). In contrast, PLGA nanospheres without the drug had no effect on cell viability in agreement with the biocompatible nature of the PLGA polymer family. Establishment of biocompatibility of the PLGA NPs is important in the development of them as drug delivery systems.
Fig. 3.
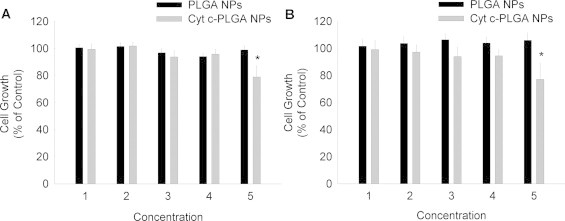
Comparison of the cell viability of HeLa cells treated with Cyt-c encapsulated in PLGA nanoparticles (NPs) vs. empty PLGA NPs after 72 (a) and 96 h (b) of incubation. The numbers 1–5 on the y-axis corresponds to 0.61, 1.21, 3.10, 6.19, and 12.38 μg/ml Cyt-c, respectively, in case of Cyt-c loaded PLGA NPs. Empty PLGA NPs were adjusted to the same PLGA concentrations as the corresponding Cyt-c-loaded PLGA NPs. Cyt-c-PLGA NPs induced a significant reduction in cell viability after 72 and 96 h of incubation for the 12.38 mg/ml protein concentration, whereas the PLGA NPs showed no significant cytotoxicity.
It has to be pointed out, however, that the effect of the drug-loaded delivery system on cell viability was too low to be clinically relevant. It is likely that PLGA NPs are not taken up effectively and thus Cyt-c is not being effectively delivered to the cell cytoplasm in agreement with recent data [41]. We can conclude from this that PLGA NPs have to be either modified with a homing ligand or release a drug coupled to a homing ligand to enable uptake by receptor-mediated endocytosis. We are currently working on transforming this system in this direction.
4. Conclusions
Nanosized delivery systems hold promise in improving protein delivery, i.e., to target tumors and inflamation. A convenient method to accomplish nanosized polymer particles is by one-step nanoprecipitation. However, encapsulation of proteins into PLGA nanospheres by nanoprecipitation was inefficient prior to our work and/or involved the solvent DMSO which irreversibly denatures most proteins [17,18,42,43]. To overcome these problems, we developed a two-step nanoprecipitation method to allow for efficient protein encapsulation into PLGA nanospheres without causing irreversible functional changes. Cell viability studies using HeLa cells demonstrate excellent biocompatibility of the PLGA nanospheres obtained. Furthermore, we demonstrate reproducible encapsulation of the model proteins lysozyme, α-chymotrypsin, and Cyt-c into PLGA nanospheres. Optimization of the processing parameters involved in the new two-step nanoprecipitation method enabled obtaining high encapsulation efficiencies. While encapsulation of lysozyme and Cyt-c via the two-step nanoprecipitation method did not lead to the formation of insoluble aggregates or activity loss, significant enzyme inactivation and formation of buffer-insoluble aggregates were observed for α-chymotrypsin. Future studies in our laboratory will be directed towards minimizing this problem.
Admittedly, as one reviewer pointed out to us, the results obtained with the therapeutic protein seem not sufficient to justify the preparative efforts. However, we feel that our work and the results obtained constitute a first significant step into the direction of solving a complex problem. Our work clearly demonstrates the feasibility of obtaining nanosized biocompatible protein delivery systems with good yield and reasonable protein stability. This should support approaches aiming at targeted protein delivery using the enhanced permeability and retention (EPR) effect to deliver pharmaceutical proteins to tumors or inflammation sites. This approach has to be augmented by targeted delivery strategies aimed at enabling endocytosis of the nanoparticles, e.g., by attaching folate to their surface.
Acknowledgements
This publication was made possible by Grant no. SC1 GM086240 from the National Institute for General Medical Sciences (NIGMS) at the National Institutes of Health (NIH) through the Support of Competitive Research (SCoRE) Program. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS. MMC and GMFF were supported by a fellowship from NIH Research Initiative for Scientific Enhancement (RISE) Program (2 R25 GM061151–11).
References
- 1.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Advanced Drug Delivery Reviews. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 2.Singh R, Lillard JW. Nanoparticle-based targeted drug delivery. Experimental and Molecular Pathology. 2009;86:215–223. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexis F, Pridgen E, Linda K, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Molecular Pharmacology. 2008;5(4):505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, Wrasidlo W, Cheresh DA. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proceedings of the National Academy of Sciences USA. 2008;105(27):9343–9348. doi: 10.1073/pnas.0803728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pathak P, Katiyar VK. Multi-functional nanoparticles and their role in cancer drug delivery—a review. Journal of Nanotechnology. 2007:1–18. [Google Scholar]
- 6.Han G, Ghosh P, Rotello VM. Functionalized gold nanoparticles for drug delivery. Nanomedicine. 2007;2(1):113–123. doi: 10.2217/17435889.2.1.113. [DOI] [PubMed] [Google Scholar]
- 7.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nature Reviews Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 8.Gelperina S, Kisich K, Iseman MD, Heifets L. The potential advantages of nanoparticle drug delivery systems in chemotherapy of tuberculosis. American Journal of Respiratory and Critical Care Medicine. 2005;172(12):1487–1490. doi: 10.1164/rccm.200504-613PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Advanced Drug Delivery Reviews. 1977;28(1):5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 10.Pridgen EM, Langer R, Farokhzad OC. Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine. 2007;2(5):669–680. doi: 10.2217/17435889.2.5.669. [DOI] [PubMed] [Google Scholar]
- 11.Langer R, David A, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 12.Fessi H, Puisieux F, Devissaguet JPh, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. International Journal of Pharmaceutics. 1989;55:R1–R4. [Google Scholar]
- 13.Bilati U, Allemann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. European Journal of Pharmaceutical Sciences. 2005;24(1):67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Pérez C, Castellanos IJ, Costantino HR, Al-Azzam W, Griebenow K. Recent trends in stabilizing protein structure upon encapsulation and release from bioerodible polymers. Journal of Pharmacy and Pharmacology. 2002;54:301–313. doi: 10.1211/0022357021778448. [DOI] [PubMed] [Google Scholar]
- 15.Barichello JM, Morishita M, Takayama K, Nagai T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Development and Industrial Pharmacy. 1999;25(4):471–476. doi: 10.1081/ddc-100102197. [DOI] [PubMed] [Google Scholar]
- 16.Bilati U, Allemann E, Doelker E. Nanoprecipitation versus emulsion-based techniques for the encapsulation of proteins into biodegradable nanoparticles and process-related stability issues. AAPS PharmSciTec. 2005;6(4):E594–604. [DOI] [PMC free article] [PubMed]
- 17.Griebenow K, Klibanov AM. Can conformational changes be responsible for solvent and excipient effects on the catalytic behavior of subtilisin Carlsberg in organic solvents? Biotechnology and Bioengineering. 1997;53(4):351–362. doi: 10.1002/(SICI)1097-0290(19970220)53:4<351::AID-BIT1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 18.Xu K, Griebenow K, Klibanov AM. Correlation between catalytic activity and secondary structure of subtilisin dissolved in organic solvents. Biotechnology and Bioengineering. 1997;56:485–491. doi: 10.1002/(SICI)1097-0290(19971205)56:5<485::AID-BIT2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 19.Giteau A, Venier-Julienne MC, Marchal S, Courthaudon JL, Sergent M, Montero-Menei C, Verdier JM, Benoit JP. Reversible protein precipitation to ensure stability during encapsulation within PLGA microspheres. European Journal of Pharmaceutics and Biopharmaceutics. 2008;70:127–136. doi: 10.1016/j.ejpb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Montalvo BL, Pacheco Y, Sosa BA, Vélez D, Sánchez G, Griebenow K. Formation of spherical protein nanoparticles without impacting protein integrity. Nanotechnology. 2008;19:465103. doi: 10.1088/0957-4484/19/46/465103. [DOI] [PubMed] [Google Scholar]
- 21.Griebenow K, Klibanov AM. On protein denaturation in aqueous-organic but not in pure organic solvents. Journal of the American Chemical Society. 1996;118:11695–11700. [Google Scholar]
- 22.Griebenow K, Klibanov AM. Can conformational changes be responsible for solvent and excipient effects on the catalytic behavior of subtilisin Carlsberg in organic solvents? Biotechnology and Bioengineering. 1997;53:351–362. doi: 10.1002/(SICI)1097-0290(19970220)53:4<351::AID-BIT1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 23.Griebenow K, Vidal M, Baéz C, Santos AM, Barletta G. Native-like enzyme properties are important for optimum activity in neat organic solvents. Journal of the American Chemical Society. 2001;123:5380–5381. doi: 10.1021/ja015889d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santra S, Kaittanis C, Perez JM. Cytochrome c encapsulating theranostic nanoparticles: a novel bifunctional system for targeted delivery of therapeutic membrane-impermeable proteins to tumors and imaging of cancer therapy. Molecular Pharmacology. 2010;7(4):1209–1222. doi: 10.1021/mp100043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber C, Coester C, Kreuter J, Langer K. Desolvation process and surface characterisation of protein nanoparticles. International Journal of Pharmaceutics. 2000;194(1):91–102. doi: 10.1016/s0378-5173(99)00370-1. [DOI] [PubMed] [Google Scholar]
- 26.Perez C, De Jesus P, Griebenow K. Preservation of lysozyme structure and function upon encapsulation and release from poly(lactic-co-glycolic) acid microspheres prepared by the water-in-oil-in-water method. International Journal of Pharmaceutics. 2002;248:193–206. doi: 10.1016/s0378-5173(02)00435-0. [DOI] [PubMed] [Google Scholar]
- 27.Castellanos IJ, Cruz G, Crespo R, Griebenow K. Encapsulation-induced aggregation and loss in activity of γ-chymotrypsin and their prevention. Journal of Controlled Release. 2002;81:307–319. doi: 10.1016/s0168-3659(02)00073-1. [DOI] [PubMed] [Google Scholar]
- 28.Castellanos IJ, Griebenow K. Improved α-chymotrypsin stability upon encapsulation in PLGA microspheres by solvent replacement. Pharmaceutical Research. 2003;20:1873–1880. doi: 10.1023/b:pham.0000003388.59659.fa. [DOI] [PubMed] [Google Scholar]
- 29.Peréz C, Griebenow K. Improved activity and stability of lysozyme at the water/methylene chloride interface: enzyme unfolding and aggregation and its prevention by polyols. Journal of Pharmacy and Pharmacology. 2001;53:1217–1226. doi: 10.1211/0022357011776667. [DOI] [PubMed] [Google Scholar]
- 30.Kim NH, Jeong MS, Choi SY, Kang JH. Peroxidase activity of cytochrome c. Bulletin of the Korean Chemical Society. 2004;25:1889–1892. [Google Scholar]
- 31.Blanco MD, Alonso MJ. Development and characterization of protein loaded poly (lactide-co-glycolide nanospheres. European Journal of Pharmaceutics and Biopharmaceutics. 1997;43:287–294. [Google Scholar]
- 32.Song CX, Labhasetwar V, Murphy H, Qu X, Humphrey WR, Shebuski RJ, Levy RJ. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. Journal of Controlled Release. 1997;43:197–212. [Google Scholar]
- 33.Chang MW, Stride E, Edirisinghe M. Controlling the thickness of hollow polymeric microspheres prepared by electrohydrodynamic atomization. Journal of the Royal Society Interface. 2010;7:S451–S460. doi: 10.1098/rsif.2010.0092.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castellanos IJ, Flores G, Griebenow K. Effect of cyclodextrins on α- chymotrypsin stability and loading in PLGA microspheres upon s/o/w encapsulation. Journal of Pharmaceutical Sciences. 2005;95(4):849–858. doi: 10.1002/jps.20512. [DOI] [PubMed] [Google Scholar]
- 35.Castellanos IJ, Al-Azzam W, Griebenow K. Effect of the covalent modification with poly(ethylene glycol) on α-chymotrypsin stability upon encapsulation in poly(lactic-co-glycolic) microspheres. Journal of Pharmaceutical Sciences. 2005;92:327–340. doi: 10.1002/jps.20243. [DOI] [PubMed] [Google Scholar]
- 36.Castellanos IJ, Crespo R, Griebenow K. Poly(ethylene glycol) as stabilizer and emulsifying agent: a novel stabilization approach preventing aggregation and inactivation of proteins upon encapsulation in bioerodible polyester microspheres. Journal of Controlled Release. 2003;88:135–145. doi: 10.1016/s0168-3659(02)00488-1. [DOI] [PubMed] [Google Scholar]
- 37.Cho K, Wang X, Nie S, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clinical Cancer Research. 2008;5:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 38.Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. Journal of Controlled Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 39.Valdez D., J -Y. Le Huérou, Gindre M., Urbach W., Waks M. Hydration and protein folding in water and in reverse micelles: compressibility and volume changes. Biophysical Journal. 2001;80:2751–2760. doi: 10.1016/S0006-3495(01)76243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gref R, Quellec P, Sanchez A, Calvo P, Dellacherie E, Alonso MJ. Development and characterization of Cyt-loaded poly(lactic acid)-poly(ethylene glycol)PEG micro- and nanoparticles. Comparison with conventional PLA particulate carriers. European Journal of Pharmaceutics and Biopharmaceutics. 2001;51:111–118. doi: 10.1016/s0939-6411(00)00143-0. [DOI] [PubMed] [Google Scholar]
- 41.Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng J-X, Kohane DS, Yeo Y. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Molecular Pharmacology. 2009;6(1):190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knubovets T, Osterhout JJ, Klibanov AM. Structure of lysozyme dissolved in neat organic solvents as assessed by NMR and CD spectroscopies. Biotechnology and Bioengineering. 1999;63:242–248. [PubMed] [Google Scholar]
- 43.Jackson M, Mantsch HH. Beware of proteins in DMSO. Biochimica et Biophysica Acta: Protein Structure and Molecular Enzymology. 1991;1078:231–235. doi: 10.1016/0167-4838(91)90563-f. [DOI] [PubMed] [Google Scholar]