

Abstract
Current vaccine approaches to combat anthrax are effective; however, they target only a single protein [the protective antigen (PA) toxin component] that is produced after spore germination. PA production is subsequently increased during later vegetative cell proliferation. Accordingly, several aspects of the vaccine strategy could be improved. The inclusion of spore-specific antigens with PA could potentially induce protection to initial stages of the disease. Moreover, adding other epitopes to the current vaccine strategy will decrease the likelihood of encountering a strain of Bacillus anthracis (emerging or engineered) that is refractory to the vaccine. Adding recombinant spore-surface antigens (e.g. BclA, ExsFA/BxpB and p5303) to PA has been shown to augment protection afforded by the latter using a challenge model employing immunosuppressed mice challenged with spores derived from the attenuated Sterne strain of B. anthracis. This report demonstrated similar augmentation utilizing guinea pigs or mice challenged with spores of the fully virulent Ames strain or a non-toxigenic but encapsulated ΔAmes strain of B. anthracis, respectively. Additionally, it was shown that immune interference did not occur if optimal amounts of antigen were administered. By administering the toxin and spore-based immunogens simultaneously, a significant adjuvant effect was also observed in some cases. Thus, these data further support the inclusion of recombinant spore antigens in next-generation anthrax vaccine strategies.
Introduction
Anthrax is an acute infection caused by the Gram-positive, spore-forming bacterium Bacillus anthracis, and is primarily a zoonotic disease. The natural route of infection leading to anthrax is ingestion of B. anthracis spores by animals (Turnbull, 2008). Human disease is generally manifested in one of three major forms, cutaneous, gastrointestinal or inhalational. More than 95 % of reported human cases worldwide are cutaneous infections. Whilst cutaneous anthrax can resolve without antibiotic intervention, as many as 20 % of untreated cases may be fatal (Turnbull, 2008). The next most common form of anthrax observed in humans is oropharyngeal/gastrointestinal anthrax, which can be caused by consuming improperly cooked meat contaminated with B. anthracis spores. Anthrax initiated by ingestion of the organism is often difficult to diagnose due to the non-specific symptoms (i.e. sore throat or abdominal pain) and may result in substantial fatality rates if left untreated (Mock & Fouet, 2001; Turnbull, 2008). An emerging new form of infection, injectional/septicaemic anthrax, has been reported among intravenous drug users in western Europe (Powell et al., 2011; Ringertz et al., 2000). The sources of the B. anthracis spores in these outbreaks were hypothesized to be contaminated heroin, probably originating from Pakistan, Afghanistan or Iran.
Inhalation of B. anthracis spores results in the most severe form of anthrax (Cote et al., 2011; Dixon et al., 1999; Mock & Fouet, 2001). As with the other forms of anthrax, differential diagnosis of inhalational anthrax can be very challenging due to its rather non-specific symptomatology (Friedlander, 1999, 2000). It is important to note that inhalational/pulmonary anthrax is not a true pneumonic disease. The lungs serve as the portal of entry of the infectious spores, but active infection generally occurs outside the alveolar spaces within lymphoid tissue. Germinated spores or vegetative bacilli are not typically found within the alveolar spaces until after systemic bacteraemia results in replicating bacilli being released back into the lungs through capillary beds (Bozue et al., 2007a; Cote et al., 2006, 2011; Glomski et al., 2007b; Heine et al., 2007; Heninger et al., 2006; Pickering et al., 2004; Ross, 1957). Inhalational anthrax has a nearly uniform fatality rate if appropriate treatment is not initiated in a timely manner (Dixon et al., 1999; Friedlander, 1999, 2000; Turnbull, 2008). Accordingly, inhalational anthrax is of predominant concern particularly when considering the impact of an attack or terrorism event involving biological agents.
Human vaccines include the human anthrax vaccine adsorbed (AVA or Biothrax-licensed vaccine in the USA), the human anthrax vaccine precipitated (AVP-licensed vaccine in the UK) and the newer recombinant protective antigen (rPA) vaccine, which is in development. PA is the toxin component responsible for translocation of the effector molecules, oedema factor and lethal factor (Baillie et al., 2004; Kudva et al., 2005; Young & Collier, 2007), and PA-based vaccines elicit predominantly anti-toxin responses (Baillie et al., 2004; Kudva et al., 2005). The anthrax toxins – lethal toxin composed of a combination of PA, and lethal factor and oedema toxin consisting of PA and oedema factor – are important virulence factors in the pathogenesis of anthrax, and are produced in large amounts by the replicating bacilli later in the course of the disease. Although effective, current vaccine strategies require a rigorous initial series of doses and annual boosters to achieve long-lasting immunity and, in general, current vaccine strategies can be somewhat reactogenic (Wasserman et al., 2003). Live attenuated vaccines (e.g. the unencapsulated Sterne or STI strains) are routinely effective at protecting animals and have been used to vaccinate humans in eastern European countries (Shlyakhov & Rubinstein, 1994; Turnbull, 1991). However, these vaccines may be too unsafe for approved use in many countries. An improved human vaccine is needed that requires fewer doses, is less reactogenic, elicits immunity targeting the early (pre-toxin) stages of infection, and could ameliorate or prevent infection by emerging or engineered strains that encode a PA refractory to anti-toxin immunity. Thus, the development of more optimal anthrax prophylactics remains a priority. Adding formaldehyde-inactivated spores (FIS) to PA-based vaccines has been reported to significantly augment the protection afforded to mice and guinea pigs challenged with virulent strains of B. anthracis (Brossier et al., 2002; Gauthier et al., 2009). Ideally, an individual or group of defined protective epitopes would be preferable for use in new vaccine formulations to obviate the use of whole killed spores. Disadvantages of the latter include the risks involved in whole-organism inactivation and the less well-defined nature of the protective immunogen(s). As the exosporium is the outermost surface of a B. anthracis spore, the exosporium has been the focus of numerous novel vaccination approaches by several laboratories. The exosporium structure surrounding the B. anthracis endospore is composed of numerous proteins. In this report, we focused on the BclA, ExsFA (BxpB) and p5303 proteins. BclA, the Bacillus collagen-like protein of B. anthracis, forms the hair-like extensions protruding from the exosporium membrane (Sylvestre et al., 2002, 2003). The BclA protein has been shown to be immunodominant (Steichen et al., 2003) and not essential for virulence (Bozue et al., 2007a; Sylvestre et al., 2002); rather, it appears to be involved in directing spores to professional phagocytes (Bozue et al., 2007a, 2007b; Oliva et al., 2008). Additionally, it has been shown that the BclA protein obscures other potentially immunostimulatory antigens (e.g. ExsK) that are located beneath the BclA ‘hair-like’ nap (Severson et al., 2009). It is important to note that, although BclA has structural similarities to human collagen, there has been no significant immunological cross-reactivity detected to date (Brahmbhatt et al., 2007). ExsFA was identified in the exosporium and has been shown to be involved in localization of the BclA protein (Redmond et al., 2004; Steichen et al., 2005; Sylvestre et al., 2005; Thompson et al., 2011; Todd et al., 2003). The putative exosporium protein p5303 was identified through an immunological screen and appears to be localized to the exosporium (Cybulski et al., 2008).
Recombinant exosporium components have been shown to augment the efficacy of a PA vaccination. Using the model of immunodeficient A/J mice challenged with a lethal dose of B. anthracis Sterne spores (Beedham et al., 2001; Flick-Smith et al., 2005; Welkos et al., 1986), Brahmbhatt, et al. (2007) demonstrated that the immune responses generated in mice receiving a single suboptimal (partially protective) injection of PA were completely protective in mice receiving a later injection (but not a concomitant injection) of BclA. These results were similar to those published earlier by Hahn et al. (2006). In these studies, the authors demonstrated that vaccination with a DNA construct encoding PA in combination with a construct encoding BclA offered superior protection against a challenge with fully virulent B. anthracis strain Ames when compared with vaccination with either BclA or PA constructs alone (Hahn et al., 2006). It has also been shown that other immunogens besides BclA may offer an advantage to vaccinated animals. Cybulski et al. (2008) observed significant protection in the Sterne vaccine strain challenge model when ExsFA or p5303 was used in vaccine formulations. Augmented protection afforded by an active anti-spore immune response has thus been reported by several laboratories. Efforts to elucidate these benefits must continue to optimize further the current anthrax vaccine strategies, as well as to increase preparedness for combating future emerging and/or engineered threats. In this report, we describe vaccine augmentation using PA administered concomitantly with recombinant spore proteins in both mouse and guinea pig models of B. anthracis infection.
Methods
Bacterial strains and culture conditions.
Spores of the wild-type Ames strain (pXO1+/pXO2+) of B. anthracis and spores of the Ames ΔbclA strain (Bozue et al., 2007a) were prepared from cultures grown in Leighton and Doi medium (Cote et al., 2006). Spores of the ΔAmes strain (pXO1−/pXO2+) of B. anthracis, derived as described earlier (Mikesell et al., 1983), were prepared on solid medium consisting of 0.8 % nutrient broth and 0.3 % yeast extract. These plates were inoculated with ΔAmes culture, grown at 37 °C overnight and then placed at room temperature in the dark until sporulation was maximal. The spores were then harvested and purified on an Omnipaque gradient, as described previously (Cote et al., 2006).
Vaccine formulations.
The vaccines discussed in this report were composed of rPA (List Biological Laboratories), recombinant spore-specific proteins produced as described earlier (Brahmbhatt et al., 2007; Cybulski et al., 2008; Mikesell et al., 1983) or a combination of the antigens. The specific amounts of antigens used in the vaccine formulations are described in the figure legend of each experiment. Aluminium hydroxide gel (AL; aluminium content per vaccination ~125 µg for mice or ~500 µg for guinea pigs) or the Sigma adjuvant system (SAS) was used as adjuvant (Sigma Aldrich). SAS is an emulsion of monophosphoryl lipid A and trehalose dicorynomycolate, which may promote mixed T helper 1 (Th1)/Th2 immune responses to vaccines, and was used as directed by the manufacturer.
Animal vaccinations and challenges.
Female BALB/c mice were obtained from the National Cancer Institute (Frederick, MD) and were approximately 6–9 weeks old at the time of initial vaccination. Hartley guinea pigs were obtained from Charles River Laboratories and were ~800 g at the time of initial vaccination. Mice received the vaccine delivered as a 200 µl dose via subcutaneous (s.c.) injection and guinea pigs received the vaccine delivered as a 500 µl s.c. dose. Animals received the initial vaccination on day 0 and booster vaccinations were given at 4-week intervals. Blood was collected from anaesthetized mice via a retro-orbital collection method every 14 days, and from anaesthetized guinea pigs via the cranial vena cava on days 30 and 120. The blood was centrifuged and serum was collected. Mice were challenged by intraperitoneal (i.p.) injection or intranasal (i.n.) instillation with wild-type Ames strain spores or ΔAmes strain spores (Cote et al., 2006, 2009; Lyons et al., 2004). Guinea pigs were challenged by intradermal (i.d.) injection, or i.n. instillation with wild-type Ames strain spores. The delivered challenge doses are indicated in the figure legends. Unless otherwise stated, all groups contained 10 animals.
The LD50 has been reported to be ~500 spores for BALB/c mice challenged i.p. (Popov et al., 2004). LD50 equivalents for Ames spores in the guinea pig infection model have been reported to be ~1.38 spores and ~1.2×105 spores when administered i.d. and i.n., respectively (Bielinska et al., 2007). LD50 determinations were performed in this study using ΔAmes spores delivered by i.p. injection or i.n. instillation to female BALB/c mice that were ~6–8 weeks old. LD50 values were calculated to be ~2×104 for i.p. injection and ~2.9×106 for i.n. instillation.
ELISA.
Serum anti-PA antibody responses were determined by a quantitative ELISA in accordance with the procedure of Little et al. (2004). The anti-spore responses of the vaccinated animals were determined using an anti-whole spore ELISA, which was a modification of previously described assays (Cote et al., 2005; Cybulski et al., 2008; Welkos et al., 2004), and utilized plates coated with antigen consisting of irradiated wild-type Ames spores or irradiated spores of the bclA deletion mutant of Ames (ΔbclA) (Bozue et al., 2007a). Polyclonal antibodies directed against spore proteins were generated in rabbits by Covance, Inc., as described previously (Cote et al., 2008; Moody et al., 2010), except that complete Freund’s adjuvant was used with the prime vaccination and incomplete Freund’s adjuvant was used in the subsequent booster(s) instead of the Ribi adjuvant system.
Statistics.
Survival rates were compared between each treatment group and control group using Fisher’s exact test. Kaplan–Meier/product-limit estimation was used to construct survival curves and to compute mean survival times. Survival curves were compared between each treatment group and control group by log rank tests. Mean times to death or to euthanasia (TTD) were compared between each treatment group and control group by t tests. LD50 equivalents for spores of the ΔAmes strain (both i.p. and i.n. challenge routes) were determined by Probit analysis. The above analyses were conducted using sas version 8.2 (SAS Institute). ELISA data were analysed by a four-parameter logistic-regression model and analysis of variance/multi-comparison t tests using GraphPad Prism version 5.00 (GraphPad Software).
Results
Impact of recombinant spore proteins in the mouse model of B. anthracis infection utilizing fully virulent Ames strain (pXO1+/pXO2+) spores
Antigen combination screenings were performed using BALB/c mice challenged with the fully virulent Ames strain. Data generated in several laboratories have suggested that inactivated spores or spore antigens may offer adjunct benefits to PA-based vaccines (Brahmbhatt et al., 2007; Brossier et al., 2002; Cote et al., 2008; Cybulski et al., 2008; Enkhtuya et al., 2006; Gauthier et al., 2009; Hahn et al., 2006; Vergis et al., 2011). We endeavoured to identify the protection afforded by specific combinations of recombinant spore proteins. An initial experiment was performed testing spore antigens (with or without suboptimal doses of PA). The experimental design was an expansion of that reported by Cybulski et al. (2008) and Brahmbhatt et al. (2007) using the A/J mouse/Sterne challenge model, but instead using BALB/c mice challenged with fully virulent Ames spores (BALB/c mouse/Ames challenge model). This approach allowed us both to characterize the impact on survival and to monitor the effects (i.e. potential immune interference) of different antigen combinations on the resulting immune response. All spore antigens were delivered as doses of ~25 µg, whilst an amount of PA shown to be suboptimal in the A/J mouse/Sterne model (50 ng) was administered per dose. The spore proteins BclA, ExsFA and p5303 were evaluated in this experiment. As shown in Table 1, all of the antigens were administered once by the s.c. route and were given as a single antigen vaccine or as part of a multi-antigen vaccine, either combined or separated. Mice were administered the vaccine proteins on day 0 and/or on day 28.
Table 1. Antigen screening performed using BALB/c mice challenged with Ames spores.
| Group no. | Primary dose | Secondary dose | Survival* | TTD (days)† |
| 1 | PBS+AL | PBS+AL | 1/10 | 3.0 (±1.2) |
| Single antigen: | ||||
| 2 | PA+AL | PBS+AL | 1/10 | 2.4 (±0.5) |
| 3 | BclA+AL | PBS+AL | 2/10 | 3.1 (±1.3) |
| 4 | ExsFA+AL | PBS+AL | 1/10 | 3.7 (±2.1) |
| 5 | P5303+AL | PBS+AL | 4/10 | 2.5 (±0.6) |
| Multi-antigen, combined: | ||||
| 6 | PA+BclA+AL | PBS+AL | 2/10 | 2.9 (±0.6) |
| 7 | PA+ExsFA+AL | PBS+AL | 2/10 | 4.0 (±4.1) |
| 8 | PA+p5303+AL | PBS+AL | 4/10‡ | 3.8 (±2.1) |
| 9 | PA+BclA+ExsFA+AL | PBS+AL | 1/10 | 4.9 (±3.7)§ |
| 10 | PA+BclA+p5303+AL | PBS+AL | 2/10 | 2.8 (±1.0) |
| 11 | PA+ExsFA+p5303+AL | PBS+AL | 3/10 | 3.9 (±2.5) |
| Multi-antigen, separated: | ||||
| 12 | PA+AL | BclA+AL | 0/10 | 3.6 (±1.2) |
| 13 | PA+AL | ExsFA+AL | 1/10 | 4.2 (±3.9) |
| 14 | PA+AL | p5303+AL | 2/10 | 3.5 (±1.7) |
| 15 | PA+AL | BclA+ExsFA+AL | 1/10 | 5.8 (±3.3)|| |
| 16 | PA+AL | BclA+p5303+AL | 1/10 | 3.9 (±2.7) |
| 17 | PA+AL | ExsFA+p5303+AL | 2/10 | 3.3 (±1.0) |
The protective efficacy of spore antigens alone and with PA. BALB/c mice were vaccinated s.c. with a suboptimal amount of PA (50 ng), 25 µg spore antigen(s) or both. The mice received one dose per antigen, given as a single antigen vaccine or as part of a multi-antigen vaccine (combined or separated). All vaccines included AL. Challenges with Ames spores of ~3.4×10 3 (or ~7 LD50 equivalents) were given 4 weeks after the last dose by the i.p. route.
Values shown are means, with sd in parentheses.
P = 0.07 when comparing the survival curve with that of group 1.
P = 0.09 when comparing TTD with that of group 1.
P = 0.01 when comparing TTD with that of group 1.
The conditions used in this initial experiment were quite stringent. Mice have proven historically to be difficult to protect with PA alone against a fully virulent challenge, despite stimulation in mice of high titres of toxin-neutralizing anti-PA antibody (Bielinska et al., 2007; Boyaka et al., 2003; Brossier et al., 2002; Flick-Smith et al., 2005; Hahn et al., 2006; Shivachandra et al., 2007; Welkos et al., 1989; Welkos & Friedlander, 1988; Williamson et al., 2005). In addition, our PA doses were purposefully selected to be suboptimal to increase the chances of revealing any anti-spore antibody contributions to protection. Mice vaccinated with PA alone succumbed as rapidly as the controls and had only a 10 % survival rate. However, some of the mice receiving the other vaccines exhibited extended TTD or enhanced overall survival rates compared with the PBS or PA-only control groups. The mortality data from this challenge experiment are depicted in Table 1. One group had a significantly delayed time to death (group 15, comprising primary vaccination with 50 ng PA followed by a day 28 vaccination of BclA and ExsFA at 25 µg each). Anti-PA ELISA titres were determined with sera collected from all 17 groups over the course of the pre-challenge vaccination period. Although the titres were low, as expected, levels significantly above those of the PBS controls were detected in some groups (data not shown). Anti-PA levels above background were detected in some of the mice that received PA concomitantly with spore antigens (e.g. group 8), and in groups given PA separately and at a different time from the spore antigen dose (groups 12–17); in general, the anti-PA titres in sera from groups 3–11 were negligible. These results suggest a possible interfering effect of the spore antigens on development of anti-PA antibodies when given at the same time, thus appearing to confirm the previous findings of Brahmbhatt et al. (2007). The overall results of this experiment were encouraging, given the stringent conditions involving single administrations of relatively low levels of antigen. The improved performance of the combination (PA and spore antigen) vaccines compared with PA alone further supported the hypothesis of several laboratories that spore components will be essential for an optimally effective novel anthrax vaccine.
Subsequent experiments were performed to determine the protection afforded to mice using vaccination conditions anticipated to be more efficacious. We sought to maximize the protection afforded by the PA+spore antigen vaccines by evaluating the timing, number and quantity of the vaccines and their components. In the first vaccine experiment, none of the groups vaccinated with spore antigens alone had been significantly protected. Even though PA alone was not efficacious, the groups that were significantly protected received PA as well as spore antigens. Thus, in later vaccine experiments, all the groups given spore antigens were also vaccinated with PA, and a more typical dose of PA (25 µg) was used.
The efficacy of vaccines delivered with two different adjuvants – AL or SAS – was compared to evaluate the roles of Th1- and Th2-biased immune responses in protection. SAS is a stable oil-in-water emulsion containing components originally formulated by Ribi Immunochem and induces mixed Th1/Th2 immune responses to vaccines. SAS contains squalene, trehalose, monophosphoryl lipid A and Tween 80 (SAS data sheet). Adjuvants containing these complex components have been shown to induce robust antibody responses as well as significant cell-mediated responses (Fox, 2009). In contrast, aluminium-based adjuvants such as AL, which are the only adjuvants licensed to date in the USA for human use, elicit mostly Th2-based humoral immunity. Several experiments were conducted to evaluate these various alterations in the vaccination scheme. There were only slight differences in survival noted in the groups of mice receiving the vaccines with different adjuvant formulations or different mixtures or numbers of doses of vaccine antigens, and these differences generally were not statistically significant. Mice receiving combination vaccines (PA+spore antigens) delivered with AL tended to respond with earlier detectable levels of PA antibodies (Fig. 1a) and slightly greater, although not significantly different, concentrations of anti-spore antibodies (Fig. 1b, c). However, these differences were transient and were no longer relevant in later study time points or after subsequent booster vaccinations (Fig. 1). Regardless of the slight differences in immune kinetics, there were no significant differences noted in protective efficacy (data not shown). Whilst antibody titres provide valuable information on immune responses during the early stages of developing a novel vaccine, it will be important to examine in more detail the impacts on cellular immune responses of a later-stage multi-antigen anthrax vaccine.
Fig. 1.

Effects of adjuvant on the resulting immune response generated from PA+spore (BclA, ExsFA and p5303) vaccinations. (a) Changes in the anti-PA IgG titres of each vaccine group during the 12-week vaccination (pre-challenge) period. The vaccines were delivered at weeks 0, 4 and 8. Sera were collected just prior to each vaccine dose. (b, c) Spore antibody ELISAs using plates coated with Ames spores (b) or ΔbclA spores (c) and sera from mice collected at week 12 post-vaccination/pre-challenge. Representative data are shown. Sera collected from mice vaccinated with a prime and boost (2×) or a prime followed by two boosts (3×) were analysed.
Evaluating potential immune interference
Titres to both PA and spore antigens were evaluated over the course of the vaccination period in all experiments performed. We confirmed results suggesting that concomitant administration of PA+spore antigens resulted in a reduced immune response to PA when PA was given at significantly lower amounts relative to spore antigens (Brahmbhatt et al., 2007). We wanted to characterize further this potential immune interference and assay the immune response when approximately equal amounts of antigen were used in vaccine formulations. In experiments in which two to three doses of vaccine antigens (PA and spore antigens) present in similar concentrations (microgram quantities) were administered, high titres of anti-PA and anti-spore antibodies were detected, and there was no obvious interference by spore antigens on the anti-PA responses (data not shown, discussed further below). PA did not have to be delivered separately from spore antigens to elicit high anti-PA titres. However, although PA appeared to be required for any degree of protection, the anti-PA antibody levels were not associated with protective immunity in mice, as shown in the first experiment and previously (Brahmbhatt et al., 2007; Brossier et al., 2002; Gauthier et al., 2009; Welkos & Friedlander, 1988). Antibody responses to spore antigens were evaluated on plates coated with irradiated wild-type Ames spores or irradiated ΔbclA spores. As detailed in representative data included in Table 2, mice receiving PA+spore antigens exhibited significant anti-spore titres. By using the ΔbclA spores as the capture antigen, the impact of antibodies directed against spore components that are beneath the external-most layer of BclA (i.e. ExsFA and p5303) could be evaluated. Before conducting these assays, ELISA titrations of rabbit anti-spore IgGs specific for these three spore exosporium antigens were carried out. As expected, whereas anti-BclA antibody had a much higher titre against the wild-type spores than the ΔbclA mutant spores, antibodies directed against the other exosporium antigens (p5303 and ExsFA) recognized the mutant spores better than wild-type spores (Fig. 2). Thus, the presence of BclA (major component of the outer ‘hair-like’ nap) partially masks recognition in the wild-type Ames spore ELISA of immune responses to the exosporium basal membrane and probably partially but not completely blocks their exposure in vivo to the immune system (Basu et al., 2007; Bozue et al., 2007b; Cybulski et al., 2008). We used antibody titres as a determinant of any potential immune interference observed by co-administering PA with recombinant spore proteins. Further development of this vaccine strategy will require evaluation of the resulting antibodies by a functional toxin-neutralization assay (Pitt et al., 1999).
Table 2. Anti-spore antibody responses: titre comparisons of vaccinated and unvaccinated groups.
Statistical analysis of variance with post-hoc Tukey’s tests for pairwise group comparisons. Titres of sera collected from vaccinated mice in a representative experiment were determined by ELISA in plates coated with wild-type (WT) Ames or ΔbclA spores.
| Week | Group compared with PBS controls* | Highest significant dilution† | |
| WT Ames plate | ΔbclA Ames plate | ||
| 4 | PA alone | <50‡ | <50‡ |
| PA+BclA+ExsF combined (3) | 3200 | 3200 | |
| PA (1), BclA+ExsF separately (2) | <50‡ | <50‡ | |
| 8 | PA alone | <50‡ | <50‡ |
| PA+BclA+ExsF combined (3) | 3 200 | ≥204 800 | |
| PA (1), BclA+ExsF separately (2) | 200 | 800 | |
| 12 | PA alone | <50‡ | <50‡ |
| PA+BclA+ExsF combined (3) | ≥204 800§ | ≥204 800 | |
| PA (1), BclA+ExsF separately (2) | 3200 | 51 200 | |
The number of total vaccine doses over the course of the entire study is shown in parentheses. Vaccinations were administered on day 0, week 4 and week 8. All vaccinations included AL. For example, the third group in each set of three received one PA dose (25 mg) on day 0 and one PA+spore antigen dose (25 µg) on week 4 and a second on week 8.
Highest dilution with A405 significantly elevated (P<0.05) compared with the PBS control (reciprocal dilution).
P>0.05 at lowest dilution tested (1 : 50).
P<0.05 at highest dilution tested (1 : 204 800).
Fig. 2.
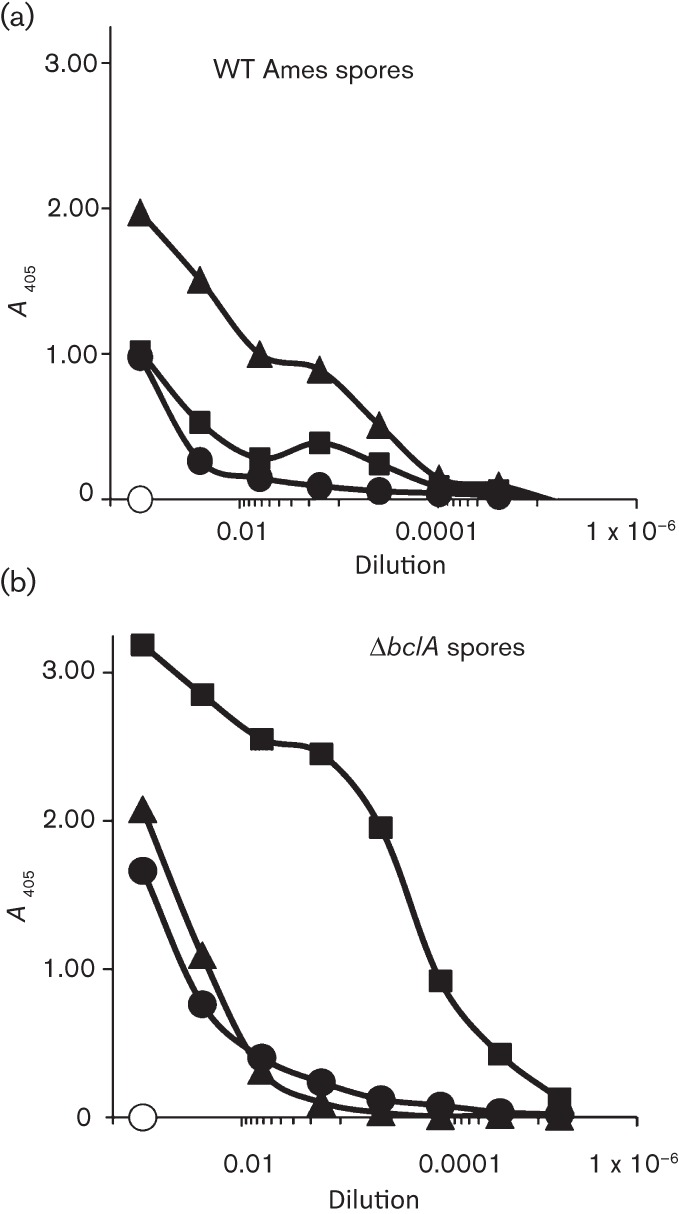
ELISA titrations of rabbit anti-spore IgGs specific for the three spore exosporium antigens BclA (▴), ExsFA (▪) and p5303 (•) were carried out on plates coated with wild-type (WT) Ames spores (a) or the BclA deletion mutant of Ames, ΔbclA (b). The negative control (○) was rabbit antibody specific for the unrelated vegetative protein CylI.
Impact of recombinant spore proteins in the mouse model of B. anthracis infection utilizing ΔAmes strain (pXO1−/pXO2+) spores
To dissect better the contribution of spore antigens in multi-antigen anthrax vaccine protection, mice were challenged with spores of the ΔAmes derivative, a pX01-cured (toxin-negative) derivative of wild-type Ames. Such toxin-negative, capsule-positive strains of B. anthracis are highly virulent for mice (Heninger et al., 2006; Welkos et al., 1986, 1993), and the absence of toxin antigens allows spore-mediated immunity to be shown more clearly. Vaccinations of the mice included a prime and booster dose of the PA+spore antigen combination vaccines, and the vaccinated mice were then challenged with ΔAmes strain spores. The spore antigens comprised BclA, ExsFA and p5303. We chose to keep PA in the vaccine formulation, in spite of the fact we were challenging with a non-toxigenic strain of B. anthracis, for the sake of consistency and also to evaluate further any potential impact on immune responses to individual vaccine components associated with concomitant antigen administration and because any later new-generation vaccine will undoubtedly include toxin antigen(s). As shown in Fig. 3(a), these data confirmed that spore antigens significantly enhanced the protection afforded by a PA vaccination (with AL) against lethal parenteral challenge with either a toxigenic or non-toxigenic but encapsulated B. anthracis Ames strain. Mice challenged by i.n. instillation were not significantly protected (Fig. 3b).
Fig. 3.
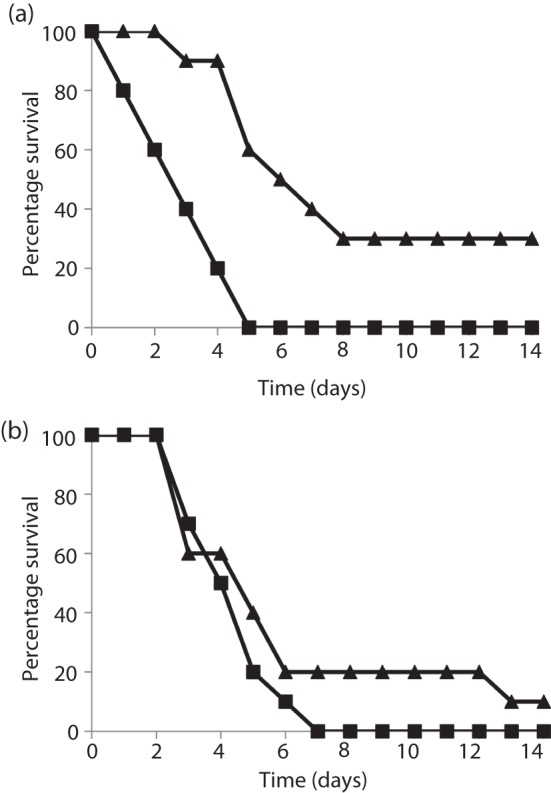
Survival results of mice vaccinated with PA alone (▪) or PA+spore antigens (▴) and challenged i.p. (a) or i.n. (b) with ΔAmes spores. All vaccines were given as two immunizations each separated by 4 weeks and all contained ~25 µg PA or 25 µg PA+10 µg BclA+10 µg ExsFA+10 µg p5303 and ~125 µg AL. Mice challenged by i.p. injection (a) received ~3×105 spores delivered in 200 µl sterile water for injections (WFI) (representing ~15 LD50 equivalents). Mice challenged through i.n. instillations (b) received ~5.2×107 spores delivered in 40 µl WFI (representing ~15 LD50 equivalents). The results from the i.p. challenge indicated that the addition of spore antigens to the vaccine afforded significant protection against ΔAmes spores (P = 0.0004 for the survival curve and P = 0.004 for TTD). The results after i.n. challenge were not statistically significant.
ELISA titrations of the sera from these mice indicated that, as observed in the previous experiments, spore antigens did not suppress the anti-PA IgG responses when given together with PA (Fig. 4a). Interestingly, the presence of the spore antigens with PA enhanced the anti-PA antibody responses (Fig. 4a). Also in agreement with our previous vaccination results, two doses of vaccine antigens elicited significantly elevated levels of serum anti-spore antibodies, as determined by ELISA analyses using wild-type spores (Fig. 4b, d) or ΔbclA spores (Fig. 4c, e).
Fig. 4.
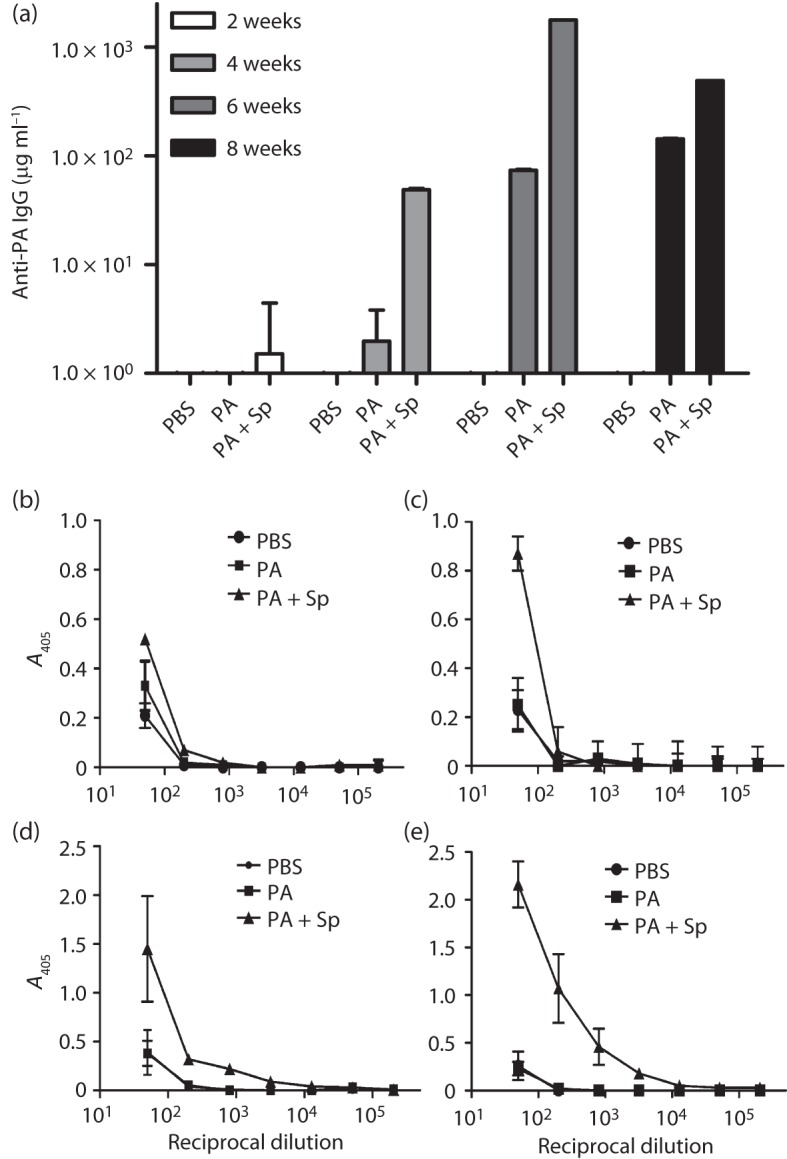
ELISAs to detect anti-PA and anti-spore antibodies in sera from vaccinated BALB/c mice. (a) Mice were vaccinated twice with PA with (+Sp) or without spore antigens on day 0 and week 4 and challenged at week 8 with ΔAmes spores. Sera were collected on weeks 2, 4, 6 and 8 and assayed on plates coated with PA. There were no statistically significant differences noted in the week 2 samples. There were significant differences between vaccines consisting of PBS or PA alone (P = 0.0009, week 4; P<0.0001, weeks 6 and 8), PBS or PA+Sp (P<0.0001 for weeks 4, 6 and 8) and PA or PA+Sp (P = 0.0009, week 2; P<0.0001, week 6). (b–d) Anti-spore antibody titres were determined for sera collected on week 2 (b, c) and week 6 (d, e) of the vaccination period and assayed on plates coated with wild-type Ames (b, d) or ΔbclA Ames (c, e) spores. The responses (b–e) of the PA+Sp vaccine group were significantly greater than those of the PBS control groups, with a mean reciprocal end-point dilution titre of 800 (P = 0.0347) for (b) and (c) and 12 800 (P = 0.007) for (d) and (e), as determined by t tests with stepdown Bonferroni adjustment for multiple comparisons. The symbols for the PBS control and PA-only groups overlapped in the graphs depicted in (c–e).
Impact of recombinant spore proteins in the guinea pig model of B. anthracis infection utilizing fully virulent Ames strain spores
Because of the difficulty associated with protecting mice against challenge with virulent strains of B. anthracis (Brossier et al., 2002; Flick-Smith et al., 2005; Gauthier et al., 2009; Hahn et al., 2006; Welkos et al., 1993), we chose additionally to test the vaccine candidates in a guinea pig model of infection. Guinea pigs received a total of three vaccinations s.c. at 4-week intervals. Guinea pigs were then challenged 4 weeks after the third vaccine dose (week 12) via challenge i.d. or i.n. with spores of the Ames strain. Regardless of the route tested, the vaccine consisting of both PA and spore-specific antigens appeared to outperform vaccination by rPA alone, and the results were suggestive of greater protection afforded by the combination compared with PA-only vaccines (Fig. 5). Serum antibody analyses revealed that, although the presence of spore antigens in the vaccine was associated with a weak and temporary inhibition of anti-PA titres early during the vaccination (Fig. 6a), anti-PA levels in the PA+spore antigen group caught up to and surpassed those in the PA-only group sera before spore challenge (Fig. 6b). This stimulatory effect of spore antigens on anti-PA titres was similar to that observed in mice (Fig. 3a, Table 2). As expected, the anti-spore antibody titres of the groups receiving PA+spore antigens were significantly higher than those of the PA-only group at both the 4 and 12 week time points (Fig. 6c–f).
Fig. 5.
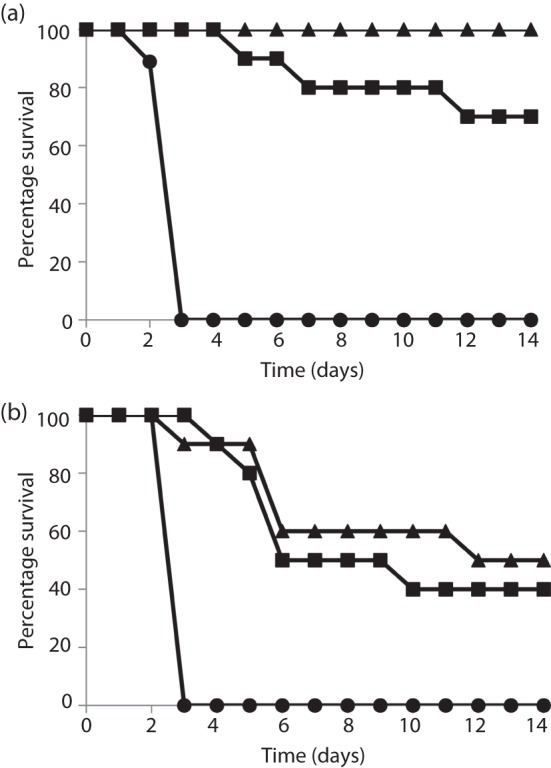
Survival results of guinea pigs vaccinated with PA alone (▪) or PA+spore antigens (▴) and challenged i.d. (a) or i.n. (b) with Ames spores. All vaccines were given as three immunizations each separated by 4 weeks and all contained ~30 µg PA or 30 µg PA+10 µg BclA+10 µg ExsF+10 µg p5303 and ~500 µg AL. The control groups (•) received PBS and AL alone. The guinea pigs challenged by i.d. injection (a) received ~156 spores delivered in 200 µl WFI and the guinea pigs challenged through i.n. instillations (b) received ~1.35×107 spores delivered in 100 µl WFI. The guinea pigs challenged i.d. were significantly protected with PA alone (P = 0.0062 for percentage survival and 0.0002 for the survival curve) and the vaccine including spore antigens was also protective (P<0.0001 for percentage survival and 0.0002 for the survival curve). The P value comparing the survival curves of PA alone with PA+spore antigens was 0.067. When challenged i.n., the animals receiving PA alone were significantly protected (P = 0.013 for TTD and P = 0.0002 for the survival curve), as were the animals receiving the PA vaccine with the addition of the spore antigens (P = 0.012 for TTD and P = 0.0002 for the survival curve). There were 10 animals per group with the exception of the negative-control groups, which contained nine animals in each group.
Fig. 6.

Titres of anti-PA and anti-spore antibodies in guinea pigs before challenge with Ames spores. The animals were vaccinated three times at 4-week intervals with PA, PA+spore antigens (PA+Sp) or PBS alone (controls). Sera were collected from each animal 4 weeks after the first dose (a, c, e) and 4 weeks after the third dose (b, d, f, just before challenge with wild-type Ames spores at week 12). ELISAs were carried out on plates coated with PA (a, b), wild-type Ames spores (c, d) or ΔbclA spores (e, f). The results are the geometric mean titres (±sem) in µg PA ml−1 and anti-spore antibody levels (mean A405 and sd). The anti-PA titres (a, b) of the PA-only group were significantly higher than those of the PA+Sp group at week 4 (P = 0.0044), but the reverse was true for sera collected just prior to challenge at week 12 (P = 0.0054). Anti-spore antibody titres in sera collected at week 4 (c, e) and week 12 (d, f) were determined in ELISAs against wild-type (c, d) or ΔbclA (e, f) spores of Ames. The responses (c–f) of the PA+Sp vaccine group were significantly greater than those of the PA-only groups with mean reciprocal end-point dilution titres of up to 51 200 (P = 0.0053) for (c), ≤12 800 (P<0.033) for (d), through all reciprocal dilutions tested (P≤0.0007) for (e) and up to 51 200 (P<0.017) for (f), as determined by t tests with stepdown Bonferroni adjustment for multiple comparisons.
Discussion
Whilst the current and next-generation anthrax vaccines are effective, they mainly target PA (Friedlander et al., 1999). However, there are several drawbacks associated with this strategy. Because these vaccine strategies are predominantly anti-toxin, essentially the bacteria are often able to infect and multiply within the host until the vaccine-induced anti-toxin effects begin to take effect. Additionally, it would be beneficial to have a multi-component anthrax vaccine capable of combating emerging or engineered threats that may be refractory to current vaccine approaches. The most promising data indicating protection attributed to early host responses specific for other components, such as the spore, have been generated by vaccinating small animals with FIS (Brossier et al., 2002; Gauthier et al., 2009). These authors demonstrated that, by targeting both the earliest phase of infection (entry of ungerminated B. anthracis spores into hosts) and also the later stage of infection (intoxication), the experimental animals were more likely to survive infection with lethal doses of B. anthracis spores.
Data from passive protection studies performed using antisera generated against whole spores have been less striking. It has been reported that passive transfer of anti-spore antibodies can significantly protect mice against challenge with a fully virulent strain of B. anthracis (Enkhtuya et al., 2006). The antibodies used were generated in rabbits against a strain of B. anthracis that was cured of both pXO1 and pXO2 to ensure that antibodies were not directed against either toxin or capsule but rather spore structures, presumably the surface-exposed exosporium layer. In contrast, Glomski et al. (2007a) could not demonstrate significant levels of passive protection when mouse immune FIS sera were transferred to naïve mice (Goossens et al., 2007). Additionally, Cote et al. (2008) demonstrated that slight protection was afforded by passive transfer of rabbit anti-irradiated-spore IgG into naïve mice, but this protection was only statistically significant when compared with completely naïve mice. When the survival was compared with mice receiving normal rabbit IgG, the observed protection was statistically insignificant, suggesting that some aspect of the protection was not spore-specific. Glomski et al. (2007a) further investigated the mechanisms associated with the supplementary protection offered by FIS when added to a PA vaccine regimen. These authors demonstrated a cellular component to this anti-spore immunity. When splenocytes harvested from FIS-immune mice were transferred to naïve mice, significant protection was achieved. Through further experimentation using transgenic animals and in vivo lymphocyte depletion methodology, it was demonstrated that interferon-γ-producing CD4 lymphocytes were, in part, responsible for the vaccine augmentation associated with FIS (Glomski et al., 2007a).
Our current report adds to the growing body of literature supporting the concept that spore-specific immunity is important when designing next-generation anthrax vaccines. Using both mouse and guinea pig models of B. anthracis infection, we demonstrated that recombinant spore proteins augment protection afforded by PA alone. The rationale for testing PA combined or separately with the spore protein stems from the observation of Brahmbhatt et al. (2007) that, when given with BclA, PA elicited lower titres of anti-PA antibodies than when PA was given alone and at a time separate from that of the spore proteins; protection was also less. Suboptimal amounts of PA were used by Brahmbhatt et al. (2007) to identify any adjunct benefits of spore antigens, as optimal amounts of PA can completely protect A/J mice against a Sterne spore challenge (Welkos & Friedlander, 1988). We wanted to confirm whether this spore antigen-associated interference held true in the BALB/c mouse/Ames spore challenge model.
We demonstrated that immune responses to PA are not necessarily hindered by the addition of spore antigens. High pre-challenge anti-PA antibody titres were observed with at least two doses of PA and with both of the adjuvants tested; however, there was no correlation between anti-PA levels and protection in the BALB/c mouse/Ames spore challenge model. These results are in agreement with previous literature on protection and immune responses to PA vaccines delivered with aluminium-based adjuvants (Brossier et al., 2002; Flick-Smith et al., 2005; Welkos & Friedlander, 1988; Welkos et al., 1989). In contrast to experiments utilizing suboptimal amounts of PA (nanogram quantities), when greater amounts of PA (microgram quantities) and/or increased numbers of vaccine doses were employed, PA and spore antigens could be delivered together without apparent interfering effects on PA responses by spore antigens. In some cases, giving the spore antigens and PA as a single vaccine resulted in an increase in the resulting anti-PA immune response (Figs 4a and 6b).
In all experiments in which BALB/c mice were challenged with fully virulent Ames strain spores, any degree of protection over the PBS controls required the addition of at least one spore antigen to the PA vaccine (Table 1 and data not shown). Neither PA alone nor spore antigens alone were significantly protective in this model. Similar results were obtained when BALB/c mice were challenged with the non-toxigenic ΔAmes strain spores (Fig. 3). Mice are difficult to protect because of their high susceptibility to most fully virulent B. anthracis strains and their sensitivity to non-toxin virulence factors (Welkos et al., 1986, 1989, 1993). Accordingly, our findings from the mouse models, whilst incremental, offer important clues to the concept of spore-specific immunogens. We also used guinea pigs to test the vaccines because of the extreme stringency associated with using fully virulent B. anthracis strains in the mouse model. Guinea pigs were partially protected (statistically significant) with PA vaccination alone; however, the addition of recombinant spore proteins improved this protection (Fig. 5). Whilst significant vaccine augmentation was observed in the mouse i.p. model and guinea pig i.d. models, the augmentation of protection afforded to these animals when they were challenged via i.n. instillation was not statistically significant. We hypothesize that this is due to the inherent differences in pathogenesis between inhalational anthrax and that of anthrax initiated through parenteral inoculation of spores. Particularly, synchronicity of infection probably plays a role in distinguishing these two disease models (Cote et al., 2011).
In summary, our data suggested that significant protection in the current mouse model requires a combination of PA and a spore antigen(s). These results agree with those reported earlier (Brossier et al., 2002; Gauthier et al., 2009; Hahn et al., 2006). The demonstration of improved protection using purified spore proteins as supplementary components of a PA vaccine provides an important proof of principle establishing spore antigen-mediated protection in animal models challenged with fully virulent B. anthracis spores.
Acknowledgements
The authors thank Ms Diana Fisher and Ms Sarah Norris for their expert statistical analyses. The research described herein was sponsored by the Defense Threat Reduction Agency JSTO-CBD plans 1.1A0010-07-RDB and CBM.VAXBT.03.10.RD.004/Medical Research/Material Command Research Plan (SLW) and the Biological Defense Research Directorate of the Naval Medical Research Center, United States Navy and the Middle Atlantic Regional Centers of Excellence (1U54 AI57168) (A. D. O’B.). Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the US Army or Department of Defense. Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Abbreviations:
- FIS
formaldehyde-inactivated spores
- i.d.
intradermal(ly)
- i.n.
intranasal(ly)
- i.p.
intraperitoneal(ly)
- rPA
recombinant protective antigen
- s.c.
subcutaneous(ly)
- TTD
time to death
- WFI
water for injections
References
- Baillie L., Townend T., Walker N., Eriksson U., Williamson D. (2004). Characterization of the human immune response to the UK anthrax vaccine. FEMS Immunol Med Microbiol 42, 267–270 10.1016/j.femsim.2004.05.011 [DOI] [PubMed] [Google Scholar]
- Basu S., Kang T. J., Chen W. H., Fenton M. J., Baillie L., Hibbs S., Cross A. S. (2007). Role of Bacillus anthracis spore structures in macrophage cytokine responses. Infect Immun 75, 2351–2358 10.1128/IAI.01982-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beedham R. J., Turnbull P. C., Williamson E. D. (2001). Passive transfer of protection against Bacillus anthracis infection in a murine model. Vaccine 19, 4409–4416 [DOI] [PubMed] [Google Scholar]
- Bielinska A. U., Janczak K. W., Landers J. J., Makidon P., Sower L. E., Peterson J. W., Baker J. R., Jr (2007). Mucosal immunization with a novel nanoemulsion-based recombinant anthrax protective antigen vaccine protects against Bacillus anthracis spore challenge. Infect Immun 75, 4020–4029 10.1128/IAI.00070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyaka P. N., Tafaro A., Fischer R., Leppla S. H., Fujihashi K., McGhee J. R. (2003). Effective mucosal immunity to anthrax: neutralizing antibodies and Th cell responses following nasal immunization with protective antigen. J Immunol 170, 5636–5643 [DOI] [PubMed] [Google Scholar]
- Bozue J., Cote C. K., Moody K. L., Welkos S. L. (2007a). Fully virulent Bacillus anthracis does not require the immunodominant protein BclA for pathogenesis. Infect Immun 75, 508–511 10.1128/IAI.01202-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozue J., Moody K. L., Cote C. K., Stiles B. G., Friedlander A. M., Welkos S. L., Hale M. L. (2007b). Bacillus anthracis spores of the bclA mutant exhibit increased adherence to epithelial cells, fibroblasts, and endothelial cells but not to macrophages. Infect Immun 75, 4498–4505 10.1128/IAI.00434-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmbhatt T. N., Darnell S. C., Carvalho H. M., Sanz P., Kang T. J., Bull R. L., Rasmussen S. B., Cross A. S., O’Brien A. D. (2007). Recombinant exosporium protein BclA of Bacillus anthracis is effective as a booster for mice primed with suboptimal amounts of protective antigen. Infect Immun 75, 5240–5247 10.1128/IAI.00884-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossier F., Levy M., Mock M. (2002). Anthrax spores make an essential contribution to vaccine efficacy. Infect Immun 70, 661–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote C. K., Rossi C. A., Kang A. S., Morrow P. R., Lee J. S., Welkos S. L. (2005). The detection of protective antigen (PA) associated with spores of Bacillus anthracis and the effects of anti-PA antibodies on spore germination and macrophage interactions. Microb Pathog 38, 209–225 10.1016/j.micpath.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Cote C. K., Van Rooijen N., Welkos S. L. (2006). Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect Immun 74, 469–480 10.1128/IAI.74.1.469-480.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote C. K., Bozue J., Moody K. L., DiMezzo T. L., Chapman C. E., Welkos S. L. (2008). Analysis of a novel spore antigen in Bacillus anthracis that contributes to spore opsonization. Microbiology 154, 619–632 10.1099/mic.0.2007/008292-0 [DOI] [PubMed] [Google Scholar]
- Cote C. K., Bozue J., Twenhafel N., Welkos S. L. (2009). Effects of altering the germination potential of Bacillus anthracis spores by exogenous means in a mouse model. J Med Microbiol 58, 816–825 10.1099/jmm.0.008656-0 [DOI] [PubMed] [Google Scholar]
- Cote C. K., Welkos S. L., Bozue J. (2011). Key aspects of the molecular and cellular basis of inhalational anthrax. Microbes Infect 13, 1146–1155 10.1016/j.micinf.2011.07.005 [DOI] [PubMed] [Google Scholar]
- Cybulski R. J., Jr, Sanz P., McDaniel D., Darnell S., Bull R. L., O’Brien A. D. (2008). Recombinant Bacillus anthracis spore proteins enhance protection of mice primed with suboptimal amounts of protective antigen. Vaccine 26, 4927–4939 10.1016/j.vaccine.2008.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon T. C., Meselson M., Guillemin J., Hanna P. C. (1999). Anthrax. N Engl J Med 341, 815–826 10.1056/NEJM199909093411107 [DOI] [PubMed] [Google Scholar]
- Enkhtuya J., Kawamoto K., Kobayashi Y., Uchida I., Rana N., Makino S. (2006). Significant passive protective effect against anthrax by antibody to Bacillus anthracis inactivated spores that lack two virulence plasmids. Microbiology 152, 3103–3110 10.1099/mic.0.28788-0 [DOI] [PubMed] [Google Scholar]
- Flick-Smith H. C., Waters E. L., Walker N. J., Miller J., Stagg A. J., Green M., Williamson E. D. (2005). Mouse model characterisation for anthrax vaccine development: comparison of one inbred and one outbred mouse strain. Microb Pathog 38, 33–40 10.1016/j.micpath.2004.10.007 [DOI] [PubMed] [Google Scholar]
- Fox C. B. (2009). Squalene emulsions for parenteral vaccine and drug delivery. Molecules 14, 3286–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander A. M. (1999). Clinical aspects, diagnosis and treatment of anthrax. J Appl Microbiol 87, 303 10.1046/j.1365-2672.1999.00896.x [DOI] [PubMed] [Google Scholar]
- Friedlander A. M. (2000). Anthrax: clinical features, pathogenesis, and potential biological warfare threat. Curr Clin Top Infect Dis 20, 335–349 [PubMed] [Google Scholar]
- Friedlander A. M., Pittman P. R., Parker G. W. (1999). Anthrax vaccine: evidence for safety and efficacy against inhalational anthrax. JAMA 282, 2104–2106 10.1001/jama.282.22.2104 [DOI] [PubMed] [Google Scholar]
- Gauthier Y. P., Tournier J. N., Paucod J. C., Corre J. P., Mock M., Goossens P. L., Vidal D. R. (2009). Efficacy of a vaccine based on protective antigen and killed spores against experimental inhalational anthrax. Infect Immun 77, 1197–1207 10.1128/IAI.01217-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomski I. J., Corre J. P., Mock M., Goossens P. L. (2007a). Cutting edge: IFN-γ-producing CD4 T lymphocytes mediate spore-induced immunity to capsulated Bacillus anthracis. J Immunol 178, 2646–2650 [DOI] [PubMed] [Google Scholar]
- Glomski I. J., Piris-Gimenez A., Huerre M., Mock M., Goossens P. L. (2007b). Primary involvement of pharynx and Peyer’s patch in inhalational and intestinal anthrax. PLoS Pathog 3, e76 10.1371/journal.ppat.0030076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens P. L., Sylvestre P., Mock M. (2007). Of spore opsonization and passive protection against anthrax. Microbiology 153, 301–302, discussion 302–304 10.1099/mic.0.2006/003210-0 [DOI] [PubMed] [Google Scholar]
- Hahn U. K., Boehm R., Beyer W. (2006). DNA vaccination against anthrax in mice—combination of anti-spore and anti-toxin components. Vaccine 24, 4569–4571 10.1016/j.vaccine.2005.08.031 [DOI] [PubMed] [Google Scholar]
- Heine H. S., Bassett J., Miller L., Hartings J. M., Ivins B. E., Pitt M. L., Fritz D., Norris S. L., Byrne W. R. (2007). Determination of antibiotic efficacy against Bacillus anthracis in a mouse aerosol challenge model. Antimicrob Agents Chemother 51, 1373–1379 10.1128/AAC.01050-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heninger S., Drysdale M., Lovchik J., Hutt J., Lipscomb M. F., Koehler T. M., Lyons C. R. (2006). Toxin-deficient mutants of Bacillus anthracis are lethal in a murine model for pulmonary anthrax. Infect Immun 74, 6067–6074 10.1128/IAI.00719-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudva I. T., Griffin R. W., Garren J. M., Calderwood S. B., John M. (2005). Identification of a protein subset of the anthrax spore immunome in humans immunized with the anthrax vaccine adsorbed preparation. Infect Immun 73, 5685–5696 10.1128/IAI.73.9.5685-5696.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S. F., Ivins B. E., Fellows P. F., Pitt M. L., Norris S. L., Andrews G. P. (2004). Defining a serological correlate of protection in rabbits for a recombinant anthrax vaccine. Vaccine 22, 422–430 10.1016/j.vaccine.2003.07.004 [DOI] [PubMed] [Google Scholar]
- Lyons C. R., Lovchik J., Hutt J., Lipscomb M. F., Wang E., Heninger S., Berliba L., Garrison K. (2004). Murine model of pulmonary anthrax: kinetics of dissemination, histopathology, and mouse strain susceptibility. Infect Immun 72, 4801–4809 10.1128/IAI.72.8.4801-4809.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikesell P., Ivins B. E., Ristroph J. D., Dreier T. M. (1983). Evidence for plasmid-mediated toxin production in Bacillus anthracis. Infect Immun 39, 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock M., Fouet A. (2001). Anthrax. Annu Rev Microbiol 55, 647–671 10.1146/annurev.micro.55.1.647 [DOI] [PubMed] [Google Scholar]
- Moody K. L., Driks A., Rother G. L., Cote C. K., Brueggemann E. E., Hines H. B., Friedlander A. M., Bozue J. (2010). Processing, assembly and localization of a Bacillus anthracis spore protein. Microbiology 156, 174–183 10.1099/mic.0.033407-0 [DOI] [PubMed] [Google Scholar]
- Oliva C. R., Swiecki M. K., Griguer C. E., Lisanby M. W., Bullard D. C., Turnbough C. L., Jr, Kearney J. F. (2008). The integrin Mac-1 (CR3) mediates internalization and directs Bacillus anthracis spores into professional phagocytes. Proc Natl Acad Sci U S A 105, 1261–1266 10.1073/pnas.0709321105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering A. K., Osorio M., Lee G. M., Grippe V. K., Bray M., Merkel T. J. (2004). Cytokine response to infection with Bacillus anthracis spores. Infect Immun 72, 6382–6389 10.1128/IAI.72.11.6382-6389.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt M. L. M., Little S., Ivins B. E., Fellows P., Boles J., Barth J., Hewetson J., Friedlander A. M. (1999). In vitro correlate of immunity in an animal model of inhalational anthrax. J Appl Microbiol 87, 304 10.1046/j.1365-2672.1999.00897.x [DOI] [PubMed] [Google Scholar]
- Popov S. G., Popova T. G., Grene E., Klotz F., Cardwell J., Bradburne C., Jama Y., Maland M., Wells J. & other authors (2004). Systemic cytokine response in murine anthrax. Cell Microbiol 6, 225–233 10.1046/j.1462-5822.2003.00358.x [DOI] [PubMed] [Google Scholar]
- Powell A. G., Crozier J. E., Hodgson H., Galloway D. J. (2011). A case of septicaemic anthrax in an intravenous drug user. BMC Infect Dis 11, 21 10.1186/1471-2334-11-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond C., Baillie L. W., Hibbs S., Moir A. J., Moir A. (2004). Identification of proteins in the exosporium of Bacillus anthracis. Microbiology 150, 355–363 10.1099/mic.0.26681-0 [DOI] [PubMed] [Google Scholar]
- Ringertz S. H., Høiby E. A., Jensenius M., Maehlen J., Caugant D. A., Myklebust A., Fossum K. (2000). Injectional anthrax in a heroin skin-popper. Lancet 356, 1574–1575 10.1016/S0140-6736(00)03133-0 [DOI] [PubMed] [Google Scholar]
- Ross J. M. (1957). The pathogenesis of anthrax following the administration of spores by the respiratory route. J Pathol Bacteriol 73, 485–494 10.1002/path.1700730219 [DOI] [Google Scholar]
- Severson K. M., Mallozzi M., Bozue J., Welkos S. L., Cote C. K., Knight K. L., Driks A. (2009). Roles of the Bacillus anthracis spore protein ExsK in exosporium maturation and germination. J Bacteriol 191, 7587–7596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivachandra S. B., Li Q., Peachman K. K., Matyas G. R., Leppla S. H., Alving C. R., Rao M., Rao V. B. (2007). Multicomponent anthrax toxin display and delivery using bacteriophage T4. Vaccine 25, 1225–1235 10.1016/j.vaccine.2006.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhov E. N., Rubinstein E. (1994). Human live anthrax vaccine in the former USSR. Vaccine 12, 727–730 10.1016/0264-410X(94)90223-2 [DOI] [PubMed] [Google Scholar]
- Steichen C., Chen P., Kearney J. F., Turnbough C. L., Jr (2003). Identification of the immunodominant protein and other proteins of the Bacillus anthracis exosporium. J Bacteriol 185, 1903–1910 10.1128/JB.185.6.1903-1910.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen C. T., Kearney J. F., Turnbough C. L., Jr (2005). Characterization of the exosporium basal layer protein BxpB of Bacillus anthracis. J Bacteriol 187, 5868–5876 10.1128/JB.187.17.5868-5876.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre P., Couture-Tosi E., Mock M. (2002). A collagen-like surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol Microbiol 45, 169–178 10.1046/j.1365-2958.2000.03000.x [DOI] [PubMed] [Google Scholar]
- Sylvestre P., Couture-Tosi E., Mock M. (2003). Polymorphism in the collagen-like region of the Bacillus anthracis BclA protein leads to variation in exosporium filament length. J Bacteriol 185, 1555–1563 10.1128/JB.185.5.1555-1563.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre P., Couture-Tosi E., Mock M. (2005). Contribution of ExsFA and ExsFB proteins to the localization of BclA on the spore surface and to the stability of the Bacillus anthracis exosporium. J Bacteriol 187, 5122–5128 10.1128/JB.187.15.5122-5128.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson B. M., Hsieh H.-Y., Spreng K. A., Stewart G. C. (2011). The co-dependence of BxpB/ExsFA and BclA for proper incorporation into the exosporium of Bacillus anthracis. Mol Microbiol 79, 799–813 10.1111/j.1365-2958.2010.07488.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd S. J., Moir A. J., Johnson M. J., Moir A. (2003). Genes of Bacillus cereus and Bacillus anthracis encoding proteins of the exosporium. J Bacteriol 185, 3373–3378 10.1128/JB.185.11.3373-3378.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull P. C. (1991). Anthrax vaccines: past, present and future. Vaccine 9, 533–539 10.1016/0264-410X(91)90237-Z [DOI] [PubMed] [Google Scholar]
- Turnbull P. (2008). Anthrax in Humans and Animals, 4th edn Geneva: WHO Press [Google Scholar]
- Vergis J. M., Cote C. K., Bozue J., Alem F., Ventura C. L., Welkos S. L., O'Brien A. D. (2011). Protection from challenge with B. anthracis Sterne or Ames after immunization with plasmid-cured B. cereus G9241 spores. In International Bacillus-ACT Meeting, p. 74, Bruges, Belgium. [Google Scholar]
- Wasserman G. M., Grabenstein J. D., Pittman P. R., Rubertone M. V., Gibbs P. P., Wang L. Z., Golder L. G. (2003). Analysis of adverse events after anthrax immunization in US Army medical personnel. J Occup Environ Med 45, 222–233 10.1097/01.jom.0000058345.05741.6b [DOI] [PubMed] [Google Scholar]
- Welkos S. L., Friedlander A. M. (1988). Comparative safety and efficacy against Bacillus anthracis of protective antigen and live vaccines in mice. Microb Pathog 5, 127–139 10.1016/0882-4010(88)90015-0 [DOI] [PubMed] [Google Scholar]
- Welkos S. L., Keener T. J., Gibbs P. H. (1986). Differences in susceptibility of inbred mice to Bacillus anthracis. Infect Immun 51, 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welkos S. L., Becker D., Friedlander A., Trotter R. (1989). Pathogenesis and host resistance to Bacillus anthracis: a mouse model. Salisbury Med Bull 68, 49–52 [Google Scholar]
- Welkos S. L., Vietri N. J., Gibbs P. H. (1993). Non-toxigenic derivatives of the Ames strain of Bacillus anthracis are fully virulent for mice: role of plasmid pX02 and chromosome in strain-dependent virulence. Microb Pathog 14, 381–388 10.1006/mpat.1993.1037 [DOI] [PubMed] [Google Scholar]
- Welkos S. L., Cote C. K., Rea K. M., Gibbs P. H. (2004). A microtiter fluorometric assay to detect the germination of Bacillus anthracis spores and the germination inhibitory effects of antibodies. J Microbiol Methods 56, 253–265 10.1016/j.mimet.2003.10.019 [DOI] [PubMed] [Google Scholar]
- Williamson E. D., Hodgson I., Walker N. J., Topping A. W., Duchars M. G., Mott J. M., Estep J., Lebutt C., Flick-Smith H. C. & other authors (2005). Immunogenicity of recombinant protective antigen and efficacy against aerosol challenge with anthrax. Infect Immun 73, 5978–5987 10.1128/IAI.73.9.5978-5987.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J. A., Collier R. J. (2007). Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem 76, 243–265 10.1146/annurev.biochem.75.103004.142728 [DOI] [PubMed] [Google Scholar]