

Abstract
Autographa californica multiple nucleopolyhedrovirus requires nuclear actin for progeny virus production and thereby encodes viral products that ensure actin’s translocation to and retention within the nucleus. Current evidence suggests that the ie0–ie1 gene complex along with five nuclear localization of actin (NLA) genes are sufficient for NLA in transient transfection experiments. Here we report that, during infection, only one of the five NLA genes, Ac102, was essential for NLA, and that AC102 had at least one other activity critical for budded virus (BV) production. Viral deletion mutants in the other four NLA genes were viable, with only two having replication phenotypes different from that of the wild type. Infection with AcΔpe38 revealed a delay in both BV production and NLA. Infection with AcΔ152 revealed a delay in BV production, but no corresponding delay in NLA. Infection with either AcΔpe38 or AcΔ152 resulted in slightly reduced BV titres. Deletion of Ac004 or he65 had no impact on actin translocation kinetics, timing of BV production or BV titres. These results implicate AC102 as a key player in baculovirus manipulation of actin.
Introduction
Actin, once thought to function exclusively in the cytoplasm, is now known to participate in many nuclear activities including transcription by all three host RNA polymerases (Visa & Percipalle, 2010; Gieni & Hendzel, 2009), chromatin remodelling (Zhao et al., 1998) and gene movement (Chuang et al., 2006; Dundr et al., 2007). Moreover, nuclear filamentous actin (F-actin) is a major requirement for nuclear reprogramming, a process that enables differentiated cells to be reversed to an embryonic state (Miyamoto et al., 2011; Miyamoto & Gurdon, 2011). In this context, it is interesting that members of the genus Alphabaculovirus, including the type species Autographa californica multiple nucleopolyhedrovirus (AcMNPV), absolutely depend on nuclear F-actin for reprogramming their host cell’s nucleus to make it suitable for progeny virus production (Wang et al., 2007; Kasman & Volkman, 2000; Lu et al., 2004; Ohkawa & Volkman, 1999; Volkman, 1988).
During AcMNPV infection, the translocation of monomeric or globular actin (G-actin) into the nucleus and the subsequent polymerization of actin within the nucleus are regulated by two different sets of viral genes (Goley et al., 2006; Ohkawa et al., 2002). Both actin polymerization and F-actin-based viral activities within the nucleus require late gene expression, including the synthesis of minor capsid proteins P78/83, BV/ODV-C42 and VP80 (Charlton & Volkman, 1991; Goley et al., 2006; Ohkawa et al., 2010; Wang et al., 2008; Li et al., 2010; Marek et al., 2011). In contrast, early genes control the initial nuclear accumulation of G-actin. Ohkawa et al. (2002) showed that expression of the AcMNPV ie0–ie1 gene complex along with Ac004, Ac152 and pe38 1 day before expression of Ac102 and he65 induced the nuclear localization of actin (NLA) in transient transfection assays.
The ie0–ie1 gene complex and its products are well-studied and have been referred to as the ‘motor’ of baculovirus infection (Efrose et al., 2010). IE1 and its splice variant IE0 are key regulators of the timing of expression of viral gene products both by transactivation and repression mechanisms (Rohrmann, 2011). Virus replication can occur in the absence of either IE1 or IE0, but not of both (Stewart et al., 2005). IE1 and three other baculovirus proteins form ‘the reticulate structure’ of the virogenic stroma where viral DNA replication takes place (Nagamine et al., 2011). We did not dissect the effects of IE1 and IE0 on nuclear accumulation of actin, but rather focused on the less well-studied NLA gene products. Of these, only HE65 and PE38 have been characterized previously. Functions ascribed to PE38 include acting as an early gene transactivator, a ubiquitin ligase, a factor in DNA replication and an enhancer of IE1-induced apoptosis (Imai et al., 2003; Kool et al., 1994; Lu & Carstens, 1993; Prikhod’ko & Miller, 1999). HE65 is an early gene product with similarity to the RNA ligase 2 nt binding pocket, but RNA ligase activity has not been confirmed experimentally (Becker & Knebel-Mörsdorf, 1993; Ho & Shuman, 2002). The remaining NLA gene products have no known functions, although insertional mutagenesis studies have indicated that Ac102 is essential for virus replication (Lu et al., 1996).
Here, we report on the significance of each NLA gene to the nuclear translocation of actin and budded virus (BV) production during AcMNPV infection in vitro, which varied from ‘absolute’ to ‘some’ to ‘none’. Deletion of Ac102 blocked both NLA and BV production. Notably, NLA using a nuclear-localization signal (NLS) in AcΔ102-transfected cells did not rescue BV production, showing that AC102 was critical for another function beyond NLA. Deletion of Ac152 both delayed BV production and caused a fourfold reduction in titre. The deletion of pe38 resulted in a similar delay in the onset of BV production with an eightfold loss in titre and, additionally, a corresponding delay in the accumulation of actin within the nucleus. Deletion of Ac004 or he65 caused no phenotype. Taken together, our results confirmed that nuclear actin is critical for AcMNPV progeny production and revealed that AC102 plays an essential role in actin translocation to the nucleus.
Results
Ac102 is essential for virus replication
Deletion mutant AcΔ102 was constructed from WOBpos, a bacmid containing the full-length genome of the E2 strain of AcMNPV, as shown in Fig. 1 (Goley et al., 2006). Transfection of AcΔ102 bacmid DNA into Sf9 cells yielded transient cytopathic effects (CPE) at 2–3 days post-transfection (p.t.) in <5 % of cells, indicative of a successful transfection. However, at 4–5 days p.t., no occlusion bodies (OBs) or CPE typically associated with AcMNPV infection were observed, in contrast to WOBpos transfections, which produced OBs in >60 % of cells. Moreover, no transmissible infectivity could be obtained from cells transfected with AcΔ102. Progeny virus production could be rescued, however, by cotransfection of the AcΔ102 bacmid together with a plasmid that expresses Ac102, demonstrating both that Ac102 is an essential gene and that deletion of Ac102 does not affect neighbouring essential genes (data not shown).
Fig. 1.
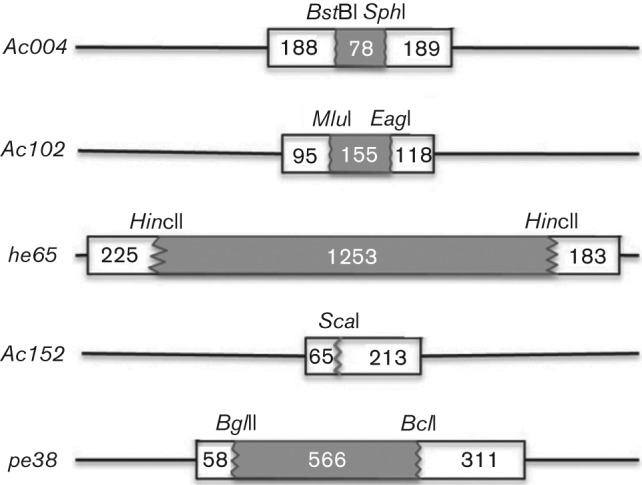
Schematic depiction of NLA deletion mutants. NLA genes were digested with the depicted restriction enzymes, deleting the shaded portion of the gene, and the 5.4 kb Blue-tet gene cluster was inserted to generate the deletion mutants. Numbers represent the base pairs deleted or between the end of the gene and the restriction site. The same restriction sites were then used when generating the FLAG-tagged rescue viruses.
AC102–FLAG and GFP–actin localization
To study AC102, we constructed Ac102-FLAG, a rescue bacmid containing Ac102 with a C-terminal FLAG epitope tag substituting for wild-type Ac102. AC102–FLAG was active in the transfection-based NLA assay (not shown), and transfection of Ac102-FLAG bacmid DNA into Sf9 cells yielded progeny BV. One-step growth curves (repeated three times) comparing parental WOBpos and Ac102-FLAG BV were very similar, revealing that the FLAG-tagged AC102 had wild-type-like activity (Fig. 2a). In localization experiments (also conducted multiple times) where TN-368 cells were infected by Ac102-FLAG BV, AC102-FLAG was localized to the nucleus, specifically to the virogenic stroma, while F-actin was translocated to the nuclear ring zone surrounding the virogenic stroma (Fig. 2b). In contrast to infected cells, in uninfected TN-368 cells cotransfected with plasmids that express Ac102-FLAG and GFP-actin, both AC102–FLAG and GFP–actin were diffuse throughout the cytoplasm (Fig. 2c), confirming the results of Ohkawa et al. (2002).
Fig. 2.
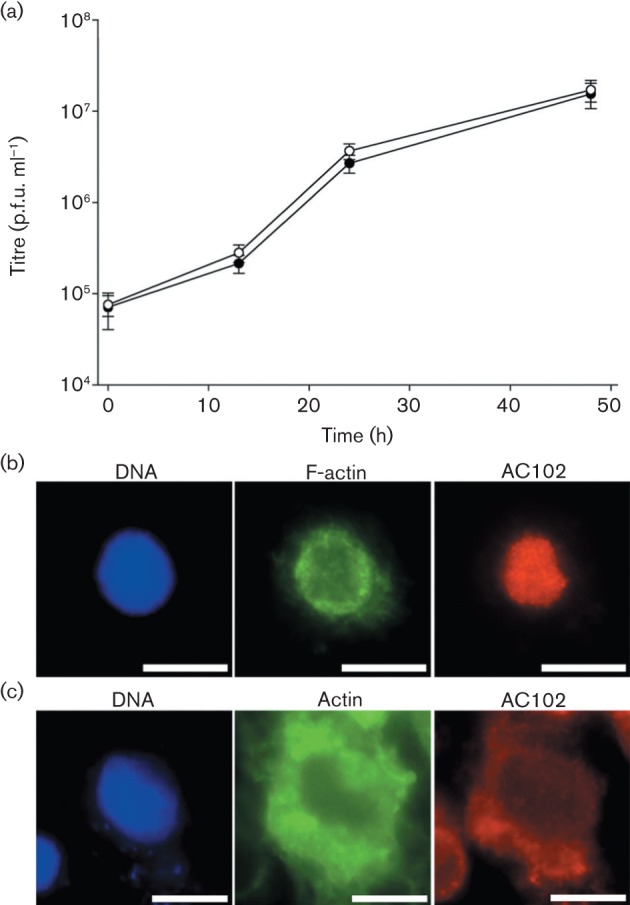
Comparative growth curves of WOBpos and Ac102-FLAG BV and localization of transiently expressed AC102–FLAG. (a) One-step growth curves of WOBpos (•) and Ac102-FLAG (○) in Sf9 cells (m.o.i. = 10). Error bars represent 1 sd. (b) Fluorescence images of AC102–FLAG (red), F-actin (green) and DNA (blue) in an Ac102-FLAG-infected TN-368 cell at 16 h p.i. (c) Fluorescence images of AC102–FLAG (red), actin (green) and DNA (blue) following transient transfection of TN-368 cells by Ac102-FLAG and GFP-actin expression plasmids. Bars, 10 µm.
To determine whether AC102 was necessary for the nuclear localization of G-actin, a recombinant bacmid carrying GFP-actin (AcWOB/GA) was constructed from WOBpos, and an Ac102 deletion bacmid (AcΔ102/GA) was constructed from AcWOB/GA. In contrast to AcWOB/GA, which produced BV that mirrored WOBpos BV in one-step growth curves conducted in triplicate (Fig. 3a), AcΔ102/GA-transfected cells did not produce BV, as expected. Nonetheless, the localization of GFP–actin could be assessed and compared following transfection. When TN-368 cells were transfected with AcWOB/GA and examined 21 h later, GFP–actin was localized to the nuclear ring zone. In contrast, cells transfected with AcΔ102/GA had little to no nuclear GFP–actin (Fig. 3b). These results, consistent in several experiments, demonstrated that AC102 was essential for virus-mediated NLA.
Fig. 3.
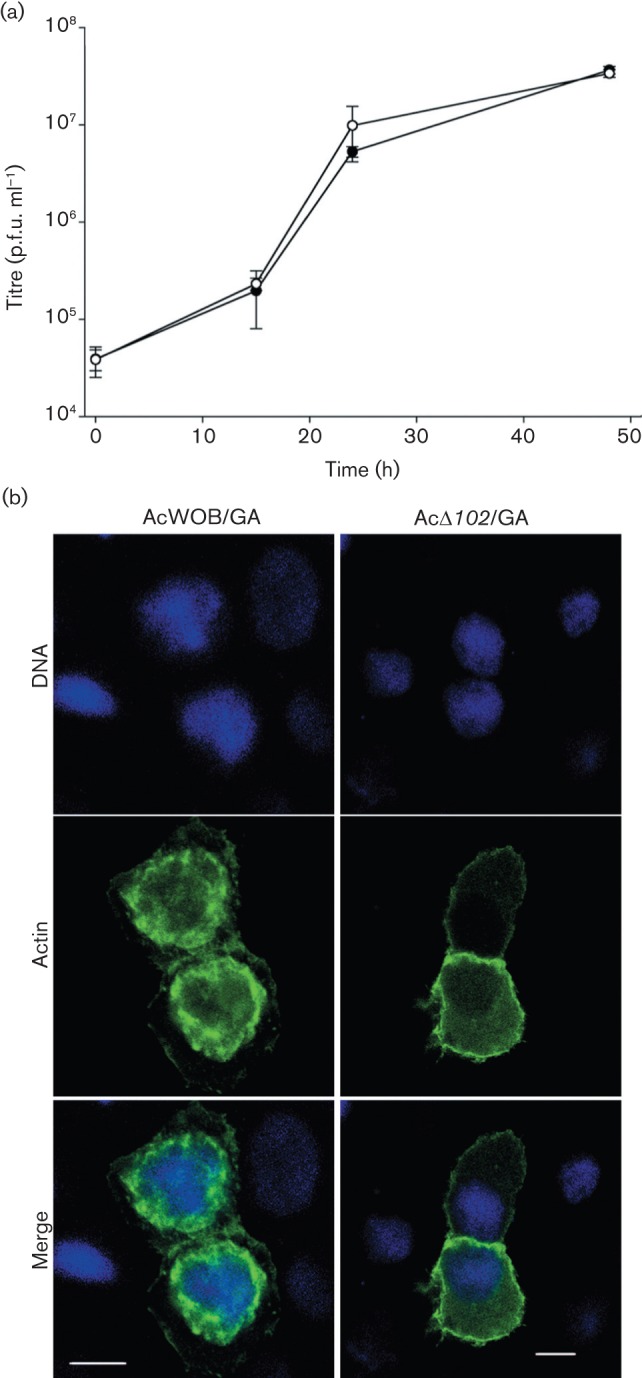
Comparative growth curves of AcWOB/GA (○) and WOBpos (•), and actin localization ± Ac102 expression in transfected cells. (a) One-step growth curves of WOBpos and AcWOB/GA in Sf9 cells (m.o.i. = 10). Error bars represent 1 sd. (b) Confocal fluorescence images of TN-368 cells transfected by AcWOB/GA or AcΔ102/GA bacmid DNA and incubated for 21 h; DNA (blue); GFP–actin (green). Bars, 10 µm.
Given that AC102 did not localize to the nucleus when expressed alone in cells and that the deletion of Ac102 was sufficient to prevent actin translocation into the nucleus, NLSs were added both to Ac102 and to GFP-actin, respectively, to determine whether the direct targeting of AC102 to the nucleus was sufficient to induce actin translocation to the nucleus and whether NLA was the only essential function of AC102. When tested in the NLA assay with the ie0–ie1 gene complex and the other NLA genes, Ac102-NLS-FLAG was active in promoting nuclear G-actin recruitment, demonstrating that the NLS tag did not interfere with AC102 function (data not shown). Moreover, when a plasmid expressing Ac102-NLS-FLAG was transfected into TN-368 cells, AC102 was localized to the nucleus (Fig. 4a). Cotransfection of Ac102-NLS-FLAG- and GFP-actin-expressing plasmids alone, however, did not result in the recruitment of G-actin to the nucleus (not shown). Thus, nuclear AC102 was not sufficient to promote nuclear G-actin accumulation; other viral factors were still required. Similarly, cotransfection of the AcΔ102 bacmid with a plasmid expressing NLS-GFP-actin ectopically producing nuclear GFP–actin did not rescue the ability of AcΔ102 to produce infectious progeny BV (Fig. 4b). These results suggested that AC102 performs other essential roles in addition to its function in promoting nuclear G-actin accumulation.
Fig. 4.
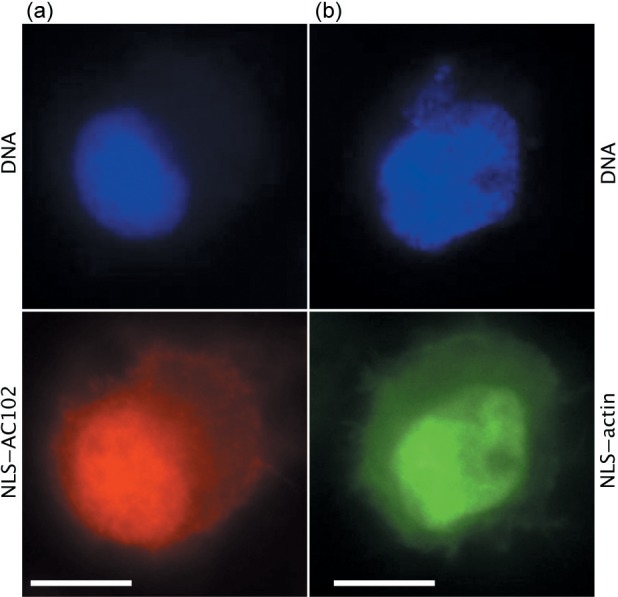
Localization of Ac102 and actin after NLS tagging. Confocal immunofluorescence images of (a) AC102-NLS-FLAG in a TN-368 cell transfected with an Ac102-NLS-FLAG expression plasmid; DNA (blue); FLAG antibody (red); and (b) NLS–GFP–actin in a High Five cell transfected with a plasmid expressing NLS-GFP-actin; DNA (blue); GFP–actin (green). Bars, 10 µm.
PE38 and AC152 affect the kinetics of BV production; only PE38 affects G-actin translocation to the nucleus significantly
To determine the impacts of pe38 and Ac152 on infectivity, deletion mutants AcΔpe38 and AcΔ152 were generated from the WOBpos background. Transfection of each bacmid individually yielded progeny BV. In comparative one-step growth curves repeated three times, AcΔpe38 BV titre was reduced eightfold at 48 h post-infection (p.i.) compared with that of WOBpos, whereas the AcΔ152 BV titre was reduced fourfold (Fig. 5a, b). To determine whether the observed differences in BV titres represented differences in the rates of BV production or differences in the time of onset of the exponential phase of BV production, one-step growth curves were performed in triplicate with hourly analysis of BV titres during the burst window from 15 to 24 h p.i. For WOBpos, the onset of exponential-phase production occurred between 19 and 20 h p.i., when the titre increased abruptly (Fig. 5c). In comparison, exponential-phase production of AcΔpe38- or AcΔ152-infected cells occurred at 23–24 h p.i., a 3–4 h delay (Fig. 5c).
Fig. 5.

Comparative growth curves of WOBpos, AcΔpe38 and AcΔ152, and actin translocation kinetics in infected cells. (a, b) One-step growth curves of WOBpos (•) and (a) AcΔpe38 (○) or (b) AcΔ152 (○) in Sf9 cells (m.o.i. = 10). Error bars represent 1 sd. (c) Comparison of WOBpos (•), AcΔpe38 (▾) and AcΔ152 (○) hourly titres in infected Sf9 cells during the onset of progeny BV production. (d) The percentage of nuclear actin-positive TN-368 cells at 12, 15 and 18 h p.i. by AcWOB/GA (black bars), AcΔpe38/GA (grey bars) or AcΔ152/GA (white bars). *Significant difference between these values and those of AcWOB/GA at a given time point (P<0.01; Student’s t-test).
To determine whether the delay in BV production was presaged by a delay in NLA, pe38 and Ac152 deletion mutants (AcΔpe38/GA and AcΔ152/GA, respectively) were constructed from AcWOB/GA. These GFP–actin-expressing bacmids were used to compare the effects of the missing genes on the localization of actin at 12, 15 and 18 h p.i. in experiments conducted in triplicate. Cell populations infected with AcWOB/GA displayed a measurable increase in the percentage of cells containing nuclear actin: 9 % at 12 h p.i., 39 % at 15 h p.i. and 60 % at 18 h p.i. (Fig. 5d). In comparison, AcΔpe38/GA-infected cells trailed behind and only registered 38.7 % positive for nuclear actin by 18 h p.i., the same percentage as the control virus at 15 h p.i. The AcΔ152/GA-infected cells showed no significant difference from the control in the number of nuclear actin-positive cells at any time point (Fig. 5d).
HE65 and AC004 are dispensable for BV production
Deletion mutants AcΔhe65 and AcΔ004 were generated from WOBpos, and both mutants produced viable progeny that were indistinguishable from WOBpos in the kinetics of BV replication in one-step growth curves (conducted in triplicate), indicating that these genes were completely dispensable for AcMNPV BV production in cell culture (Fig. 6a, b). Additionally, neither deletion mutant had any demonstrable effect on actin translocation into the nucleus (not shown). Nonetheless, both the genes were fused to 3′ FLAG epitope tags to locate their respective products during infection. In cells infected by either Ache65-FLAG or Ac004-FLAG, the gene products were localized to the cytoplasm and remained there throughout infection (Fig. 6c, d).
Fig. 6.
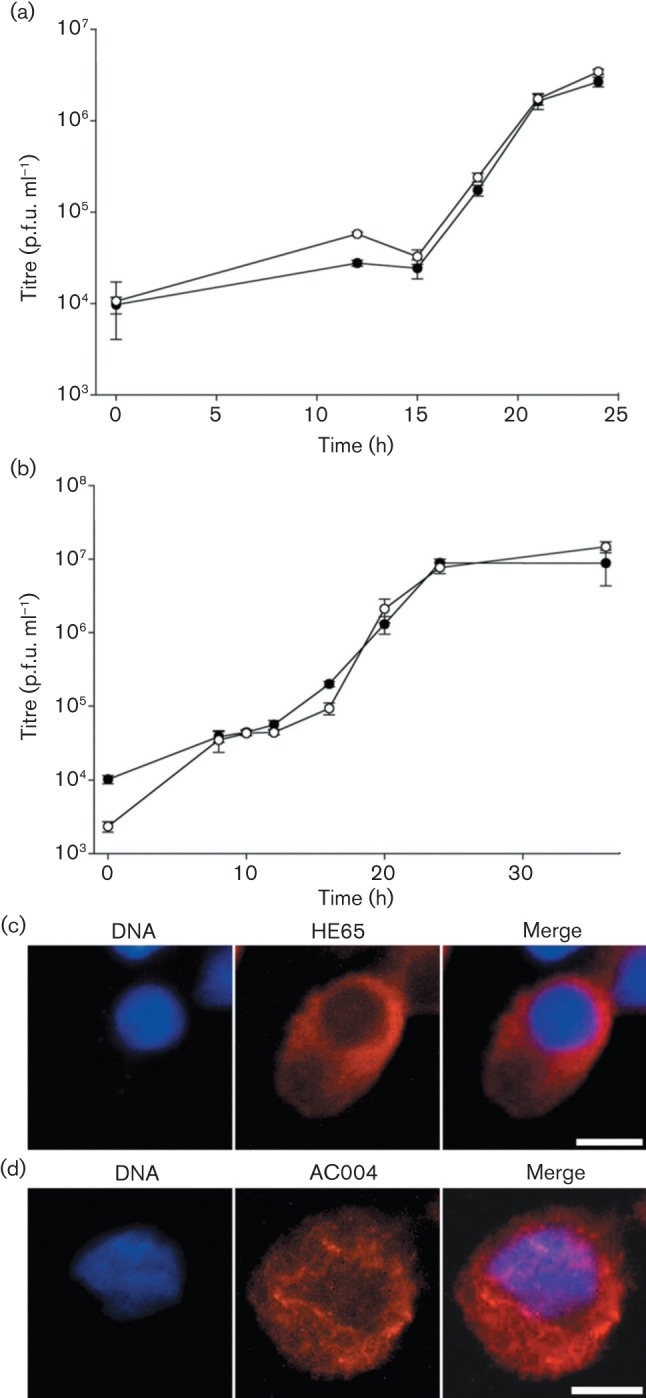
Comparative growth curves of WOBpos, AcΔhe65 and AcΔ004, and localization of HE65 and AC004. (a, b) One-step growth curves of WOBpos (•) and (a) AcΔhe65 (○) or (b) AcΔ004 (○) in Sf9 cells (m.o.i. = 10). Error bars represent 1 sd. Indirect immunofluorescence of (c) HE65–FLAG at 24 h p.i. by Ache65-FLAG and (d) AC004–FLAG at 4 h p.i. by Ac004-FLAG, both in TN-368 cells and detected by FLAG antibody (red), in relation to DNA (blue), with the merged image also shown. Bars, 10 µm.
Discussion
Translocation of host actin from the cytoplasm to the nucleus is fundamental to AcMNPV’s infection strategy. While Ohkawa et al. (2002) reported that the ie0–ie1 gene complex and five additional AcMNPV early genes were needed to translocate actin to the nucleus in transient transfection experiments, we found that only one of the five, Ac102, was essential for this activity during infection. Curiously, while Ac102 could be replaced by he65 in transient transfection assays (Ohkawa et al., 2002), Ac102 was essential for infection, whereas he65 was not. Actin can be induced to localize to the nucleus by the addition of an N-terminal NLS, where it is capable of functioning normally (Hofmann et al., 2009; Posern et al., 2002). We found, however, that when nuclear actin was supplied in AcΔ102-transfected cells by ectopic expression of NLS–actin, BV production was not rescued. These results demonstrated that Ac102 had a second critical function in addition to mediating actin translocation to the nucleus, which explains the discrepancy in transfection and infection results.
Of the 46 currently available complete, annotated lepidopteran baculovirus genomes, only one, Leucania separata multiple nucleopolyhedrovirus (LsMNPV), does not contain a clearly identifiable Ac102 homologue. LsMNPV does contain clearly identifiable p78/83 and arif-1 (actin rearrangement-inducing factor 1) genes, however, suggesting that, even in the absence of an Ac102 homologue, actin may still play an important role in LsMNPV biology. As for Ac102, the gene is part of a conserved, essential region of the genome (Lu et al., 1996), and evidence suggests that the Ac102 gene product becomes incorporated into occlusion-derived virus (ODV) but, curiously, not BV (Braunagel et al., 2003; Wang et al., 2010). There are no non-baculovirus homologues or recognizable motifs to provide clues to its function.
Our results concerning deletion of pe38 confirmed and extended those of Milks et al. (2003). We confirmed the 4 h delay in the onset of AcMNPV BV production and the eightfold reduction in BV titre. Additionally, we recorded a 3–4 h delay in the nuclear accumulation of actin in AcΔpe38-infected cells, which could account for the 4 h lag in BV production, given that BV production requires nuclear actin (Ohkawa & Volkman, 1999). The deletion of pe38 leads to a delay in the expression of several viral genes, including some NLA genes (Jiang et al., 2006). However, PE38 is required for nuclear G-actin accumulation in transient transfection assays even when all NLA genes are under the control of constitutive promoters, showing that PE38’s NLA role involves an activity other than controlling the expression of other NLA genes (Ohkawa et al., 2002). One possible NLA function of PE38 could involve its ubiquitin ligase activity (Imai et al., 2003). For example, PE38 could ubiquitinate the actin-specific nuclear export protein exportin-6, ultimately facilitating the nuclear accumulation of actin (Stüven et al., 2003). Another possibility involves PE38’s role in enhancing IE1-induced apoptosis (Prikhod’ko & Miller, 1999), a stress that could lead to the accumulation of actin within the nucleus (Luchetti et al., 2002), even though apoptosis is ultimately blocked (Clem, 2007).
Deletion of Ac152 led to a 4 h delay in the onset of BV production and a fourfold reduction in virus titre overall. There was a suggestion of a corresponding delay in nuclear actin accumulation at 15 h p.i., but the putative lag was not statistically significant (see Fig. 5d). More experimentation is needed to determine whether actin accumulation is delayed between 12 and 15 h in AcΔ152-infected cells.
Neither the kinetics of nuclear actin translocation nor BV production were altered by the deletion of he65 or Ac004. Moreover, HE65 and AC004 both remained in the cytoplasm throughout infection (although there is a possibility that the FLAG tags interfered with proper localization). Regardless, under our experimental conditions, the functions of these two proteins were either not essential or not unique.
Studies of the many roles of nuclear actin are now becoming mainstream and nuclear actin has gained new respect as a scientific topic. Understanding how baculoviruses accumulate and deploy nuclear actin for their various viral purposes could complement and provide context for these studies. Whilst the exact mechanism remains to be determined, our studies identified AC102 as a major player in the transport and accumulation of actin during infection.
Methods
Cell culture.
Growth curves were performed using Spodoptera frugiperda Sf9 cells, whereas TN-368 or High Five cells were used for microscopy, as has been described previously (Ohkawa et al., 2002). Sf9 cells were cultured at 27 °C in Grace’s insect medium (JRH Biosciences) supplemented with 10 % FBS (Gibco-BRL). TN-368 and High Five cells were cultured at 27 °C in Grace’s insect medium supplemented with 10 % FBS, lactalbumin (Gibco-BRL) and yeastolate hydrolysate (Gibco-BRL). Medium for cells grown in suspension culture was further supplemented with 0.1 % pluronic F-68 (Sigma).
WOBpos and derivatives.
WOBpos is an E2 strain AcMNPV bacmid that contains a kanamycin-resistance cassette (Goley et al., 2006). All constructs used in this study were derived from WOBpos and were confirmed by restriction analysis and DNA sequencing. Bacmid DNA was isolated from bacteria using a Qiagen Large-Construct maxiprep kit. To isolate mutant viruses, bacmid DNA was transfected into Sf9 cells using Cellfectin (Invitrogen). Cells were plated in a 35 mm dish and incubated with 2 µg bacmid DNA pre-mixed with Cellfectin reagent for 30 min. After a 5 h incubation, the transfection medium was replaced with fresh medium. Cells were inspected at 2–3 days p.t. for the presence of occlusions to determine the transfection efficiency, and again at 4–5 days p.t. for evidence of progeny production (e.g. an increase in the percentage of occlusion-positive cells). The culture medium was collected and progeny virus was amplified in 1×107 Sf9 cells.
Mutant virus construction.
NLA deletion plasmids were constructed by inserting a lacZ reporter gene and a tetracycline-resistance cassette (Blue-tet; 5.4 kb combined) into the central portion of the gene of interest. These deletion plasmids were linearized and electroporated into Escherichia coli BW25113/pKD46 electrocompetent cells along with WOBpos DNA. Positive mutant bacmids were generated by homologous recombination and isolated by plating transformed E. coli on medium containing kanamycin and tetracycline. Specifically, as depicted in Fig. 1, to generate an Ac102 deletion plasmid, the KpnI E fragment of AcMNPV in pBSKS+ (Stratagene) was digested with EagI and MluI, blunted with Klenow and ligated to Blue-tet, deleting 41 % of the coding region. For AcΔpe38, the SphI J fragment in pUC18 (Roche) was subcloned to remove a BglII site, then digested with BclI and BglII, blunted and ligated to Blue-tet, deleting 61 % of the coding region. Because of the small size of Ac152, a single cut with ScaI was made in the SphI J fragment of AcMNPV in pUC18, and Blue-tet was ligated to these blunt ends. To delete Ac004, an approximately 3 kb fragment from EcoRI to SalI of the HindIII F fragment of AcMNPV was cloned into pBSKS+, then subcloned to remove a BstBI site. The resulting plasmid was digested with SphI and BstBI, blunted and ligated to Blue-tet, deleting the central 17 % of Ac004. To generate AcΔhe65, he65 ± approximately 200 bp up- and downstream was removed from the KpnI D fragment of AcMNPV in pBSKS+ by EcoRI digestion. The resulting fragment cloned into pBSKS+ was digested with HincII, blunted, and ligated to Blue-tet, deleting 76 % of the coding region.
Due to the close proximity of both Ac152 and pe38 to known transactivator ie-2, ie-2 expression was tested by quantitative RT-PCR and found to be the same in cells infected by WOBpos, AcΔpe38 or AcΔ152 (data not shown).
AcWOB/GA construction.
The eGFP Bombyx mori A4 actin gene was removed from a modified Clontech vector (Ohkawa et al., 2002) by digestion with NheI and XbaI and inserted into pIE1 T.V.3 (Jarvis et al., 1996) that had been digested with XbaI, putting GFP-actin under control of the AcMNPV ie1 promoter and the Bombyx mori nucleopolyhedrovirus (BmNPV) hr5 enhancer. The fragment containing the hr5 enhancer sequence, ie1 promoter and GFP-actin gene was subsequently removed from pIE1 T.V.3 by BlpI digestion, then blunted with Klenow, digested by SpeI and ligated to pWOBGent3 (Ohkawa et al., 2010) that had been cut with SmaI and XbaI. AcWOB/GA was generated by transforming electrocompetent E. coli with WOBpos bacmid DNA and the linearized plasmid ie1GA/WOBGent3, and selecting for recombinants on medium that contained kanamycin and gentamicin.
Growth curves.
One-step growth curves were performed by incubating 2×106 Sf9 cells in 6 cm dishes for 45 min, removing the medium and infecting the cells with BV at an m.o.i. of 10. After 1 h, the viral inoculum was removed, cells were rinsed twice in Grace’s medium and 4 ml fresh medium was added. Cells were incubated at 27 °C and, at specified time points, 30 µl medium was collected from each dish. Virus titres were determined using immunoplaque assays, as described previously (Volkman & Goldsmith, 1981).
FLAG and NLS tagging.
For localization studies, 3′ FLAG-tagged fusions were constructed for Ac004, Ac102 and he65 using the pFLAG-CMV-5a vector (Sigma). pFLAG-CMV-5a was modified by adding a chloramphenicol-resistance cassette (CAT) (from pKD3; GenBank accession no. AY048742) into the blunted BsgI restriction site in the hGH poly(A) region to generate pFLAG-TA.CAT. Each NLA gene was PCR-amplified with primers containing a 5′ BamHI site and a 3′ KpnI site to allow insertion into pFLAG-TA.CAT in frame with the FLAG tag. This resulted in NLA-FLAG genes followed by a CAT cassette 200 bp downstream. To make rescue plasmids, the NLA-FLAG gene and CAT cassette were removed from pFLAG-TA.CAT by digesting with BlpI (downstream of the CAT cassette), blunting and digesting with the upstream restriction enzyme used to make the deletion plasmids (e.g. EagI for Ac102). The NLA gene was then digested from the appropriate AcMNPV fragment (described above) with the downstream site used to make the deletion plasmid, blunted and then digested at the upstream restriction site used to make the deletion plasmid, allowing for the in-frame ligation of the NLA-FLAG gene/CAT cassette. This resulted in a rescue plasmid that contained the NLA-FLAG gene, followed by the CAT cassette, followed by a short repeat of the 3′ end of the NLA gene. The rescue plasmids were electroporated into E. coli along with DNA from the deletion bacmid to generate FLAG-tagged rescue bacmids with the FLAG fusions being expressed behind the native viral gene promoters.
For transfection assays, each NLA-FLAG gene was removed from the FLAG vector and inserted into pACThr3 behind a B. mori actin promoter preceded by a BmNPV hr3 enhancer sequence (from Ohkawa et al., 2002). For Ac004-FLAG, the gene was removed from pFLAG-CMV-5a by BstXI digestion, blunting and BamHI digestion, and ligated into pACThr3 that had been digested with XbaI, blunted and digested with BamHI. The remaining NLA-FLAG genes were removed from pFLAG-CMV-5a with AvaI and BamHI digestion and ligated into pACThr3 digested with XhoI and BamHI. For the addition of an NLS to Ac102, an oligonucleotide containing an SV40 NLS was generated by annealing single-stranded oligomers 5′-GATCCGGTACCCCCAAGAAGAAGCGTAAGGTGGAGGACCGTAC-3′ and 3′-GCCATGGGGGTTCTTCTTCGCATTCCACCTCCTGG-5′, which creates an NLS with BamHI- and KpnI-compatible overhangs. This oligo was cloned into pFLAG-TA.CAT (which had been digested with BamHI and KpnI). The resulting plasmid could be cut by BamHI and KpnI, and ligated to Ac102 (which was amplified with primers containing a 5′ BamHI site and 3′ KpnI site, as above). The resulting Ac102-NLS-FLAG gene was removed from this plasmid by BbsI digestion, blunting and BamHI digestion, and ligated to pIE1/153A (Lu et al., 1997), which had been digested with NotI, blunted and digested with BamHI.
N-terminal NLS additions to actin have previously been demonstrated to move actin to the nucleus while still allowing actin to assemble into the cytoskeleton (Hofmann et al., 2009; Posern et al., 2002). To generate actin with an NLS tag, pACThr3/GA (from Ohkawa et al., 2002) was digested with BamHI and NcoI, and ligated to an oligonucleotide generated by annealing single-stranded oligomers 5′-GATCCATGGGTGGTGGTCCCAAGAAGAAGCGTAAGGTGGAGGAC-3′ and 3′-GTACCCACCACCAGGGTTCTTCTTCGCATTCCACCTCCTGGTAC-5′. When expressed, this construct generates a methionine–triple glycine–NLS (MGGGPKKKRKVED) in frame with GFP–actin in pACThr3.
Fluorescence microscopy
The NLA transfection assay.
TN-368 cells (4×104) were applied to a 22×22 mm coverslip and allowed to settle for 30 min. At day 0, cells were transfected with Ac004, Ac152 and pe38 expressed from pIE1/153A (5 µg of each plasmid) along with 0.5 µg pIE1actGA (Ohkawa et al., 2002), using Cellfectin reagent as per the manufacturer’s protocols. At day 1 p.t., control and experimental cells were transfected with 5 µg pIE1/153A expressing Ac102 or he65. Cells were fixed, stained and evaluated for the presence of nuclear actin on a Zeiss Axiophot photomicroscope on day 3 p.t. All FLAG- and NLS-tagged NLA gene products were tested for function in this assay.
NLA localization.
To determine the localization of NLA gene products, 2×105 TN-368 cells were plated on a 22×22 mm coverslip, allowed to attach for 45 min, the medium removed then inoculated with AcNLA-FLAG BV at an m.o.i. of 10. After a 1 h adsorption period, the inoculum was removed and fresh medium was added. The coverslips were placed at 27 °C for the designated times p.i., then fixed in 2 % paraformaldehyde fixative in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH 6.9) for 10 min, rinsed in PHEM for 5 min, solubilized in 0.15 % Triton X-100 in PHEM for 10 min, rinsed twice in PHEM for 5 min, incubated in normal goat serum in PHEM for 5 min then in mouse M2 anti-FLAG antibody (1 : 200 in PHEM; Sigma) for 25 min, stained with DAPI (1 µg ml−1 in PHEM; Sigma) for 30 s, rinsed twice in PHEM for 5 min, stained with TRITC-conjugated goat anti-mouse antibody (1 : 100 in PHEM; Zymed) and, when necessary, FITC–phalloidin (1 : 100 in PHEM; Sigma) for 45 min, rinsed twice in PHEM for 5 min and then mounted onto glass slides in ProLong antifade mounting medium (Molecular Probes). Coverslips were viewed on a Zeiss Axiophot photomicroscope equipped for fluorescence microscopy.
To determine the localization of AC102 in the absence of other viral genes, 1×105 TN-368 cells were applied to a 22×22 mm coverslip and allowed to attach for 45 min, then transfected with 5 µg Ac102-FLAG or Ac102-NLS-FLAG in pIE1/153A, with or without 0.5 µg pIE1actGA or pACThr3/NLS-GA, pre-mixed for 10 min in Cellfectin reagent. After a 5 h incubation period, fresh medium was added and cells were allowed to incubate for 2 days then fixed and stained as described above. Coverslips were viewed on a Zeiss Axiophot photomicroscope equipped for fluorescence microscopy.
Actin localization.
To test for NLA in AcWOB/GA- or AcΔ102/GA-transfected cells, 1×105 TN-368 cells were plated on a 22×22 mm coverslip and allowed to attach for 45 min, then transfected with bacmid DNA (2 µg) that had been pre-mixed with Cellfectin reagent for 30 min at 27 °C. After a 5 h incubation, fresh medium was added and cells were incubated for 21 h at 27 °C. The coverslips were then immersed in 2 % paraformaldehyde fixative in PHEM buffer for 10 min, rinsed in PHEM for 5 min, stained with 1 µg Hoechst dye ml−1 for 15 min, rinsed twice in PHEM for 5 min, and then mounted onto glass slides in ProLong antifade mounting medium. Coverslips were viewed on a Zeiss confocal laser-scanning microscope.
To determine the localization of actin during the course of infection, 1.75×105 TN-368 cells were applied to 22×22 mm coverslips and allowed to attach for 45 min, then inoculated with AcWOB/GA, AcΔpe38/GA or AcΔ152/GA at an m.o.i. of 10. The viral inoculum was removed after 1 h and fresh medium was added. The cells were incubated at 27 °C and processed at 12, 15 or 18 h p.i. Coverslips were fixed in 2 % paraformaldehyde in PHEM for 10 min, washed in PHEM for 5 min, stained with Hoechst dye (1 µg ml−1) for 15 min, washed twice in PHEM for 5 min, and mounted onto glass slides with ProLong antifade mounting medium. For statistical randomization, coverslips were marked with three horizontal and three vertical lines (generating nine points of intersection), and images were captured at these intersection points on a Zeiss Axiophot photomicroscope equipped for fluorescence microscopy. Images were scored for the total number of cells (based on the number of nuclei visible), and for the number of nuclear actin-positive cells. With a mean of 25–30 cells per field of view, nine images per slide and triplicate repeats, approximately 700–800 cells were scored for each virus at each time point.
Acknowledgements
The authors wish to thank Annette Rowe, Jadyn Damon, Vinh Dao, Helen Hwang and Kavita Rangan for their valuable contributions to this study. The authors also wish to thank Steve Ruzin and Denise Schichnes at the UC Berkeley Biological Imaging Facility for technical assistance. Financial support was provided by federal HATCH funds, Regional Research funds, various independent donors to L. E. V., and NIH/NIGMS grant R01 GM059609 to M. D. W.
References
- Becker D., Knebel-Mörsdorf D. (1993). Sequence and temporal appearance of the early transcribed baculovirus gene HE65. J Virol 67, 5867–5872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunagel S. C., Russell W. K., Rosas-Acosta G., Russell D. H., Summers M. D. (2003). Determination of the protein composition of the occlusion-derived virus of Autographa californica nucleopolyhedrovirus. Proc Natl Acad Sci U S A 100, 9797–9802 10.1073/pnas.1733972100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton C. A., Volkman L. E. (1991). Sequential rearrangement and nuclear polymerization of actin in baculovirus-infected Spodoptera frugiperda cells. J Virol 65, 1219–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C. H., Carpenter A. E., Fuchsova B., Johnson T., de Lanerolle P., Belmont A. S. (2006). Long-range directional movement of an interphase chromosome site. Curr Biol 16, 825–831 10.1016/j.cub.2006.03.059 [DOI] [PubMed] [Google Scholar]
- Clem R. J. (2007). Baculoviruses and apoptosis: a diversity of genes and responses. Curr Drug Targets 8, 1069–1074 10.2174/138945007782151405 [DOI] [PubMed] [Google Scholar]
- Dundr M., Ospina J. K., Sung M. H., John S., Upender M., Ried T., Hager G. L., Matera A. G. (2007). Actin-dependent intranuclear repositioning of an active gene locus in vivo. J Cell Biol 179, 1095–1103 10.1083/jcb.200710058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efrose R., Swevers L., Iatrou K. (2010). Baculoviruses deficient in ie1 gene function abrogate viral gene expression in transduced mammalian cells. Virology 406, 293–301 10.1016/j.virol.2010.07.020 [DOI] [PubMed] [Google Scholar]
- Gieni R. S., Hendzel M. J. (2009). Actin dynamics and functions in the interphase nucleus: moving toward an understanding of nuclear polymeric actin. Biochem Cell Biol 87, 283–306 10.1139/O08-133 [DOI] [PubMed] [Google Scholar]
- Goley E. D., Ohkawa T., Mancuso J., Woodruff J. B., D’Alessio J. A., Cande W. Z., Volkman L. E., Welch M. D. (2006). Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science 314, 464–467 10.1126/science.1133348 [DOI] [PubMed] [Google Scholar]
- Ho C. K., Shuman S. (2002). Bacteriophage T4 RNA ligase 2 (gp24.1) exemplifies a family of RNA ligases found in all phylogenetic domains. Proc Natl Acad Sci U S A 99, 12709–12714 10.1073/pnas.192184699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann W. A., Arduini A., Nicol S. M., Camacho C. J., Lessard J. L., Fuller-Pace F. V., de Lanerolle P. (2009). SUMOylation of nuclear actin. J Cell Biol 186, 193–200 10.1083/jcb.200905016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai N., Matsuda N., Tanaka K., Nakano A., Matsumoto S., Kang W. (2003). Ubiquitin ligase activities of Bombyx mori nucleopolyhedrovirus RING finger proteins. J Virol 77, 923–930 10.1128/JVI.77.2.923-930.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis D. L., Weinkauf C., Guarino L. A. (1996). Immediate-early baculovirus vectors for foreign gene expression in transformed or infected insect cells. Protein Expr Purif 8, 191–203 10.1006/prep.1996.0092 [DOI] [PubMed] [Google Scholar]
- Jiang S. S., Chang I. S., Huang L. W., Chen P. C., Wen C. C., Liu S. C., Chien L. C., Lin C. Y., Hsiung C. A., Juang J. L. (2006). Temporal transcription program of recombinant Autographa californica multiple nucleopolyhedrosis virus. J Virol 80, 8989–8999 10.1128/JVI.01158-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasman L. M., Volkman L. E. (2000). Filamentous actin is required for lepidopteran nucleopolyhedrovirus progeny production. J Gen Virol 81, 1881–1888 [DOI] [PubMed] [Google Scholar]
- Kool M., Ahrens C. H., Goldbach R. W., Rohrmann G. F., Vlak J. M. (1994). Identification of genes involved in DNA replication of the Autographa californica baculovirus. Proc Natl Acad Sci U S A 91, 11212–11216 10.1073/pnas.91.23.11212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Wang Y., Bai H., Wang Q., Song J., Zhou Y., Wu C., Chen X. (2010). The putative pocket protein binding site of Autographa californica nucleopolyhedrovirus BV/ODV-C42 is required for virus-induced nuclear actin polymerization. J Virol 84, 7857–7868 10.1128/JVI.00174-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A., Carstens E. B. (1993). Immediate-early baculovirus genes transactivate the p143 gene promoter of Autographa californica nuclear polyhedrosis virus. Virology 195, 710–718 10.1006/viro.1993.1422 [DOI] [PubMed] [Google Scholar]
- Lu A., Craig A., Casselman R., Carstens E. B. (1996). Nucleotide sequence, insertional mutagenesis, and transcriptional mapping of a conserved region of the baculovirus Autographa californica nuclear polyhedrosis virus (map unit 64.8–66.9). Can J Microbiol 42, 1267–1273 10.1139/m96-165 [DOI] [PubMed] [Google Scholar]
- Lu M., Farrell P. J., Johnson R., Iatrou K. (1997). A baculovirus (Bombyx mori nuclear polyhedrosis virus) repeat element functions as a powerful constitutive enhancer in transfected insect cells. J Biol Chem 272, 30724–30728 10.1074/jbc.272.49.30724 [DOI] [PubMed] [Google Scholar]
- Lu S., Ge G., Qi Y. (2004). Ha-VP39 binding to actin and the influence of F-actin on assembly of progeny virions. Arch Virol 149, 2187–2198 10.1007/s00705-004-0361-4 [DOI] [PubMed] [Google Scholar]
- Luchetti F., Burattini S., Ferri P., Papa S., Falcieri E. (2002). Actin involvement in apoptotic chromatin changes of hemopoietic cells undergoing hyperthermia. Apoptosis 7, 143–152 10.1023/A:1014362415047 [DOI] [PubMed] [Google Scholar]
- Marek M., Merten O. W., Galibert L., Vlak J. M., van Oers M. M. (2011). Baculovirus VP80 protein and the F-actin cytoskeleton interact and connect the viral replication factory with the nuclear periphery. J Virol 85, 5350–5362 10.1128/JVI.00035-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milks M. L., Washburn J. O., Willis L. G., Volkman L. E., Theilmann D. A. (2003). Deletion of pe38 attenuates AcMNPV genome replication, budded virus production, and virulence in Heliothis virescens. Virology 310, 224–234 10.1016/S0042-6822(03)00143-0 [DOI] [PubMed] [Google Scholar]
- Miyamoto K., Gurdon J. B. (2011). Nuclear actin and transcriptional activation. Commun Integr Biol 4, 582–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K., Pasque V., Jullien J., Gurdon J. B. (2011). Nuclear actin polymerization is required for transcriptional reprogramming of Oct4 by oocytes. Genes Dev 25, 946–958 10.1101/gad.615211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamine T., Abe A., Suzuki T., Dohmae N., Matsumoto S. (2011). Co-expression of four baculovirus proteins, IE1, LEF3, P143, and PP31, elicits a cellular chromatin-containing reticulate structure in the nuclei of uninfected cells. Virology 417, 188–195 10.1016/j.virol.2011.06.001 [DOI] [PubMed] [Google Scholar]
- Ohkawa T., Volkman L. E. (1999). Nuclear F-actin is required for AcMNPV nucleocapsid morphogenesis. Virology 264, 1–4 10.1006/viro.1999.0008 [DOI] [PubMed] [Google Scholar]
- Ohkawa T., Rowe A. R., Volkman L. E. (2002). Identification of six Autographa californica multicapsid nucleopolyhedrovirus early genes that mediate nuclear localization of G-actin. J Virol 76, 12281–12289 10.1128/JVI.76.23.12281-12289.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa T., Volkman L. E., Welch M. D. (2010). Actin-based motility drives baculovirus transit to the nucleus and cell surface. J Cell Biol 190, 187–195 10.1083/jcb.201001162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G., Sotiropoulos A., Treisman R. (2002). Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol Biol Cell 13, 4167–4178 10.1091/mbc.02-05-0068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prikhod’ko E. A., Miller L. K. (1999). The baculovirus PE38 protein augments apoptosis induced by transactivator IE1. J Virol 73, 6691–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmann G. F. (2011). Baculovirus Molecular Biology, 2nd edn Bethesda, MD: National Library of Medicine (US), National Center for Biotechnology Information [Google Scholar]
- Stewart T. M., Huijskens I., Willis L. G., Theilmann D. A. (2005). The Autographa californica multiple nucleopolyhedrovirus ie0-ie1 gene complex is essential for wild-type virus replication, but either IE0 or IE1 can support virus growth. J Virol 79, 4619–4629 10.1128/JVI.79.8.4619-4629.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stüven T., Hartmann E., Görlich D. (2003). Exportin 6: a novel nuclear export receptor that is specific for profilin·actin complexes. EMBO J 22, 5928–5940 10.1093/emboj/cdg565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visa N., Percipalle P. (2010). Nuclear functions of actin. Cold Spring Harb Perspect Biol 2, a000620 10.1101/cshperspect.a000620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman L. E. (1988). Autographa californica MNPV nucleocapsid assembly: inhibition by cytochalasin D. Virology 163, 547–553 10.1016/0042-6822(88)90295-4 [DOI] [PubMed] [Google Scholar]
- Volkman L. E., Goldsmith P. A. (1981). Baculovirus bioassay not dependent upon polyhedra production. J Gen Virol 56, 203–206 10.1099/0022-1317-56-1-203 [DOI] [PubMed] [Google Scholar]
- Wang Q., Liang C., Song J., Chen X. (2007). HA2 from the Helicoverpa armigera nucleopolyhedrovirus: a WASP-related protein that activates Arp2/3-induced actin filament formation. Virus Res 127, 81–87 10.1016/j.virusres.2007.03.021 [DOI] [PubMed] [Google Scholar]
- Wang Y., Wang Q., Liang C., Song J., Li N., Shi H., Chen X. (2008). Autographa californica multiple nucleopolyhedrovirus nucleocapsid protein BV/ODV-C42 mediates the nuclear entry of P78/83. J Virol 82, 4554–4561 10.1128/JVI.02510-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Deng F., Hou D., Zhao Y., Guo L., Wang H., Hu Z. (2010). Proteomics of the Autographa californica nucleopolyhedrovirus budded virions. J Virol 84, 7233–7242 10.1128/JVI.00040-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K., Wang W., Rando O. J., Xue Y., Swiderek K., Kuo A., Crabtree G. R. (1998). Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 95, 625–636 10.1016/S0092-8674(00)81633-5 [DOI] [PubMed] [Google Scholar]