

Abstract
A series of potent amide non-urea inhibitors of soluble epoxide hydrolase (sEH) is disclosed. The inhibition of soluble epoxide hydrolase leads to elevated levels of epoxyeicosatrienoic acids (EETs), and thus inhibitors of sEH represent one of a novel approach to the development of vasodilatory and anti-inflammatory drugs. Structure-activities studies guided optimization of a lead compound, identified through high-throughput screening, gave rise to sub-nanomolar inhibitors of human sEH with stability in human liver microsomal assay suitable for preclinical development.
Keywords: Soluble epoxide hydrolase, Non-urea inhibitors, Hypertension, Human liver microsomes stability
Soluble epoxide hydrolase (sEH) is a bifunctional homodimeric enzyme with hydrolase and phosphatase activity detected in various species ranging from plants to mammals.1 In humans it is mostly located in liver, kidney, intestinal and vascular tissues.2 The sEH enzyme is selective for aliphatic epoxides of fatty acids, and one of the most important substrates is epoxyeicosatrienoic acid (EET).3 EETs are one of the metabolic derivatives of arachidonic acid.4 The epoxygenase CYP2 enzymes catalyse the epoxydation of olefin bonds of arachidonic acid generating EETs.5 EETs exhibit vasodilatory effects in various arteries6,7 and possess anti-inflammatory properties.8 sEH mediates the addition of water to EETs, converting them to the corresponding diols – dihydroxyeicosatrienoic acids (DHETs), which show abolished or diminished biological activity . The inhibition of this enzyme, therefore may represent a promising therapeutic strategy since it would lead to elevated levels of EETs, which could then have beneficial therapeutic effects on blood pressure and inflammation.9
The most explored sEH inhibitors to date are urea-based compounds and numerous structure-activity relationship (SAR) studies led to discovery of several potent urea-based sEH inhibitors with IC50s in the lower nanomolar range.10,11 However, the urea-based inhibitors often suffer from poor solubility and stability, which hinders their pharmacological use in vivo.12,13
Amides, carbamates and thioureas have been evaluated as alternative pharmacophores in an attempt to improve physical properties of urea-based inhibitors.10,11 In this report we disclose our efforts towards designing novel non-urea, amide-based inhibitors of sEH.
Previously we identified through high-throughput screening (HTS)14 several hits with a low micromolar to low nanomolar potency. A majority of these compounds were urea-based inhibitors. However, several were non-ureas and among them the most potent was the derivative of isonipecotic acid, with substantial activity of 20 nM. Our initial SAR studies15 led to discovery of compound 1 with an IC50 of 7.9 nM (Figure 1). Based on its potent inhibition of sEH activity, we continued our evaluation of the right-hand side (2,4,6-trimethylphenyl ring) of 1, and this follow-up SAR study revealed compound 2, with an IC50 of 1.6 nM.16
Figure 1.
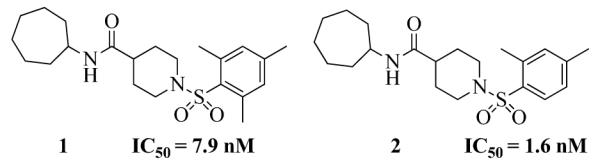
Chemical structures of non-urea inhibitors
Herein we report modifications of the left-hand side of non-urea inhibitor 2. In particular, we were focused on the replacement of the cycloheptyl moiety of the 2, following the SAR reported by Rose et al.17, and guided by docking model of the previously reported protein-inhibitor complex and X-ray cocrystal structure of human sEH and 1.
The X-ray crystallographic structure of human sEH and an inhibitor 4-(3-cyclohexylureido)-carboxylic acid complex (PDB code: 1ZD3) revealed the catalytic pocket and key structural features required to inhibit sEH enzyme. Two tyrosine (Tyr383 and Tyr466) and one aspartic acid (Asp335) residues, located in the hydrolase catalytic pocket of sEH, are involved in degradation of EET - tyrosine residues act as hydrogen bond donors to promote the epoxide ring opening by Asp335.18,19 The urea group of 4-(3-cyclohexylureido)-carboxylic acid fits in the hydrolase catalytic pocket and the carbonyl oxygen of the urea moiety is engaged in a hydrogen bond interaction with Tyr381 and Tyr466 while the N–H acts as a hydrogen bond donor to Asp335 The co-crystal structure of 1 and human sEH was determined (See Supplementary info) to guide the design of more potent inhibitors. Our close examination of the structure revealed that the amide moiety of 1 is positioned in the same fashion as the urea group in urea-based inhibitors, where the amide moiety, instead of urea group, is involved in hydrogen bonding with tyrosine and aspartic acid residues in the catalytic pocket of sEH. We noticed that the 2,4,6-trimethylphenyl ring on the right-hand side of this inhibitor occupies the smaller of the two lipophilic pockets in the sEH active site, while the cycloheptyl moiety on the left-hand side is directed towards the large deep pocket that opens toward solvent, which allows access for further structural modifications (Figure 2). We therefore hypothesized that the left-hand segment in 1 could be optimized to further improve the potency of this group of non-urea sEH inhibitors.
Figure 2.
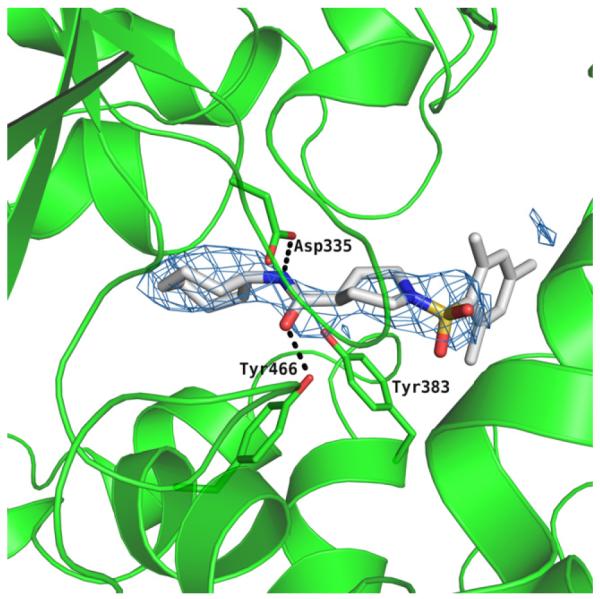
The active site of the hydrolase domain of human sEH complexed with 1. The Fo-Fc electron density map is contoured at 3σ. Hydrogen bonds are indicated by dashes. The structure reveals that the 2,4,6-trimethylphenyl group is directed into the hydrophobic environment, whereas the cycloheptyl moiety is exposed toward the solvent. Coordinates of the complex deposited as 4HAI with the Protein Data Bank. The image was produced using PYMOL.
Scheme 1 outlines the synthetic route used to form the left-hand side library of non-urea amide sEH inhibitors. Sulfonamide 5 was prepared from two commercially available starting materials, methyl isonipecotate 3 and 2,4-dimethylbenzenesulfonyl chloride 4. Saponification of this methyl ester with LiOH afforded acid 6. EDC peptide coupling reactions of 6 with various commercially available amines provided analogs 7-1 to 7-46.
Scheme 1.

Reagents and conditions: (a) Et3,N, CH2Cl2, rt, 24 h; 89% (b) LiOH, THF/H2O, rt, 24 h; 91% (c) R-NH2, EDC, DMAP, CH2C12, rt, 24 h; 65-92%.
A sensitive fluorescent based assay was employed to determine IC50 values of these sEH inhibitors. In short, cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyloxyran-2-yl) methyl carbonate (CMNPC) was used as the fluorescent substrate. Human sEH (1 nM) was incubated with the inhibitor for 5 min in pH 7.0 Bis–Tris/HCl buffer (25 mM) containing 0.1 mg/mL of BSA at 30 °C prior to substrate introduction ([S] = 5 μM). Activity was determined by monitoring the appearance of 6-methoxy-2-naphthaldehyde over 10 min by fluorescence detection with an excitation wavelength of 330 nm and an emission wavelength of 465 nm. Reported IC50 values are the average of the three replicates with at least two datum points above and at least two below the IC50.20 In Table 1 are summarized the biological results.
Table 1.
The biological results for sulfonamide analogs 7-1 to 7-46.
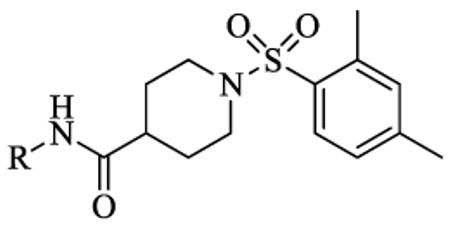
| |||||
|---|---|---|---|---|---|
| Compound | R | IC50a,b(nM) | Compound | R | IC50(nM) |
| 7-1 |
|
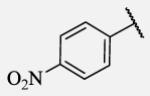
18 | 7-24 |
|
4.6 |
| 7-2 |
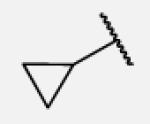
|
6900 | 7-25 |
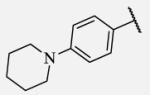
|
29 |
| 7-3 |
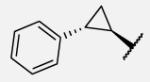
|
5.2 | 7-26 |
|
102 |
| 7-4 |
|
263 | 7-27 |
|
41 |
| 7-5 |
|
5.8 | 7-28 |
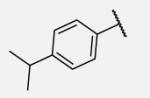
|
2.8 |
| 7-6 |
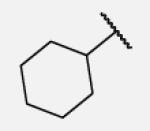
|
1.7 | 7-29 |
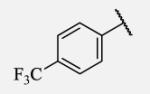
|
6.9 |
| 7-7 |
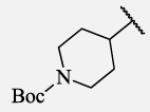
|
1.1 | 7-30 |
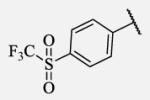
|
8.7 |
| 7-8 |
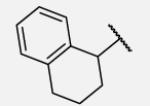
|
0.6 | 7-31 |
|
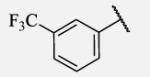
20 |
| 7-9 |
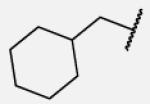
|
1.2 | 7-32 |
|
25 |
| 7-10 |
|
0.4 | 7-33 |
|
20 |
| 7-11 |
|
8.5 | 7-34 |
|
17 |
| 7-12 |
|
23 | 7-35 |
|
12 |
| 7-13 |
|
20 | 7-36 |
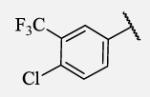
|
43 |
| 7-14 |
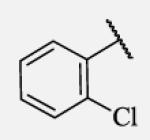
|
250 | 7-37 |
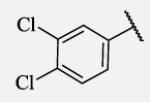
|
6.7 |
| 7-15 |
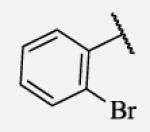
|
30 | 7-38 |
|
1.6 |
| 7-16 |
|
640 | 7-39 |
|
290 |
| 7-17 |
|
2200 | 7-40 |
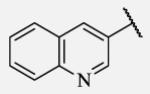
|
45 |
| 7-18 |
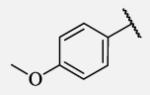
|
25 | 7-41 |
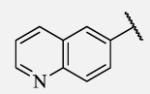
|
78 |
| 7-19 |
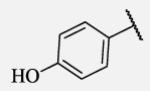
|
55 | 7-42 |
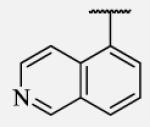
|
8.3 |
| 7-20 |
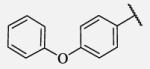
|
6.0 | 7-43 |
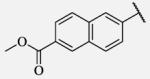
|
2.3 |
| 7-21 |
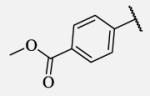
|
13 | 7-44 |
|
23 |
| 7-22 |
|
110 | 7-45 |
|
0.6 |
| 7-23 |
|
5.2 | 7-46 |
|
30000 |
Reported IC50 values are the average of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20% suggesting that differences of two fold or greater are significant.
t-AUCB that has an IC50 between 1 and 2 nM was used as positive control.
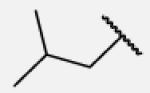
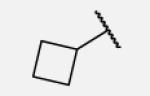
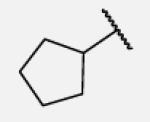
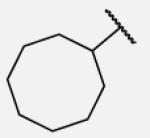
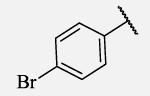
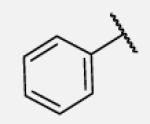
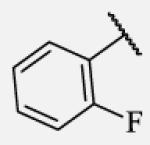
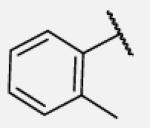
Previous studies on urea-based inhibitors containing piperidine moiety have shown that the hydrophobic cycloalkyl groups on the left-hand side of the molecules are positively correlated with inhibitory potency.17 Among this set of analogs we identified several inhibitors possessing improved or similar potency comparing to the lead compound 2, specifically compound 7-10 showed an IC50 of 0.4 nM, the most potent amide non-urea sEH inhibitor reported to date. Replacement of cycloalkyl ring with a more compact phenyl ring (compound 7-12), resulted in 15-fold drop in potency against the human sEH. However, introduction of the phenyl ring allowed us access to electronically and sterically diverse structures, and as well attachment of various polar groups, which could in turn improve physical and pharmacokinetic properties. Placement of fluorine or bromine in the ortho position did not significantly change the potency of the non-urea inhibitors (7-13 and 7-15), while chlorine and methyl group decreased the potency for 10 and 30-fold, respectively (7-14 and 7-16).
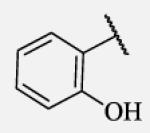
Polar hydroxyl group in ortho position showed clear negative effect on potency in non-urea based compounds (7-17). Although the para substitution is generally tolerated, placement of polar substituents resulted in less potent inhibitors.
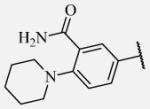
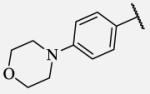
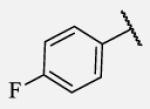
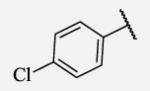
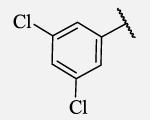
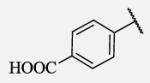
Placement of methoxy group in para position (compound 7-18) did not significantly changed the potency comparing to compound 7-12, while introduction of hydroxyl group in the same position (compound 7-19 can be observed as a metabolite of 7-18) led to 2-fold decreased potency presumably because of unfavorable electron character and increased polarity. Similar results, but with more drastically change in potency can be observed based on results for methyl ester compound 7-21 and its corresponding carboxylic acid compound 7-22. We synthesized the 4-trifluoromethoxyphenyl analog 7-23, since it has been reported previously that this moiety is more metabolically stable replacement for cycloalkyl rings.12 We observed 4-fold increase in potency for this compound compared with compound 7-12, thus it was selected for further pharmacokinetic studies. Interestingly, the analog 7-24, showed 5-fold increased activity comparing to phenyl compound 7-12, despite the presence of the high polarity nitro functionality. The metabolic stability for this inhibitor was evaluated as well. We also introduced a basic nitrogen (piperidine and morpholine rings in para position; analogs 7-25, 7-26 and 7-27) in order to allow formulation of the inhibitor as a salt. These modifications did not improve the potency, similar to other polar substituents in this position. On the other hand, the inhibition potencies increased when small non-polar para or meta substituents were added (7-28, 7-29, 7-30 and 7-31). Since it has been known that halogens can enhance polarity and decrease the rate of metabolism degradation due to their electron withdrawing effect on the aromatic ring, we decided to prepare a set of analogs containing various halogens in different position on the left-hand side phenyl moiety. The fluorinated, chlorinated and brominated para-phenyl compounds (7-32, 7-33 and 7-34, respectively) did not show significant improvement in activity compared to compound 7-12. Placement of two chlorine atoms in meta, and meta and para positions showed a 2-fold and 3-fold lower IC50 against human sEH enzyme, 7-35 and 7-37 respectively.
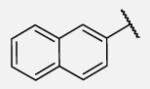
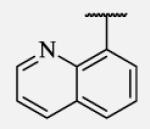
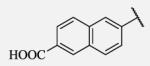
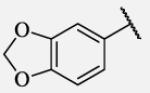
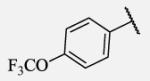
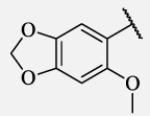
Future rational optimization of these meta, para disubstituted halogens might yield additional more potent compounds. Inclusion of 2-naphthalene on the left side of the molecule 7-38 resulted in high potency against the human enzyme, which is already shown in recent literature17, thus, we decided to test in vitro metabolic profile for this compound. We tried to introduce a nitrogen in this moiety in order to improve physical properties and the ease of formulation. Various aminoquinolines were attached via different position to central non-urea moiety. 5-Aminoquinoline derivative 7-39 led to 5-fold lower potency against sEH, 3-aminoquinoline derivative 7-40 showed 30-fold diminished potency, while 6- and 8-aminoquinoline analogs 7-41 and 7-42, led to even more drastically decreased potency, 50-fold and 180-fold, respectively. The poor performance of aminoquinolines returned our attention to 2-naphthalene moiety, where we tried to introduce polar group in position 6. Methylester analog 7-43 showed slight decrease in activity, while corresponding carboxylic acid 7-44 had 15-folds lower inhibition then the 2-naphthalene analog. Interestingly, 3,4-methylenedioxybenzene analog 7-45 led us to discovery of subnano molar potent inhibitor of human sEH enzyme.
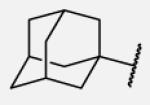
The selected non-urea sEH inhibitors were profiled in human liver microsomal assay22 as a predictor of in vivo oxidative metabolism (Table 2). Our results from this assay indicated that although compounds with hydrophobic cycloalkyl substituents on the right-side of the non-urea piperidine scaffold such as cyclohexyl, methylcyclohexyl, cycloheptyl, cyclooctyl or adamantyl were more potent inhibitors of sEH, compared to compounds with aromatic moiety, these compounds showed poor metabolic profile in human liver microsomal assay. Evaluation of the in vitro metabolic stability for aromatic compounds revealed intermediate metabolic profiles for compounds with para-substitution (compounds 7-23 and 7-24), with the exception of the carboxylic acid derivative 7-44, which demonstrated excellent in vitro metabolic stability in human liver microsomes.
Table 2.
Stability in human liver microsomes (t1/2) for selected compounds
| Compound | hLM t1/2 (min)a | CLint, app(mL/min/kg)b | Compound | hLM t1/2 (min)a | CLint, app(mL/min/kg)b |
|---|---|---|---|---|---|
| 2 | 5.5 | 220 | 7-24 | 180 | 7.0 |
| 7-3 | 14 | 90 | 7-37 | 25 | 50 |
| 7-6 | 14 | 90 | 7-38 | 36 | 35 |
| 7-9 | 2.4 | 520 | 7-42 | 8.7 | 140 |
| 7-11 | 3.7 | 340 | 7-44 | 220 | 5.6 |
| 7-20 | 11 | 120 | 7-45 | 36 | 35 |
| 7-23 | 46 | 28 |
Data represents averages of duplicate determination. hLMt1/2 is the half life in human liver microsomes.
CLint, app is apparent intrinsic clearance.
In summary, our structure-activity relationship of the lead 2 reveals some particular structural requirements to the left-hand side part of the piperidine amide-based sEH inhibitors. A varying degree of bulky, nonpolar cycloalkyl rings are well tolerated in this region by target enzyme. In contrast, proper substitution on the phenyl ring is crucial for attaining good potency, emphasizing the importance of the small nonpolar groups and halogens in para position as a recognition element for sEH, suggesting that left-hand side phenyl is in a relatively close proximity to a several hydrophobic residues located in the large, non-polar pocket of sEH that opens towards solvent, and probably participating in a π-stacking interaction with them. The above observations are corroborated by the costructure of 1 with sEH.
While low nanomolar and sub-nanomolar potencies were achieved, the majority of tested inhibitors in this non-urea based series were still not as stable in human liver microsome assay as some other non-urea sEH inhibitors previously reported.21 However, compound 7-4423 showed good stability and it will be further explored in various in vivo experiments. Our future work will focus as well on possible combinations of these data obtained from this SAR study and combinations of related fragments that would be used for future design of highly potent inhibitors with improved pharmacokinetic profiles.
Supplementary Material
Acknowledgments
This work was supported in part by NIEHS R01 ES002710 and NIH R01 HL 107887 (MEN). Preliminary structural work was performed at the Center for Advanced Microstructures and Devices (Baton Rouge), funded in part by the Louisiana Governors’ Biotechnology Initiative. The work includes research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by grants from the National Center for Research Resources (5P41RR015301-10) and the National Institute of General Medical Sciences (8 P41 GM103403-10) from the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank Dr. David Neau for assistance with data collection. BDH is a George and Judy Senior fellow of the American Asthma Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Newman JW, Morisseau C, Hammock BD. Prog. Lipid. Res. 2005;44:1. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- (2).Hammock BD, Grant D, Storms D. In: Comprehensive Toxicology. Sipes I, McQueen C, Gandolfi A, editors. Oxford; Pergamon: 1997. p. 283. [Google Scholar]
- (3).Imig JD, Hammock BD. Nat. Rev. Drug Discov. 2009;8:794. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chacos N, Falck JR, Wixtrom C, Capdevila J. Biochem. Biophys. Res. Commun. 1982;104:916. doi: 10.1016/0006-291x(82)91336-5. [DOI] [PubMed] [Google Scholar]
- (5).Spector AA, Fang X, Snyder GD, Weintraub NL. Prog. Lipid Res. 2004;43:55. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- (6).Larsen BT, Gutterman DD, Hatoum OA. Eur. J. Clin. Invest. 2006;36:293. doi: 10.1111/j.1365-2362.2006.01634.x. [DOI] [PubMed] [Google Scholar]
- (7).Capdevila JH, Falck JR, Harris RC. Journal of lipid research. 2000;41:163. [PubMed] [Google Scholar]
- (8).Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Science. 1999;285:1276. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ. Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- (10).Marino JP., Jr. Curr. Top. Med. Chem. 2009;9:452. doi: 10.2174/156802609788340805. [DOI] [PubMed] [Google Scholar]
- (11).Shen HC. Expert Opin. Ther. Pat. 2010;20:941. doi: 10.1517/13543776.2010.484804. [DOI] [PubMed] [Google Scholar]
- (12).Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. J. Med. Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Watanabe T, Schulz D, Morisseau C, Hammock BD. Anal. Chim. Acta. 2006;559:37. doi: 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pubchem. 2008. AID:1026.
- (15).Xie Y, Liu Y, Gong G, Smith DH, Yan F, Rinderspacher A, Feng Y, Zhu Z, Li X, Deng SX, Branden L, Vidovic D, Chung C, Schurer S, Morisseau C, Hammock BD, Landry DW. Bioorg. Med. Chem. Lett. 2009;19:2354. doi: 10.1016/j.bmcl.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pecic S, Deng SX, Morisseau C, Hammock BD, Landry DW. Bioorg. Med. Chem. Lett. 2012;22:601. doi: 10.1016/j.bmcl.2011.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. J. Med. Chem. 2010;53:7067. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gomez GA, Morisseau C, Hammock BD, Christianson DW. Biochemistry. 2004;43:4716. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- (19).Gomez GA, Morisseau C, Hammock BD, Christianson DW. Protein Sci. 2006;15:58. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal. Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Taylor SJ, Soleymanzadeh F, Eldrup AB, Farrow NA, Muegge I, Kukulka A, Kabcenell AK, De Lombaert S. Bioorg. Med. Chem. Lett. 2009;19:5864. doi: 10.1016/j.bmcl.2009.08.074. [DOI] [PubMed] [Google Scholar]
- (22).In vitro human liver microsomal metabolic stability: Microsomal stability was assessed in pooled human liver microsomes (Celsis, Edison, NJ). All reactions were carried out for 90min at 37°C in an NADPH-generating system consisting of glucose 6-phosphate, glucose 6-phosphate dehydrogenase, and NADP+ (Sigma, St. Louis, MO). Positive control incubations proceeded with 7-ethoxycoumarin as the substrate. Reactions were terminated by adding methanol. The mixtures were centrifuged and the supernatants were evaporated. The residues were reconstituted in mobile phase (85% ACN; 15% H2O) and subjected to LC/MS analysis.
- (23).Analytical data for the representative compounds. Compound 7–10: 1H NMR (400 MHz, CDCl3): δ 7.78–7.76 (d, J = 8 Hz, 1H) 7.09 (s, 1H), 7.07 (s, 1H), 3.96–3.89 (m, 1H), 3.71–3.66 (d, J = 9.6 Hz, 2H), 2.69 (t, J = 12 Hz, 2H), 2.55 (s, 3H), 2.35 (s, 3H), 2.10–2.03 (m, 1H), 1.86–1.82 (d, J = 10.8 Hz, 2H), 1.79-1.68 (m, 4H), 1.66-1.42 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 172.5, 143.7, 138.1, 133.7, 133.1, 130.6, 126.8, 49.6, 44.9, 43.0, 32.8, 28.8, 27.5, 25.9, 24.1, 21.6, 20.8; ESI-MS (M++H): 407; Compound 7-44: 1H NMR (400 MHz, DMSO-d6): δ 10.27 (s, 1H), 8.45 (s, 1H), 8.34 (s, 1H), 7.99–7.97 (d, J = 9.2 Hz, 1H), 7.88-7.85 (d, J = 10 Hz, 1H), 7.82-7.80 (d, J = 8.8, 1H), 7.68-7.66 (d, J = 8.4 Hz, 1H), 7.61-7.59 (d, J = 8.4 Hz, 1H), 7.24 (s, 1H) 7.20-7.18 (d, J = 7.6 Hz, 1H), 3.65-3.62 (d, J = 12.4 Hz, 2H), 2.67 (t, J = 11.6 Hz, 2H), 2.53 (s, 3H), 2.35 (s, 3H), 1.92-1.90 (d, J = 11.2 Hz, 2H), 1.69-1.59 (dd, J = 11.6 and 9.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.8, 168.0, 143.9, 139.5, 137.7, 136.3, 134.2, 134.0, 133.4, 131.0, 130.7, 130.5, 129.3, 127.4, 127.2, 121.2, 115.5, 115.2, 45.1, 28.7, 21.6, 21.5, 20.9; ESI-MS (M-+H): 465.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.