

Abstract
Interleukin (IL)-33 is an IL-1 family cytokine that displays dual function: a cytokine via its receptor, T1/ST2 or a chromatin-binding factor within the nucleus. Functionally, it promotes Th2-associated immunity by enhancing the activation and survival of several cell types. However, the pathways regulating IL-33 expression are still unclear. While several cells display constitutive expression of IL-33, we showed previously that mast cells expressed low levels of IL-33 constitutively but that IL-33 was induced upon IgE-mediated activation. This was mediated via a calcium-dependent mechanism. Here, we define the pathway through which this inducible IL-33 is regulated. Importantly, this pathway does not alter expression in cells with high constitutive IL-33 expression, like epithelial cells or fibroblasts. Our data shows that, upstream of calcium, inhibition of PI3K and Sphk activity decreases inducible IL-33 expression to IgE/antigen activation. In addition, expression of Sphk1 shRNA prevents upregulation of IL-33 expression. Downstream of calcium, NFAT activity is necessary and sufficient for inducible IL-33 expression. We also demonstrate calcium-dependent transcription from two regions of the IL-33 gene that contain putative NFAT-binding sites, one upstream of exon 1 and one upstream of the start site. Interestingly, we show that blocking other calcium pathways, including IP3R, or NF-κB inhibits IgE-driven IL-1β, another IL-1 family cytokine, but have no influence on inducible IL-33 expression. In summary, our data demonstrates cell-specific differences in the regulation of IL-33 expression and defines a pathway critical for the expression of inducible IL-33 by mast cells upon their activation.
Introduction
Interleukin-33 (IL-33, IL-1F11, NF-HEV) belongs to the IL-1 cytokine family, which also includes IL-1α, IL-1β, IL-1Ra (IL-1 receptor antagonist) and IL-18 (1). It was originally described as a nuclear located transcriptional regulator that associated with chromatin via a helix-turn-helix-like motif in its amino-terminus (2, 3). However, in 2005, IL-33 was reported as the ligand for the receptor T1/ST2 (DER4, Fit-1) (1). Since then, T1/ST2, in association with the IL-1R accessory protein (IL-1RAcP) (4, 5), has been shown to facilitate a diverse range of immunoregulatory influences in response to IL-33, that include stimulating Th1/Th2-associated (1, 6–8) and pro-inflammatory cytokine production (9), as well as survival (6, 8), migration (6, 10), and adhesion (6, 8) of T1/ST2-expressing cells. Furthermore, IL-33 has become increasingly established as functionally important in a variety of Th2-associated immune responses including allergic airway inflammation (11–13), helminth expulsion (14), allergic conjunctivitis (15), and anaphylaxis (16). However, many of these studies utilized administration of exogenous recombinant IL-33 and consequently very little is known about the pathways that regulate IL-33 expression.
Several cells types have been shown to exhibit constitutive expression of IL-33; these include endothelial cells, epithelial cells, fibroblasts, and pancreatic stellate cells (17–20). In these studies, IL-33 was present predominantly within the cell nucleus. In the context of regulation, IL-33 levels were increased in endothelial cells when they became super-confluent, but were then downregulated upon pro-inflammatory cytokine stimulation (17). Conversely, IL-33 expression in pancreatic stellate cells was upregulated upon stimulation by either TNF-α or IL-1β, and also in fibroblasts by the combination of TLR3 stimulation and TGFβ (18). Similarly, TLR3 stimulation increased IL-33 expression in mucosal epithelial cells; this TLR3 stimulation was shown to be NF-κB dependent, as a potent NF-κB-pathway inhibitor, BAY 11-7082, blocked this increased expression (21).
More recently, IL-33 has been found to be expressed in an inducible fashion by some immune cells, including dendritic cells (22), monocytes/macrophages (23–26), and mast cells (25, 27). Here, while IL-33 is induced upon cell activation, constitutive expression of IL-33 appears to be low or absent. For example, our previous work demonstrated that, while unstimulated mast cells possess very low basal IL-33 expression, antigen-mediated cross-linking of IgE on FcεRI led to significant increases in mRNA and protein (27). Interestingly, like TLR3, the IL-33 receptor T1/ST2 also signals via NF-κB (as well as MAP kinases) (1) and yet IL-33 did not promote further IL-33 in mast cells, despite being a potent inducer of several other mast cell-derived cytokines (28–30). Instead, IL-33 expression in mast cells was calcium sensitive, since EDTA addition prevented IgE-mediated expression and ionomycin was sufficient to induce expression. However, the molecular mechanism behind this calcium-sensitive inducible IL-33 expression in mast cells remains unclear, and we therefore wanted to elucidate this pathway. While there are many pathways that can lead to calcium mobilization in mast cells upon cross-linking of FcεRI, two major pathways that have been shown to regulate mast cell responses are 1) sphingosine-1-phosphate (S1P) generated via the sphingosine kinases (Sphks) and 2) inositol trisphosphate (IP3) binding to its receptor, IP3R (31). Furthermore, mast cells express two isoforms of Sphk, Sphk1 and Sphk2, that have been shown to exert differential effects on cytokine production and degranulation of mast cells (32). This is regulated by phosphatidylinositol 3-kinase (PI3K), where PI3K is required for full activation of Sphk1 but not for Sphk2 (33).
In this study, we show that inducible IL-33 expression by mast cells upon cross-linking of FcεRI is dependent on a PI3K-Sphk1/S1P-Calcium-NFAT pathway. Furthermore, NF-κB is dispensable for this IL-33 expression but is required for concurrent IL-1β expression. We also identify two regulatory regions upstream of the il33 transcription start site that contain putative NFAT binding sites and support calcium-driven transcription. However, in cell types that exhibit constitutive expression, activation of this calcium-dependent pathway had little to no effect on IL-33 mRNA levels, suggesting that IL-33, in addition to having dual functions, also possesses dual pathways that regulate its expression.
Materials and Methods
Cell lines and Reagents
N,N-Dimethyl- d-erythro-sphingosine (DMS) was obtained from BioMol. 2-Aminoethoxydiphenyl borate (2APB), LY294002, Cyclosporine A (CsA), ionomycin, mouse IgE (SPE-7 clone) and dinitrophenyl (DNP)-human serum albumin (DNP-HSA) were obtained from Sigma-Aldrich. Wortmannin, BAY 11-7085 and inhibitor of NFAT-Calcineurin association-6 (INCA-6) were obtained from CalBiochem. W146 (trifluoroacetate salt) and JTE-013 were purchased from Cayman Chemical. Taqman gene expression primers and probes were obtained from Applied Biosystems. Primers were obtained from Invitrogen. pLG.3-basic and pLG.3-control plasmid, LDH assay and dual-glo luciferase assay were obtained from Promega. Fluo-4 was obtained from Invitrogen. Anti-mouse CD16/32 (2.4G2), APC-anti-mouse CD117 (c-kit), PE-anti-mouse FcεRI (MAR-1) and FITC-anti-rat IgG2a were obtained from BD Biosciences. APC-anti-rat IgG F(ab′) was from eBioscience. pGFP-V-RS plasmids with or without scrambled, Sphk1 or Sphk2 shRNA were purchased from Origene AMAXA mouse dendritic cell transfection buffer was obtained from Lonza. Monoclonal anti-mouse IL-33 antibody (Clone 396118) and anti-rat IgG2a isotype antibody were obtained from R&D Systems. Fixation & Permeabilization buffers were from eBioscience or R&D.
The mouse fibroblast cell line, NIH3T3, was provided by Dr. Christian Stehlik (Northwestern University). The mouse endothelial cell line, HEVa, was provided by Dr. Joan Cook-Mills (Northwestern University). The mouse mast cell line, MC/9, and the mouse intestinal epithelial cell line, CMT93, were purchased from ATCC.
Bone Marrow Derived Mast Cells (BMMCs) generation and activation
Bone marrow-derived mast cells (BMMC) were obtained by flushing bone marrow from femurs using complete RPMI media, 10% FBS, penicillin/streptomycin, L-glutamine and non-essential amino acid. Cells were then cultured in mast cell growth media (complete RPMI media + 30ng/ml recombinant mouse IL-3) for 5 weeks. Mast cell phenotype was determined by flow cytometry for c-kit+/FcεRI+ cells (>95%).
Mast cells were primed with 1μg/ml DNP-specific IgE overnight and then activated with 0.5μg/ml DNP-HSA or 0.25μM ionomycin for 4 hours for gene expression or 24 hours for protein expression. In some experiments, mast cells were pre-treated with inhibitors 30 minutes prior to DNP-HSA or ionomycin activation.
Real Time RT-PCR
RNA from treated cells was generated by RNeasy RNA isolation kit (Qiagen). cDNA was synthesized by qScript cDNA SuperMix (Quanta Bioscience). Gene expression was determined by real-time PCR using an ABI 7500 Thermal Cycler (Applied Biosystems) and specific Taqman probes (Applied Biosystems) for each gene of interest. β-actin expression was used as an internal control and changes in the cycle threshold values determined, as previously described (34).
Intracellular staining
BMMCs or MC/9 cells were activated with IgE/DNP or 1μM ionomycin with or without inhibitors treatment for 24 hours. Cells were stained with APC-anti-mouse CD117 following by fixing and permeabilizing with Fixation & Permeabilization Buffer (eBioscience or R&D). Cells were then stained with monoclonal anti-mouse IL-33 or isotype antibody, followed by FITC-anti-rat IgG2a or APC-anti-rat IgG F(ab′)2.
Calcium mobilization assay
BMMCs were primed with 1μg/ml DNP-specific IgE overnight. Cells were then incubated with calcium-free buffer (HBSS, 2mM Probenecid and 0.1% BSA) containing 1.5μM fluo-4 for 30 minutes and transferred to calcium-containing buffer (HBSS, 2mM Probenecid, 0.1% BSA and 1.8mM CaCl2). Cells were treated with different inhibitors and calcium flux was measured 2 minutes prior to 0.5μg/ml DNP-HSA or DMSO stimulation. Cells were also stimulated with 0.25μM ionomycin to confirm the ability to induce calcium flux in the presence of absence of inhibitors. Calcium flux was analyzed by flow cytometry. The effect of all inhibitors on calcium flux upon IgE activation of mast cells is shown in Supplementary Fig. 1.
Construction of il-33 regulatory region reporter assays
Putative NFAT binding sites were determined using Patch public 1.0 software (Biobase). The region between −1600 to −1 of il33 upstream of exon 1 (il-33-exon) was cloned from C57BL/6 genomic DNA using the following primers: forward, 5′-AGCAAATCTTTTAGTCATAGATGC-3′; and reverse, 5′-AACACAGTCGCGCTTCAG-3′. HindIII and XhoI restriction enzyme sites were introduced to the ends of il-33-exon using primers: forward, 5′-CTCGAGAGCAAATCTTTTAGTCAT-3′; and reverse, 5′-AAGCTTAACACAGTCGCGC-3′. The region between −1000 to +100 within il33 intron 1 relative to the ATG transcriptional start site (il-33-ATG) was cloned from C57BL/6 genomic DNA using the following primers: forward, 5′-AAGGTTTCTCCCGCTGCG-3′; and reverse, 5′-AAGGACCAGGGCTTCGCCT-3′. HindIII and XhoI restriction enzyme sites were introduced to the ends of il-33-ATG by the following primers: forward, 5′-CTCGAGGGTTTCTCCCGC-3′; and reverse, 5′-AAGCTT GGACCAG GGCTTC-3′. il-33-exon and il-33-ATG were cloned into the pGL.3-basic plasmid (Promega) using HindIII and XhoI.
BMMC Transfection
BMMCs were electroporated by AMAXA Nucleofector II (Lonza) using mouse dendritic cell buffer (Lonza) and program C-005. Cells were suspended in mast cell culture medium with 30ng/ml IL-3 immediately after electroporation.
Luciferase assays
1×106 BMMCs were cotransfected with 1μg of il-33-exon, il-33-ATG or pGL.3-basic (mock) with 0.8μg of pRNL-SV40 plasmid (Promega) to control for efficiency of transfection. 24 hours after transfection, cells were lysed with lysis buffer (Promega) and luciferase activity analyzed using the Dual-Glo Luciferase Assay System (Promega). The Firefly luciferase activity was normalized to the Renilla luciferase activity. The Firefly/Renilla ration was expressed as the relative luciferase unit (RLU). The fold induction above the control group (unstimulated) was determined based on changes in the RLU values.
shRNA plasmid transfection
2×106 BMMCs were electroporated with 4μg scrambled shRNA-, Sphk1 shRNA- or Sphk2 shRNA-expressing plasmids that co-express eGFP as indicated by AMAXA system as previously described. Three days after transfection, cells were split into 2 wells (1ml/well) with extra 30ng/ml IL-3. Seven days after transfection, mast cells were primed with DNP-IgEovernight and stimulated with DNP-HSA for 24 hours. IL-33 expression after IgE-mediated activation was determined in eGFP+ cells versus eGFP− cells by intracellular flow staining
NFAT overexpression
A plasmid expressing a constitutively active NFAT-c1 mutant (caNFAT-c1) that co-expressed eGFP was a gift from Dr. Neil Clipstone (Loyola University, Chicago) and previously described (35). BMMCs were transfected with 200ng of caNFAT-c1 plasmid or control plasmid by AMAXA system as previously described. IL-33 protein expression in eGFP+ cells versus eGFP− cells was analyzed by intracellular staining 24 hours after transfection.
Statistical analysis
Statistics were performed on GraphPad Prism 4 software (GraphPad) by using the 2-tailed Student t test or 2 way ANOVA, as appropriate, to determine significance.
Results
IL-33 expression is induced by calcium in mast cells but not in several other cell types
Since we previously demonstrated that ionomycin was sufficient to upregulate IL-33 gene expression in mast cells (27), we first explored if IL-33 gene expression was calcium dependent in other cell types that had been previously reported to express IL-33. A murine fibroblast cell line (NIH3T3), endothelial cell line (HEVa), and mucosal epithelial cell line (CMT93) were tested, in addition to an immortalized murine mast cell line (MC/9) and primary BMMCs that served as positive controls. Cells were stimulated with 0.25μM ionomycin for 4 hours and IL-33 mRNA expression analyzed by real-time PCR. We found that, as predicted, resting mast cells (both BMMC and MC/9) express very low levels of IL-33 compared to the fibroblast, endothelial, or epithelial cells (Fig. 1A). Upon ionomycin stimulation, however, IL-33 expression was significantly increased in mast cells and HEVa endothelial cells, but not in either fibroblasts or epithelial cells. Interestingly, the relative expression levels were higher in epithelial and endothelial cells than in fibroblasts, as seen from the 2−ΔCt values. Intracellular staining showed that the IL-33 produced by mast cells was present in both the nucleus and cytoplasm, although predominantly in the cytoplasm (Fig. 1B), which is in contrast to the predominantly nuclear IL-33 localization in endothelial or epithelial cells (17, 19) but is similar to IL-33 localization seen in monocytes (23). These results indicated that the regulation of IL-33 by calcium mobilization is cell-type dependent and that the localization of IL-33 in mast cells after activation is predominantly cytoplasmic.
Figure 1. Ionomycin-induced IL-33 expression is cell-type dependent.
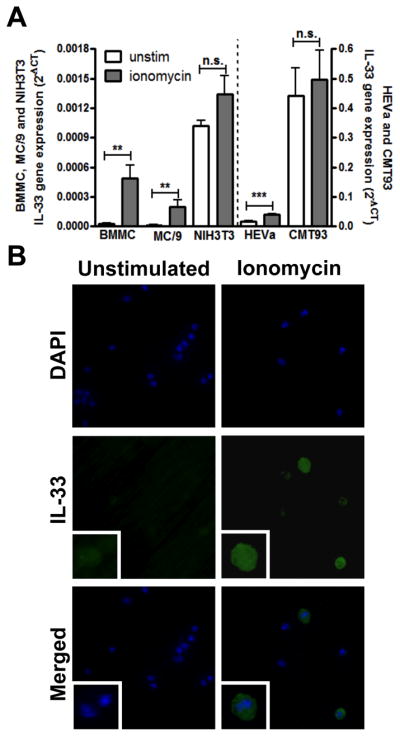
A. BMMC, MC/9, NIH3T3, HEVa and CMT93 cells were stimulated with 0.25 μM ionomycin (gray bar) for 4 hours and IL-33 mRNA expression was analyzed by real-time RT-PCR. B. BMMCs with or without 1 μM ionomycin stimulation were intracellularly stained with FITC-anti-IL-33 and mounted with DAPI–containing buffer. Blue, DAPI staining for nucleus. Green, IL-33 staining. Images are representative of 3 independent experiments. Gene expression is pooled from 3 independent experiments, n=3; *p<0.05, ***p<0.001.
Sphk/S1P regulates IL-33 gene expression in mast cells
Since calcium was necessary to induce IL-33 expression in mast cells upon FcεRI crosslinking (30), we wanted to determine which pathways immediately downstream of FcεRI were required for calcium-mediated upregulation of IL-33. Since both IP3R and Sphks are downstream of FcεRI signaling and previously shown to regulate the expression of several cytokines from mast cells (36), we tested whether activation of either of these signaling mediators was necessary. BMMCs were primed with DNP-specific IgE overnight and treated with different doses of N,N- Dimethyl-d-erythro-sphingosine (DMS), a potent inhibitor of the Sphks (37) or 2-Aminoethoxydiphenyl borate (2APB), an inhibitor of several calcium-dependent pathways including IP3R, 30 minutes prior to DNP-HSA activation. The blockade of IgE-mediated calcium flux by DMS is shown in Supplemental Fig. 1A and by 2-APB in Supplemental Fig. 1B. While DMS can also inhibit protein kinase C (PKC) activity (38), we used a relatively low concentration of DMS that has been shown to inhibit Sphk function without significantly affecting PKC activity (39). Cells were harvested 4 hours after stimulation and IL-33 expression was analyzed. At the concentrations used, both inhibitors did not affect cell viability, as determined by LDH release assay (data not shown). We found that IL-33 gene expression was inhibited by DMS and not by 2APB (Fig. 2A), indicating a role for the calcium-activation pathway involving Sphks, but not IP3R or the other pathways affected by 2APB. DMS decreased IL-33 gene expression in a dose-dependent manner (Fig. 2B) while 2APB did not affect IL-33 gene expression at any dose investigated (data not shown). DMS had no effect on ionomycin-stimulated IL-33 gene expression, as expected since ionomycin induces calcium mobilization directly and does not require Sphk activation, (Fig. 2C). In contrast to IL-33, IL-1β was inhibited by 2APB but not by DMS (Fig. 2D), confirming that IP3R activation from FcεRI is required for expression of some cytokines, including other IL-1 family members. We conclude that IL-33 gene expression is therefore likely regulated by Sphk-S1P-induced Ca2+ mobilization.
Figure 2. IL-33 expression in mast cells is regulated by Sphk-induced calcium mobilization.
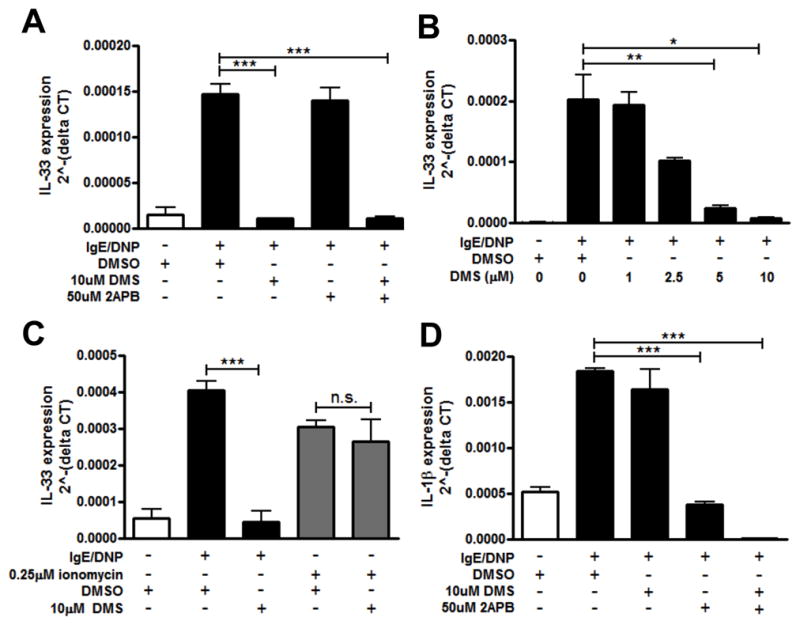
BMMCs were pretreated with 10μM DMS, or 50μM 2-APB, or both 30 minutes prior to activation by IgE/DNP (black bar). IL-33 (A) and IL-1β (D) gene expression was analyzed by real-time RT-PCR. B. IL-33 expression was inhibited by DMS in a dose-dependent manner. C. IL-33 expression was analyzed in BMMCs pretreated with 10μM DMS and activated with IgE/DNP (black bar) or 0.25μM ionomycin (gray bar). n=3 from 3 independent experiments; *p<0.05, **p<0.01, ***p<0.001.
Autocrine effects of S1P regulate IL-33 gene expression in mast cells
S1P generated downstream of Sphk activation has been shown to be released by mast cells and act via S1P receptors expressed on the surface of mast cells in an autocrine manner to further increase calcium signaling (40). Mast cells express two S1P receptors, S1PR1 and S1PR2 (32); we therefore used two specific inhibitors of S1PR1 and S1PR2 (W146 and JTE-013, respectively), to determine if such autocrine effects through the S1P receptors influenced IL-33 gene expression. BMMCs were treated and activated as previously describe and cell viability was determined by LDH assay. IL-33 gene expression was inhibited by both W146 and JTE-013 (Fig. 3), although the highest dose of JTE-013 did slightly decrease mast cell viability, as determined by LDH release (data not shown). IL-6 secretion was also inhibited by both W146 and JTE-013 (data not shown), in agreement with previously published findings (41). These data indicate that S1P-mediated autocrine effects are also important for IL-33 expression.
Figure 3. S1PR-mediated autocrine effects are required for IL-33 gene expression in mast cells.
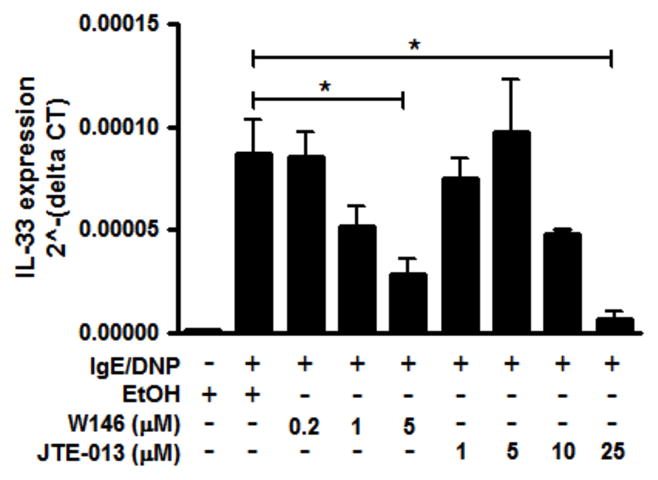
BMMCs were pretreated with different doses of W146, or JTE-013, 30 minutes prior to activation by IgE/DNP. IL-33 gene expression was analyzed by real-time RT-PCR. Data represented 5 individual experiments. n=3; *p<0.05
PI3K is important for IL-33 gene expression in mast cells
Since mast cells have been shown to express two different sphingosine kinases, Sphk1 and Sphk2 (42), and phosphoinositide 3-kinase (PI3K) is required for Sphk1 activation upon FcεRI crosslinking but not for Sphk2 activation (33), we next investigated the effects of two inhibitors of PI3K activity, LY294002 and wortmannin. BMMCs were treated, activated and analyzed as previously described and the effects on IgE-mediated calcium flux by wortmannin is shown in Supplemental Fig. 1C and by LY294002 in Supplemental Fig. 1D. Both PI3K inhibitors suppressed IL-33 gene expression in a dose-dependent manner in response to IgE/antigen activation (Fig. 4A) without inducing cell death (data not shown). Supporting this result, we also determined that IL-33 protein induction in mast cells was inhibited by PI3K inhibition upon IgE/DNP stimulation using intracellular staining (Fig. 4B). However, ionomycin stimulation bypassed the inhibitory effects of blocking PI3K and induced IL-33 expression even in the presence of the PI3K inhibitors (Fig. 4C). Accordingly, IL-33 expression by mast cells upon activation of FcεRI is dependent upon PI3K, again supporting a role for Sphk1.
Figure 4. IL-33 expression in mast cells is dependent on PI3K activity.

BMMCs were pretreated with different doses of PI3K inhibitors, LY294002 and wortmannin, 30 minutes prior to IgE/DNP (A and C, black bar) and 0.25μM ionomycin (C, gray bar) activation. IL-33 expression was analyzed by real-time PCR. (B) BMMCs pretreated with 10μM LY294002 or 100nM Wortmannin were activated with IgE/DNP for 24 hours. IL-33 protein expression was detected by intracellular staining. Gray line, isotype control; black dot, unstimulated; gray fill, IgE/DNP stimulation; black line, LY294002 treatment; black dash, wortmannin treatment. n=3 from 2-3 individual experiments; *p<0.05, **p<0.01, ***p<0.001, n.s. = no significant difference.
Sphk1, but not Sphk2, shRNA inhibited IL-33 expression upon IgE/DNP stimulation
DMS, the SphK inhibitor we used in Figure 2, is not selective for either Sphk1 or Sphk2 and so does not distinguish which Sphk was required for the calcium-mediated inducible IL-33 expression from mast cells while inhibition of PI3K may influence other signaling pathways. To better define the role of Sphk1 over Sphk2, we therefore utilized shRNA knockdown of Sphk1 or Sphk2 in BMMC using plasmids that expressed scrambled, Sphk1, or Sphk2 shRNA with eGFP co-expression to detect shRNA induction in transfected cells. Plasmid-transfected cells were gated on GFP+ cells; while scrambled shRNA or Sphk2 shRNA had no effect on IL-33 induction, cells that expressed Sphk1 shRNA failed to upregulate IL-33 upon IgE/DNP activation (Fig. 5), indicating that Sphk1 was required for IL-33 expression in mast cells.
Figure 5. Sphk1 shRNA inhibited IL-33 upregulation upon IgE/DNP activation.

BMMCs were transfected with scrambled (top), Sphk1 (middle) or Sphk2 (bottom) shRNA plasmids that also express GFP for 7 days and activated with IgE/DNP. Cells transfected with shRNA plasmids were gated on GFP+ cells. IL-33 protein expression of shRNA transfected cells (GFP+ gated) was analyzed by intracellular staining 24 hours after stimulation. Data is representative of 4 independent experiments.
NFAT regulates IL-33 gene expression in mast cells
Since nuclear factor of activated T cell (NFAT) is a critical calcium-sensitive transcription factor downstream of calcium induction, we next wanted to examine the role of NFAT in the induction of IL-33 expression. BMMCs were pre-treated using Inhibitor of NFAT-Calcineurin interaction-6 (INCA-6), which is a specific peptide that interrupts the interaction between NFAT and calcineurin, and Cyclosporine A (CsA); BMMCs were then activated by IgE/antigen or ionomycin as previously described. The absence of effect on IgE-mediated calcium flux by CsA is shown in Supplemental Fig. 1E and by INCA-6 in Supplemental Fig. 1F. IL-33 gene expression was significantly decreased in a dose-dependent manner by both inhibitors (Fig. 6A) without influencing cell viability (data not shown). INCA-6 and CsA also inhibited IL-33 gene expression upon ionomycin activation (Fig. 6B) as predicted, since NFAT activation lies downstream of calcium mobilization. Using intracellular staining, we observed that CsA and INCA-6 also blocked the upregulation of intracellular IL-33 protein in response to ionomycin (data not shown). Furthermore, to determine if NFAT activity is sufficient to induce IL-33 expression, we overexpressed a constitutively activated NFAT mutant (caNFAT-c1) in BMMCs. This caNFAT-c1 mutant contains an IRES-GFP, allowing for selection of transfected cells based on GFP expression. Overexpression of active NFAT was sufficient to increase IL-33 protein expression (Fig. 6C). These results indicate that IL-33 gene expression by mast cell is regulated by activation of the Ca2+-sensitive transcription factor NFAT.
Figure 6. IL-33 expression in mast cells is dependent on transcription factor NFAT.

BMMCs pretreated with different doses of INCA-6 or CsA are activated with IgE/DNP (A, black bar) or 0.25 μM ionomycin (B, gray bar) for 4 hours. IL-33 gene expression was analyzed by real-time PCR. C. BMMCs were transfected with constitutively active NFATc1 mutant (caNFATc1). Gray line, caNFATc1-; gray fill, caNFATc1+, based on GFP expression. Data represented 2–3 individual experiments. n=3; *p<0.05, **p<0.01, ***p<0.001.
NF-κB is dispensable for IL-33 gene expression in mast cells
Although it is well established that calcium induces NFAT activation, calcium can also induce NF-κB activation (43). Additionally, the expression of constitutive IL-33 has been shown to be NF-κB driven in mucosal epithelial cells (21) and the other IL-1 family cytokines, IL-1β and IL-18, are regulated by NF-κB (44). We therefore wanted to examine if NF-κB was also needed for IL-33 expression by mast cells. Pretreatment of BMMC with BAY 11-7085, the NF-κB inhibitor previously shown to block IL-33 expression in mucosal epithelial cells (21), 30 minutes prior to activation by cross-linking of the FcεRI did not affect IL-33 expression (Fig. 7A). Conversely, IL-1β expression was significantly inhibited in a dose-dependent manner (Fig. 7B). Taken together, these findings further support differences between the pathways regulating constitutive and inducible IL-33, as well as different pathways in the regulation of the IL-1 family cytokines upon FcεRI-mediated activation of mast cells.
Figure 7. NF-κB is dispensable for IL-33 expression in mast cells.
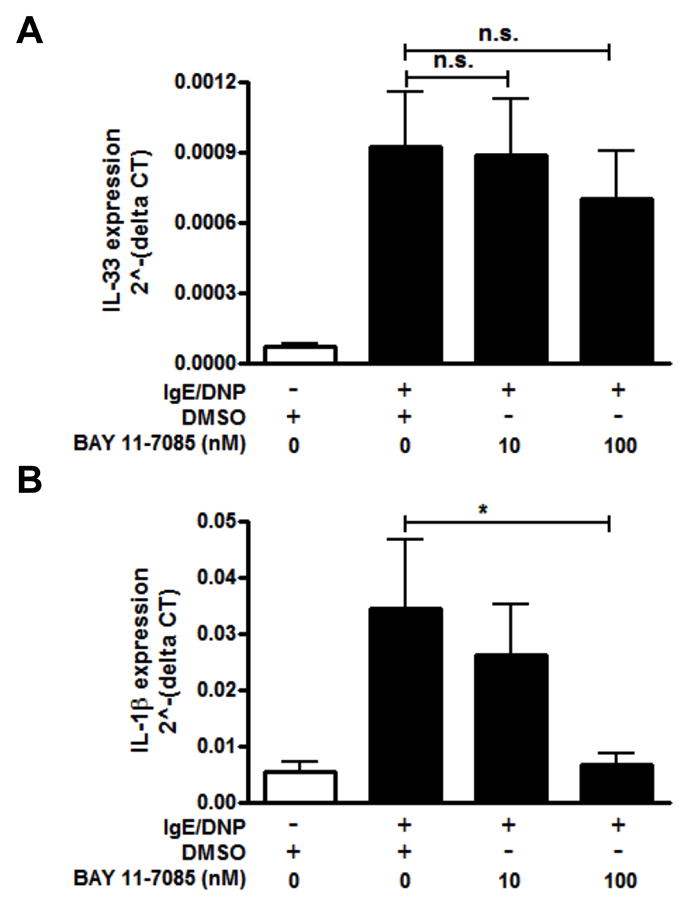
BMMCs were pretreated with different doses of NF-κB inhibitor, BAY 11-7085, and activated with IgE/DNP for 4 hours. IL-33 (A) and IL-1β (B) gene expression were analyzed by real-time PCR. n=3 from 2 independent experiments; **p<0.01, ***p<0.001, n.s. = no significant difference.
Regulatory regions of il33 support calcium-dependent transcription
We next wanted to determine if there were any regulatory regions of il33 that specifically conferred responsiveness to ionomycin. The ATG start codon of il33 is located at exon 2 (45); it has been previously shown that some genes can contain regulatory regions within the intronic region between exon 1 and exon 2 when the ATG is located at exon 2 (46, 47). For this reason, we cloned both the upstream region from exon 1 of il33 (base pairs −1600 to −1 from the start of exon 1; il-33-exon) and the intron 1 of il33 (base pairs −1000 to +100 from the ATG starting site of exon 2; il-33-ATG) into the pGL.3-basic luciferase plasmid (Promega) and investigated the ability of these regions to support luciferase expression that would indicate which region contained regulatory potential. BMMC’s were transfected using AMAXA Nucleofector II with co-transfection of a Renilla-expressing control plasmid (pGL.3-RNL) to control for efficiency. Transfected cells were activated with 0.25μM ionomycin for 24 hours in the presence of vehicle, 10μM INCA-6 or 1.5μM CsA and luciferase activity was analyzed. Interestingly, both il-33-exon (Fig. 8A) and il-33-ATG (Fig. 8B) supported ionomycin-induced transcription and this was inhibited by blockade of NFAT activation, suggesting these regions may contain NFAT regulatory sites. Indeed, using Patch public 1.0 software (Biobase), we identified several putative NFAT binding sites (TTTCC, GGGAA, TGCTGA, or TCAGCA) within these two regions.
Figure 8. Two regulatory regions of il33 support calcium-dependent transcription.

The region between −1600 to −1 of il33 upstream according to exon 1 (il-33-exon) and the region between −1000 to +100 within il33 intron 1 according to ATG transcriptional start site (il-33-ATG) were cloned into pGL.3-basic luciferase reporter plasmid. BMMCs were transfected with pGL.3-basic (mock), il-33-exon (A), or il-33-ATG (B) together with pGL.3-RNL as a transfection efficiency control. Cells were activated with 0.25μM ionomycin 24 hours in the presence of vehicle, 10μM INCA-6 or 1.5μM CsA after transfection. Luciferase activity was analyzed 24 hours after stimulation.. Data represented 4–5 independent experiments. **p<0.01, ***p<0.001.
Discussion
IL-33 is being increasingly recognized as regulating immune responses in infection, allergic, autoimmune, and chronic inflammatory diseases from both patient studies and animal models (13). Consequently, the IL-33/ST2 pathway may have potential as a therapeutic target in various diseases (48). Despite this wealth of information on its functions, how IL-33 expression is regulated has remained unclear. Knowing the mechanisms of regulation may help target IL-33 in specific therapeutic ways.
From several studies that have investigated the cellular expression of IL-33, there would appear to be two prevailing findings. In some cells, such as epithelial cells, endothelial cells, and fibroblasts, IL-33 is constitutively expressed at high basal levels and is located within the nuclear compartment (17, 20, 21, 49). Conversely, in mast cells (as well as dendritic cells and monocytes or macrophages) IL-33 is induced upon cellular activation and, from the studies that have explored cellular location, this expression would seem to be predominately cytoplasmic (22–27). In our previous study, we saw no evidence for cleavage of IL-33 in mast cells (30) and so the cytoplasmic location is rather surprising, since full-length IL-33 has a chromatin-binding domain (3), likely responsible for its nuclear localization in most cells. We did determine that the intracellular detection of IL-33 in mast cells did not require brefeldin A and so IL-33 was likely not being transported via the Golgi apparatus, a finding that is consistent with IL-33 lacking a signal peptide sequence required for this pathway (50). However, mast cells are well recognized for containing several unique granules and vesicles and so one possibility is that IL-33 is exported via one of these unique mechanisms. However, despite this remaining area of query, the inducible nature of IL-33 expression in mast cells has allowed us to define the necessary pathways that lead to IL-33 transcription in these cells.
Interestingly, our findings seem to support divergent signaling mechanisms for regulating the expression of inducible versus constitutive IL-33. Previous findings had demonstrated the importance of TLR3-mediated NF-κB in regulating IL-33 levels in cell types with constitutive expression (18, 21). However, TLR3 stimulation failed to induce IL-33 in mast cells (data not shown) while the induction of IL-33 that did occur in response to FcεRI-mediated activation was not influenced by NF-κB inhibition. Conversely, IL-1β expression that occurred upon activation was highly NF-κB dependent. Consequently, our data would suggest that the expression of the IL-1 family members that occurs in mast cells are dependent on different signaling pathways mediated downstream of FcεRI.
Our study represents the first to define the cellular signaling pathways requires for induction of IL-33 in inflammatory cells and has defined this as being regulated via NFAT. Interestingly, IL-33 itself has also been suggested to require calcium for its effects on mast cells since calcium chelation diminished IL-33 driven activation (16). However, the effect of IL-33 on many mast cell-derived cytokines, as well as prostaglandins and leukotrienes, were predominantly NF-κB dependent. Interestingly, IL-5 and IL-13 were not NF-κB dependent, suggesting that alternative signaling pathways from T1/ST2 regulate these cytokines. We previously demonstrated that IL-33 activation did not increase IL-33 expression and that T1/ST2 is actually shed from the mast cell surface upon FcεRI-mediated activation (30). Consequently, while signaling from T1/ST2 may be capable of inducing a calcium-associated response, this seems insufficient to activate the pathway needed for inducible IL-33 in mast cells. Therefore, while mast cells are capable of upregulating IL-33 upon antigen-triggered activation, this is unlikely to serve any autocrine or paracrine amplification influences due to reduction of surface ST2 expression and the lack of NFAT-mediated signals if binding was to occur. Andrade and colleagues have demonstrated that IL-33 treatment delivered concurrently with antigen activation leads to an altered or stronger mast cell response, including NFAT activation (51). This was also dependent upon NF-κB, rather being a direct influence of IL-33 and ST2 on NFAT. Consequently, we postulate a dual role for IL-33 whereby the release of constitutive IL-33 from epithelium or other cells during antigen exposure may enhance immediate inflammatory responses, while mast cell-derived IL-33 occurs later and mediates an alternative function, such as the recruitment of inflammatory cells after FcεRI-mediated activation that we previously demonstrated in a model of anaphylaxis (30).
In defining the pathway from FcεRI to IL-33 expression, we chose to specifically explore the S1P and IP3 pathways due to the extensive evidence for these being critical pathways involved in IgE-mediated activation (36). However, other pathways may also contribute to mobilizing calcium in mast cells. The calcium release activated calcium (CRAC) channel is a store-operated channel and opened by the emptying of internal calcium stores (52). It has been shown that CRAC channels could prolong cytosolic increase of calcium and regulate degranulation, chemokine and cytokine secretion in mast cells (53, 54). Our findings would support this pathway as not being necessary since 2-APB, which can activate CRAC channels at concentrations of less than 10 μM or inhibit at more than 50 μM (55), did not alter IL-33 expression at the 50μM dose upon IgE/DNP activation. Indeed, 2APB has been shown to exert potent effects independently of IP3R (55, 56). However, the lack of effects of 2APB on expression of IL-33 would suggest that these alternative pathways most likely do not participate. Interestingly, in addition to divergence in the critical transcription factors, induction of IL-33 and IL-1β was also divergent at the level of the proximal signaling pathways required, since IL-33 was sensitive to the Sphks inhibitor DMS, while IL-1β was blocked by 2-APB inhibition of the IP3-IP3R pathway.
Furthermore, our findings suggest that induction of IL-33 is dependent on Sphk1 rather than Sphk2. However, some contrasting results for the roles of Sphk1 and Sphk2 in mast cells have been reported which may relate to the approach taken. In BMMCs, Sphk1 has been proposed to regulate antigen-induce calcium mobilization, degranulation, and migration (42); furthermore, Sphk1-deficient mice show less responsiveness in a systemic anaphylactic model (57). In contrast, Sphk2 and not Sphk1 was shown to modulate calcium influx and downstream signaling in fetal liver-derived mast cells (58). In adult mice, it has been suggested that compensatory pathways exist in the SphK single knockouts (42). We observed no differences in IL-33 expression in mast cells derived from bone marrow of either the Sphk1 or Sphk2 knockout (a gift from Dr Juan Rivera, National Institutes of Health) (Supplemental Fig 2), suggesting such compensation may also impact IL-33. However, we believe that the combination of evidence demonstrating the requirement for the Sphks generally and also PI3K and shRNA supports our conclusion that Sphk1 is mostly likely the critical pathway to IL-33 expression.
Our data also demonstrates that NFAT activity is important for IL-33 expression. Using bioinformatics-based analysis, we identified two potential regulatory regions, located upstream of exon 1 and within intron 1, which contained putative NFAT binding sites. Interestingly, while we show both regions support ionomycin-driven transcription, the region upstream of il33 exon 1 had a higher capability to induce luciferase activity compared to the intron 1, suggesting that this region may be a primary regulatory region of IL-33 expression while intron 1 may play some enhancing role. Mast cells are known to express four members of the NFAT family: NFAT1–4 (59) and different NFAT members have been shown to modulate unique mast cell responses (60, 61). Which specific NFAT members and which NFAT binding sites are required for IL-33 expression in mast cells are future areas of focus. However, the inhibition of luciferase activity by INCA-6 and CsA suggests this is the critical pathway regulating inducible IL-33 expression.
In summary, we have elucidated a pathway that regulates IL-33 expression in mast cells (as shown in Fig. 9). Here, our data argues that cross-linking of the IgE receptors activates Sphk1, generates S1P, and induces calcium mobilization. This enhances NFAT activity that binds to regulatory regions of il33 and induces IL-33 expression. S1P autocrine effects are required for this pathway. Importantly, our studies support this pathway being critical for inducible IL-33, such as seen in mast cells upon their activation, but not for the constitutive IL-33 observed in other cells, including fibroblasts and epithelial cells.
Figure 9. Pathway for inducible expression of IL-33 in mast cells.
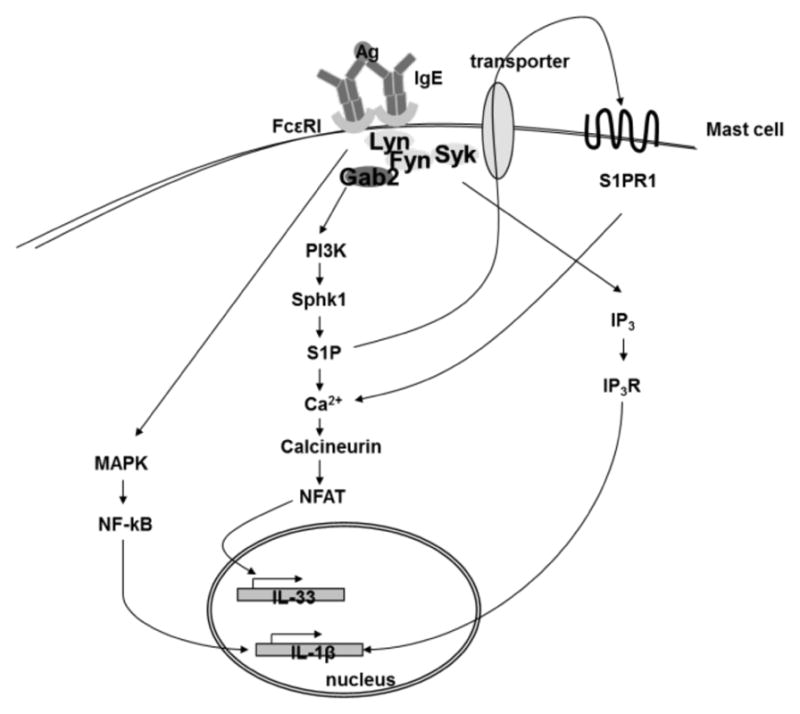
IL-33 expression in mast cells upon cross-linking of IgE receptors is regulated by a PI3K-Sphk1-S1P-NFAT pathway. NF-κB is dispensable for the expression of IL-33 and, instead regulates IL-1β. S1P receptor also participates in regulating IL-33 expression.
Supplementary Material
Acknowledgments
We would like to gratefully acknowledge Drs Joan Cook-Mills, Christian Stehlik and Neil Clipstone for providing reagents and Dr Juan Rivera for mouse bone marrow. We also acknowledge Miller Scientific Communications for editing services.
Footnotes
Supported by NIH/NIAID Grant R56AI092136
References
- 1.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, Brandtzaeg P, Erard M, Haraldsen G, Girard JP. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. 2003;163:69–79. doi: 10.1016/S0002-9440(10)63631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282–7. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179:2551–5. doi: 10.4049/jimmunol.179.4.2551. [DOI] [PubMed] [Google Scholar]
- 5.Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci U S A. 2007;104:18660–5. doi: 10.1073/pnas.0705939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:1526–34. doi: 10.1182/blood-2008-05-157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–30. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 8.Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, Saito H, Galli SJ, Nakae S. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. 2007;87:971–8. doi: 10.1038/labinvest.3700663. [DOI] [PubMed] [Google Scholar]
- 9.Moulin D, Donze O, Talabot-Ayer D, Mezin F, Palmer G, Gabay C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine. 2007;40:216–25. doi: 10.1016/j.cyto.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007;37:2779–86. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- 11.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, Hoshino T, Fujimoto J, Nakanishi K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 12.Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, Komai-Koma M, Pitman N, Li Y, Niedbala W, McKenzie AN, Teixeira MM, Liew FY, Xu D. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 13.Oboki K, Ohno T, Kajiwara N, Saito H, Nakae S. IL-33 and IL-33 receptors in host defense and diseases. Allergol Int. 59:143–60. doi: 10.2332/allergolint.10-RAI-0186. [DOI] [PubMed] [Google Scholar]
- 14.Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol. 2008;180:2443–9. doi: 10.4049/jimmunol.180.4.2443. [DOI] [PubMed] [Google Scholar]
- 15.Matsuba-Kitamura S, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Taki Y, Muto T, Ikeda T, Mimura O, Nakanishi K. Contribution of IL-33 to induction and augmentation of experimental allergic conjunctivitis. Int Immunol. 22:479–89. doi: 10.1093/intimm/dxq035. [DOI] [PubMed] [Google Scholar]
- 16.Pushparaj PN, Tay HK, H’Ng SC, Pitman N, Xu D, McKenzie A, Liew FY, Melendez AJ. The cytokine interleukin-33 mediates anaphylactic shock. Proc Natl Acad Sci U S A. 2009;106:9773–8. doi: 10.1073/pnas.0901206106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Kuchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, De Angelis PM, Scott H, Haraldsen G. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. 2008;173:1229–42. doi: 10.2353/ajpath.2008.080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarstrom C, Loos T, Kasprzycka M, Sorensen DR, Nilsen HR, Kuchler AM, Vatn MH, Haraldsen G. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. 2010;177:2804–15. doi: 10.2353/ajpath.2010.100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:G821–32. doi: 10.1152/ajpgi.00178.2010. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Lu R, Zhao G, Pflugfelder SC, Li DQ. TLR-mediated induction of pro-allergic cytokine IL-33 in ocular mucosal epithelium. Int J Biochem Cell Biol. 2011;43:1383–91. doi: 10.1016/j.biocel.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanagawa Y, Suzuki M, Matsumoto M, Togashi H. Prostaglandin E(2) enhances IL-33 production by dendritic cells. Immunol Lett. 2011;141:55–60. doi: 10.1016/j.imlet.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Nile CJ, Barksby E, Jitprasertwong P, Preshaw PM, Taylor JJ. Expression and regulation of interleukin-33 in human monocytes. Immunology. 2010;130:172–80. doi: 10.1111/j.1365-2567.2009.03221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 2009;284:19420–6. doi: 10.1074/jbc.M901744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohno T, Oboki K, Kajiwara N, Morii E, Aozasa K, Flavell RA, Okumura K, Saito H, Nakae S. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J Immunol. 2009;183:7890–7. doi: 10.4049/jimmunol.0802449. [DOI] [PubMed] [Google Scholar]
- 26.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–3. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One. 5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–4. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 29.Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, Okayama Y, Akira S, Saito H, Galli SJ, Nakae S. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol. 2007;82:1481–90. doi: 10.1189/jlb.0407200. [DOI] [PubMed] [Google Scholar]
- 30.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009;228:149–69. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olivera A. Unraveling the complexities of sphingosine-1-phosphate function: the mast cell model. Prostaglandins Other Lipid Mediat. 2008;86:1–11. doi: 10.1016/j.prostaglandins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivera A, Urtz N, Mizugishi K, Yamashita Y, Gilfillan AM, Furumoto Y, Gu H, Proia RL, Baumruker T, Rivera J. IgE-dependent activation of sphingosine kinases 1 and 2 and secretion of sphingosine 1-phosphate requires Fyn kinase and contributes to mast cell responses. J Biol Chem. 2006;281:2515–25. doi: 10.1074/jbc.M508931200. [DOI] [PubMed] [Google Scholar]
- 34.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neal JW, Clipstone NA. A constitutively active NFATc1 mutant induces a transformed phenotype in 3T3-L1 fibroblasts. J Biol Chem. 2003;278:17246–54. doi: 10.1074/jbc.M300528200. [DOI] [PubMed] [Google Scholar]
- 36.Rivera J, Olivera A. A current understanding of Fc epsilon RI-dependent mast cell activation. Curr Allergy Asthma Rep. 2008;8:14–20. doi: 10.1007/s11882-008-0004-z. [DOI] [PubMed] [Google Scholar]
- 37.Edsall LC, Van Brocklyn JR, Cuvillier O, Kleuser B, Spiegel S. N,N-Dimethylsphingosine is a potent competitive inhibitor of sphingosine kinase but not of protein kinase C: modulation of cellular levels of sphingosine 1-phosphate and ceramide. Biochemistry. 1998;37:12892–8. doi: 10.1021/bi980744d. [DOI] [PubMed] [Google Scholar]
- 38.Michaud J, Kohno M, Proia RL, Hla T. Normal acute and chronic inflammatory responses in sphingosine kinase 1 knockout mice. FEBS Lett. 2006;580:4607–12. doi: 10.1016/j.febslet.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 39.Igarashi Y, Hakomori S, Toyokuni T, Dean B, Fujita S, Sugimoto M, Ogawa T, el-Ghendy K, Racker E. Effect of chemically well-defined sphingosine and its N-methyl derivatives on protein kinase C and src kinase activities. Biochemistry. 1989;28:6796–800. doi: 10.1021/bi00443a002. [DOI] [PubMed] [Google Scholar]
- 40.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–63. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oskeritzian CA, Price MM, Hait NC, Kapitonov D, Falanga YT, Morales JK, Ryan JJ, Milstien S, Spiegel S. Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J Exp Med. 207:465–74. doi: 10.1084/jem.20091513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pushparaj PN, Manikandan J, Tay HK, H’Ng SC, Kumar SD, Pfeilschifter J, Huwiler A, Melendez AJ. Sphingosine kinase 1 is pivotal for Fc epsilon RI-mediated mast cell signaling and functional responses in vitro and in vivo. J Immunol. 2009;183:221–7. doi: 10.4049/jimmunol.0803430. [DOI] [PubMed] [Google Scholar]
- 43.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–6. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 44.Ferrero-Miliani L, Nielsen OH, Andersen PS, Girardin SE. Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1beta generation. Clin Exp Immunol. 2007;147:227–35. doi: 10.1111/j.1365-2249.2006.03261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talabot-Ayer D, Calo N, Vigne S, Lamacchia C, Gabay C, Palmer G. The mouse interleukin (Il)33 gene is expressed in a cell type- and stimulus-dependent manner from two alternative promoters. J Leukoc Biol. 2012;91:119–25. doi: 10.1189/jlb.0811425. [DOI] [PubMed] [Google Scholar]
- 46.Feo S, Antona V, Barbieri G, Passantino R, Cali L, Giallongo A. Transcription of the human beta enolase gene (ENO-3) is regulated by an intronic muscle-specific enhancer that binds myocyte-specific enhancer factor 2 proteins and ubiquitous G-rich-box binding factors. Mol Cell Biol. 1995;15:5991–6002. doi: 10.1128/mcb.15.11.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutton JR, Antonellis A, Carney TJ, Rodrigues FS, Pavan WJ, Ward A, Kelsh RN. An evolutionarily conserved intronic region controls the spatiotemporal expression of the transcription factor Sox10. BMC Dev Biol. 2008;8:105. doi: 10.1186/1471-213X-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JH, Wang LC, Yu HH, Lin YT, Yang YH, Chiang BL. Type I IL-1 receptor (IL-1RI) as potential new therapeutic target for bronchial asthma. Mediators Inflamm. 2010:567351. doi: 10.1155/2010/567351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saidi S, Bouri F, Lencel P, Duplomb L, Baud’huin M, Delplace S, Leterme D, Miellot F, Heymann D, Hardouin P, Palmer G, Magne D. IL-33 is expressed in human osteoblasts, but has no direct effect on bone remodeling. Cytokine. 2011;53:347–54. doi: 10.1016/j.cyto.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 50.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 51.Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol. 2011;41:760–72. doi: 10.1002/eji.201040718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Capite J, Parekh AB. CRAC channels and Ca2+ signaling in mast cells. Immunol Rev. 2009;231:45–58. doi: 10.1111/j.1600-065X.2009.00808.x. [DOI] [PubMed] [Google Scholar]
- 53.Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–8. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 55.Prakriya M, Lewis RS. Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Broad LM, Braun FJ, Lievremont JP, Bird GS, Kurosaki T, Putney JW., Jr Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J Biol Chem. 2001;276:15945–52. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- 57.Olivera A, Eisner C, Kitamura Y, Dillahunt S, Allende L, Tuymetova G, Watford W, Meylan F, Diesner SC, Li L, Schnermann J, Proia RL, Rivera J. Sphingosine kinase 1 and sphingosine-1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. J Clin Invest. 120:1429–40. doi: 10.1172/JCI40659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olivera A, Mizugishi K, Tikhonova A, Ciaccia L, Odom S, Proia RL, Rivera J. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity. 2007;26:287–97. doi: 10.1016/j.immuni.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 59.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–32. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 60.Monticelli S, Solymar DC, Rao A. Role of NFAT proteins in IL13 gene transcription in mast cells. J Biol Chem. 2004;279:36210–8. doi: 10.1074/jbc.M406354200. [DOI] [PubMed] [Google Scholar]
- 61.Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, Tertilt C, Bopp T, Heib V, Becker M, Taube C, Schild H, Schmitt E, Stassen M. Specific and redundant roles for NFAT transcription factors in the expression of mast cell-derived cytokines. J Immunol. 2006;177:6667–74. doi: 10.4049/jimmunol.177.10.6667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.