

Abstract
Staphylococcus aureus is the leading cause of nosocomial infections and a major cause of community-acquired infections. Biofilm formation is a key virulence determinant in certain types of S. aureus infection, especially those involving inserted medical devices. We found in a previous study that the calcium chelators sodium citrate and EGTA inhibit biofilm formation in certain strains of S. aureus but actually augment biofilm formation in other strains. Even two closely related strains, Newman and 10833, exhibited strikingly different biofilm phenotypes in the presence of calcium chelators, in that biofilm formation was inhibited in Newman but augmented in 10833. We also found that the surface protein clumping factor B (ClfB) plays a role in this phenomenon. In this study, we confirm that ClfB is required for biofilm formation under calcium-depleted conditions. We investigated the post-translational regulation of ClfB-mediated biofilm formation and found evidence that both calcium and the protease aureolysin disrupt established ClfB-dependent biofilms. Finally, we investigated the genetic basis for the biofilm-negative phenotype in strain Newman versus the biofilm-positive phenotype in strain 10833 under calcium-depleted conditions and found that strain 10833 contains a deletion that results in a stop codon within the aureolysin gene (aur). When 10833 expressed Newman aur, surface-associated ClfB and the ability to form a biofilm in chelating conditions was lost. Thus, the positive effect of chelating agents on biofilm formation in certain strains can be explained by increased ClfB activity in the absence of calcium and the discrepancy in the response of strains 10833 and Newman can be explained by point mutations in aur. This study reveals a previously unknown, to our knowledge, role for ClfB in biofilm formation and underscores the potential for striking phenotypic variability resulting from minor differences in strain background.
Introduction
Staphylococcus aureus is the causative agent of a diverse array of acute and chronic infections and an important virulence factor is its capacity to form tenacious biofilms on surfaces. Surfaces that can be colonized by S. aureus biofilms range from medical implants, such as catheters and orthopaedic implants, to a variety of host tissue as exemplified by osteomyelitis and infective endocarditis. An estimated 250 000–500 000 infections related to intravenous catheter use, 1–6 per 1000 catheter days, occur every year in the USA (Crnich & Maki, 2005; Maki et al., 2006; Ramos et al., 2011). S. aureus is one of the most common isolates (20 %) and is associated with greater health care costs than any other species isolated (Walz et al., 2010). Biofilm formation on the intraluminal or extraluminal surface of the catheter leads to these infections. S. aureus encodes a variety of components that can play a role in the establishment of a biofilm, depending on strain background and environmental conditions. One of the major factors is poly-N-acetylglucosamine (PNAG), which is encoded by the intercellular adhesin locus (Cramton et al., 1999). Other factors include proteinaceous adhesins and extracellular DNA.
We found in a separate study (Abraham et al., 2012), that subinhibitory concentrations of the chelating agents trisodium citrate and EGTA effectively reduce biofilm formation in a subset of S. aureus isolates, whereas in some strains, they do not exert an appreciable effect, and in other strains they actually significantly augment biofilm formation. The finding that chelating agents can augment biofilm formation in some strains may have important clinical implications as citrate is used in catheter lock solutions such as Citra-Lock and DuraLock-C (although the FDA does not approve citrate for this use in the USA (O’Grady et al., 2011). We also found in this study, that clumping factor B (ClfB) plays a role in chelator-induced biofilm formation. ClfB is a 150 kDa protein with a C-terminal LPXTG sorting motif, which is used to covalently anchor the MSCRAMM (microbial surface component recognizing adhesive matrix molecule) to the cell wall peptidoglycan (Schneewind et al., 1993). The N-terminal A domain mediates fibrinogen binding and subsequently can lead to bacterial adherence to immobilized fibrinogen, blood clots, conditioned biomaterial ex vivo and thrombin-damaged heart valves in a rat model of infective endocarditis (Entenza et al., 2000; François et al., 2000; McDevitt et al., 1994; Ní Eidhin et al., 1998; O’Brien et al., 2002; Vaudaux et al., 1995). The A domain of ClfB contains a short SLAVA motif, which is a putative protease recognition sequence (McDevitt et al., 1994). The A region also appears to contain an abbreviated EF-Hand domain (Ní Eidhin et al., 1998). EF-Hand domains bind calcium ions (Ca2+) specifically (Michiels et al., 2002). Ca2+-binding has been shown to interfere with the binding activity of a number of EF-Hand domain-containing proteins and inhibits the ability of clumping factor A (ClfA) and ClfB to bind fibrinogen and the ability of the biofilm associated protein (Bap) to contribute to biofilm formation (Arrizubieta et al., 2004; Ní Eidhin et al., 1998; O’Connell et al., 1998).
Despite previous reports that suggest ClfB mediates clumping only in the presence of fibrinogen (Miajlovic et al., 2007; Ní Eidhin et al., 1998), we confirmed in this study that fibrinogen is not required for ClfB-mediated aggregation and that ClfB is an important mediator of biofilm formation in the presence of EGTA and trisodium citrate. We sought to confirm our hypothesis that chelators induce biofilm formation through the removal of divalent cations and found that calcium disrupts biofilms pre-formed in EGTA. We also sought to determine the basis for the very different effect of EGTA on biofilm formation in strain Newman versus 10833 and found that ClfB-dependent biofilms are disrupted by the metalloprotease aureolysin, that 10833 aur contains a null mutation and that ClfB accumulates on the surface of 10833 in the absence of aureolysin activity.
Methods
Bacterial strains and media.
Escherichia coli strain CH3-Blue (Bioline) was used for cloning plasmids and was grown in Luria–Bertani medium containing 100 µg ampicillin ml−1 for selection of the plasmid. S. aureus RN4220 is a restriction-deficient strain derived from the laboratory strain NCTC 8325 (Kreiswirth et al., 1983). Two ATCC deposits were made of derivatives of the same original coagulase-producing isolate from a case of osteomyelitis by two different people. Strain Newman was deposited by E. S. Duthrie in 1950 and given the strain designation ATCC 13420 (now referred to as NCTC 8178), and Newman D2c (NCTC 10833) was deposited by J. Hawiger in 1972 and given the strain designation ATCC 25904 (Grundmeier et al., 2004). From here on, we refer to NCTC 8178 as Newman and NCTC 10833 as 10833. Newman fails to produce a substantial biofilm in the presence of EGTA. Strain 10833 produces a stronger biofilm in the presence of EGTA and USA300 JE2, a USA300 LAC community-acquired meticillin-resistant S. aureus strain isolated from the Los Angeles County Jail that has been cured of the three plasmids (Kennedy et al., 2008), produces stronger biofilms in the presence of EGTA. Strains NE163, NE551, NE186, NE543 and NE391 from the Nebraska Transposon Mutant Library were obtained from the NARSA repository and carry the bursa aurealis mariner-based, erythromycin resistance-expressing transposon within the genes encoding aureolysin, fibronectin-/fibrinogen-binding protein, fibrinogen binding protein A, ClfA and ClfB, respectively. The transposon was transduced to Newman and 10833 using phage 80 as described by Kasatiya & Baldwin (1967). For complementation of the mutation, aur from 10833 or Newman was cloned into the IPTG-inducible vector pCL15 (Luong & Lee, 2006). Primers aur-pCL15Fwd (5′-GTGAGGAAATTTTCAAGATATG-3′) and aur-pCL15Rev (5′-TTACTCCACGCCTACTTCATTCC-3′) were used to amplify the aur gene from 10833 or Newman genomic DNA. The PCR product was digested with BamHI and KpnI and ligated to pCL15 to produce Newman (pNaur) or 10833 (p1aur) aur-expressing pCL15. Plasmids were purified and transformed into RN4220 and selected on tryptic soy agar (TSA) containing chloramphenicol (cm) before the constructs were transduced into strains 10833 and Newman by phage 80α. Strains harbouring aur-pCL15 were cultured in the presence of 10 µg cm ml−1 and 1 mM IPTG.
The clfB gene was replaced with an erythromycin (erm) resistance cassette in S. aureus strain RN4220 by homologous recombination using the pMAD vector (kindly provided by Michel Débarbouillé and Maryvonne Arnaud, Pasteur Institute, Paris, France) as described by Arnaud et al. (2004). Genomic DNA from strain SA113 was used as a template for PCR using primers clfBSoe1 (5′-AGATCTGCACAAGGTAAGTTTGTGGAAGGAT-3′) and clfBSoe4 (5′-AGATCTCCTTGTTCAATTCAGCAATGAGTTGATCG-3′) and the PCR product was cloned into pCR4-TOPO (Invitrogen). The resulting construct was amplified using primers clfBSoe2 (5′-CTCGAGCGTCTAATCGAATACTTATTCTGCTTATTCGAC-3′) and clfBSoe3 (5′-CAGAATAAGTATTCGATTAGACGCTCGAGCAAATGCAACTTTATTTGGTGCAATGATGGC-3′), digested with XhoI, and ligated to an erm resistance cassette excised from plasmid pSC57 with XhoI. The resulting plasmid was digested with BamHI and the region surrounding the clfB gene and the intervening erm cassette were ligated to pMAD. This construct was electroporated into strain RN4220 (Lee, 1993) and homologous recombination was selected for as described by Arnaud et al. (2004). Once gene replacements in RN4220 were confirmed by PCR, the mutations were then transduced into S. aureus strains 10833 and Newman by using phage 80α and transductants were selected on TSA plates containing 10 µg erm ml−1.
Deletion of the ica locus to produce strains 10833Δica : : tet, NewmanΔica : : tet, 10833Δica : : tetΔclfB : : erm and NewmanΔica : : tetΔclfB : : erm was performed by phage transduction from SA113Δica : : tet (Cramton et al., 1999). All S. aureus strains used in this study were grown aerobically at 37 °C on tryptic soy broth (TSB) or agar plates supplemented with the appropriate antibiotic.
Static biofilm assay.
Biofilm assays were performed essentially as described by Christensen et al. (1985). Briefly, overnight planktonic cultures of S. aureus were diluted to a final OD600 of 0.015 in fresh medium and 200 µl of the culture was added into individual wells of a 96-well tissue culture-treated polystyrene plates (Cellstar, Greiner Bio-one). The cells were grown in TSB supplemented with 1 % glucose (TSBG) or medium supplemented with 12.5 mM EGTA or 12.5 mM trisodium citrate (these concentrations were previously determined to be 50 % of the MIC for these compounds). The plates were incubated at 37 °C for 18 h, spent medium was removed, and the wells were washed once with 200 µl sterile water, dried and stained with safranin. The biofilms were visually inspected and quantified by dissolving the stain with 200 µl 33 % (v/v) acetic acid and determining the A415 using a 96-well plate spectrophotometer (BioTek 800 Plate reader). Replica plates were used to determine total growth (biofilm and planktonic) at 595 nm. Results for these experiments were compiled from three biological replicates each of which contained five technical replicates.
Antiserum inhibition assay of biofilm formation.
Overnight cultures of each strain of S. aureus were diluted to a final OD600 of 0.015 in TSBG or TSBG+12.5 mM EGTA containing varying concentrations of either ClfB antiserum or rabbit non-immune sera (0–16.0 µg ml−1) and added to wells of a 96-well plate. ClfB antiserum was raised against recombinant, purified N-terminal ClfB domains N1,2,3 (amino acids 44–542) (Ní Eidhin et al., 1998) and was kindly provided by Clarissa Pozzi and Gerald Pier, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA. The plate was incubated at 4 °C for 2 h to allow antiserum binding, followed by 37 °C for 18 h. Non-adherent cells were removed by washing with water and adherent bacteria were stained with safranin. The percentage inhibition of biofilm formation was determined using the formula: [1−Biofilm415 nm (with antiserum)/Biofilm415 nm (without antiserum)]×100 (Hussain et al., 1997). To measure clumping, overnight cultures of strains 10833 and Newman were diluted to a final OD600 of 0.015 in 2 ml TSBG or TSBG containing 12.5 mM EGTA. Either no serum, 4 µg ClfB-specific antiserum ml−1 or 4 µg non-immune rabbit serum ml−1 was added. The cultures were incubated, without shaking, at 37 °C for 18 h. Tubes were lightly vortexed and visually inspected for clumping.
Western blot analysis of ClfB.
The ClfB antiserum was used to detect bacterial surface-associated ClfB. Cultures of 10833, 10833aur : : Tn, 10833aur : : Tn+p1aur, 10833aur : : Tn+pNaur, Newman, Newmanaur : : Tn, Newmanaur : : Tn+p1aur and Newmanaur : : Tn+pNaur were grown overnight in six-well tissue culture plates in TSBG+12.5 mM EGTA. Bacteria (planktonic and biofilm) were collected, and cell wall-associated proteins including ClfB were released from the bacterial surface by lysostaphin, separated by SDS-PAGE and analysed by Western blot using the ClfB antiserum as described by McAleese et al. (2001).
Dispersal/protection assay.
Biofilms were pre-established on individual wells of 96-well plates as described above. After 18 h of stationary incubation, the spent medium was removed and replaced with an equivalent volume of varying concentrations of CaCl2, MgCl2 or MnCl2, or purified aureolysin (Axxora LLC), diluted in deionized water and allowed to incubate for another 18 h at 37 °C. The biofilms retained on the plate were washed and stained with safranin as described above.
Sequence analysis of 10833 and Newman aur.
Genomic DNA was isolated from 10833 or Newman using the DNeasy tissue kit (Qiagen). The aur gene was amplified by PCR using a pair of external (5′-CGATTATTGCGTCTTACATAGTTG-3′ and 5′-AGACAACCCTCACACTCCTCTC-3′) and a pair of internal primers (5′-GTATTGACGGTGGATTTAGCC-3′ and 5′-GCACGTTGCTCATCTTTTACG-3′). Sequence translation and alignment were performed using the StarORF open reading frame finder tool (http://web.mit.edu/star/orf/).
Statistical analysis.
Statistical tests were performed using Student’s t-test and P<0.05 was considered significant.
Results
ClfB antiserum inhibits biofilm formation in 10833 and USA300 JE2
We found previously (Abraham et al., 2012) that a subset of S. aureus strains produce strong biofilms in the presence of the chelating agents EGTA and trisodium citrate. We also found that if we deleted clfB in these strains, the ability to produce a biofilm under chelating conditions was nearly completely abolished. This result was surprising because a role for ClfB in biofilm formation has never been established and because its capacity to promote aggregation was previously thought to depend upon the presence of fibrinogen. Therefore, to confirm the requirement for ClfB under chelating conditions, we determined the effects of an antiserum specific for ClfB on biofilm formation in the presence of EGTA. Strains 10833 and USA300 JE2 are strains that form biofilms in EGTA, whereas biofilm formation by strain Newman is inhibited by EGTA. These three strains were cultured in either TSBG or TSBG+12.5 mM EGTA containing ClfB antiserum or non-immune serum. Biofilm formation by 10833 in EGTA (Fig. 1) was inhibited by increasing concentrations of the ClfB antiserum and concentrations greater than 4.0 µg ml−1 dramatically inhibited biofilm formation (>65 % inhibition, P<0.0001). To confirm that ClfB antiserum was not affecting growth, total growth (biofilm and planktonic) was determined at 595 nm (Fig. 1, green dots). The ClfB antiserum did not affect growth of this strain in EGTA at any concentration tested. In contrast, biofilms produced by this strain in TSBG alone remained robust and intact at every concentration of the ClfB antiserum tested. The non-immune control serum did not inhibit biofilms produced in TSBG or EGTA. Strain Newman (Fig. S1a, available with the online version of this paper) fails to form significant biofilms in TSBG containing EGTA; therefore, we only tested the effect of the antiserum on biofilm formation in TSBG alone. Biofilms produced by Newman in TSBG were not affected by the ClfB antiserum. Fig. S1(b) illustrates that results obtained using strain USA300 JE2 were similar to 10833. Because ClfB has previously been associated with aggregation and clumping, we repeated these experiments in culture tubes to observe clumping and found similar results, that the antiserum inhibited aggregation of 10833 and USA300 JE2 when these strains were cultured in TSBG containing EGTA (Fig. S2). We also tested the role of other known fibronectin- and fibrinogen-binding proteins in biofilm formation in the presence of EGTA and found that only ClfB played a role, whereas deletion of the genes encoding ClfA, the fibrinogen/fibronectin-binding protein and fibrinogen-binding protein A did not affect biofilm formation in EGTA (Fig. S3). Together, these results confirm that biofilms formed in EGTA are mediated by ClfB, that clumping in EGTA depends upon ClfB and that these ClfB-mediated effects do not depend on fibrinogen. The results also suggest that biofilms formed in TSBG alone are mediated by factors other than ClfB.
Fig. 1.
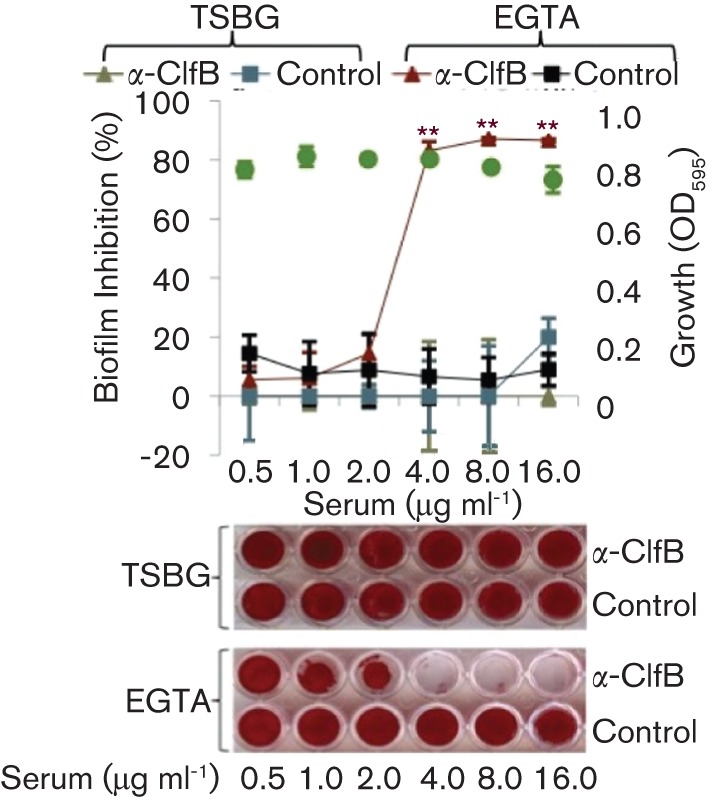
Biofilm formation in EGTA is inhibited by ClfB antiserum (α-ClfB). Strain 10883 was cultured in 96-well polystyrene plates in TSBG+12.5 mM EGTA and treated with varying concentrations of either ClfB antiserum or rabbit non-immune serum (0.5–16.0 µg ml−1) or in the absence of any added serum. Non-adherent cells were removed and adherent bacteria were stained with safranin. For quantitative data the safranin was solubilized and the A415 was recorded. The percentage inhibition of biofilm formation was determined using the formula: [1−Biofilm415 nm (with antiserum)/Biofilm415 nm (without antiserum)]×100. Representative biofilm images are shown below the graph. Growth (OD595, green dots) was assessed in the presence of the α-ClfB antibody. **P<0.0001 as determined by Student’s t-test. Values shown are mean; error bars represent ±sd.
The intercellular adhesin locus does not influence biofilm formation in EGTA
To confirm that components other than ClfB, such as PNAG, play a role in biofilm formation in the absence of EGTA, we analysed biofilm formation by 10833 mutants that lacked either the intercellular adhesin locus (ica) or both ica and clfB. As shown in Fig. 2, deletion of ica alone did not affect biofilm formation in the presence of EGTA but the clfB deletion prevented biofilm formation. However, in the absence of EGTA, the ica deletion partially abrogated biofilm formation and the additional clfB deletion had no effect. This supports our hypothesis that in the absence of the chelating agent EGTA, ClfB is not required for biofilm formation but other biofilm components, such as PNAG, play a role instead.
Fig. 2.
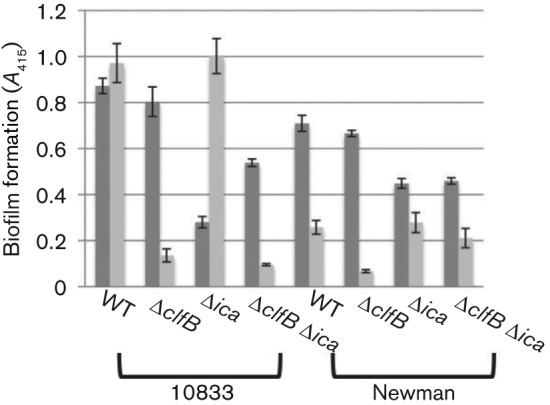
Deletion of the intercellular adhesin locus abrogates biofilm formation in the absence of EGTA. Strains 10833, Newman and their respective ica and clfB single and double deletion mutants were tested for biofilm formation in TSBG (dark grey) and TSBG+12.5 mM EGTA (light grey). Biofilms were stained with safranin, the safranin was solubilized, and A415 was recorded. Results were averaged from three biological replicates, each with four technical replicates. Bars represent mean±sd.
Aureolysin inhibits biofilm formation in EGTA
The Zn2+-dependent metalloprotease aureolysin has been shown previously to cleave ClfB between Ser197 and Leu198, or between Ala199 and Val200 within the SLAVA motif, thereby preventing the protein from binding immobilized fibrinogen (McAleese et al., 2001; Perkins et al., 2001). Therefore, we hypothesized that the difference in the capacities for biofilm formation by 10833 and Newman in EGTA was related to aureolysin-mediated ClfB degradation. We tested aur transposon mutants for biofilm formation under chelating and non-chelating conditions. In strain 10833, which already produces a strong biofilm in EGTA, a transposon insertion into aur did not affect the biofilm-forming ability; however, in strain Newman, the aur mutation resulted in a substantial increase (P<0.0001) in biofilm in EGTA and trisodium citrate (Fig. 3). These results suggest that in strain 10833, aureolysin does not affect biofilm formation under chelating conditions but that in strain Newman, it significantly inhibits it. We hypothesized that aureolysin had the capacity to cleave ClfB from the cell surface and disrupt 10833 biofilms but that due to low expression levels or possibly a mutation that affected function, it did not perform this function in this strain. To test this hypothesis, 10833, 10833aur : : Tn and Newmanaur : : Tn biofilms were pre-established in TSBG or TSBG containing 12.5 mM EGTA. After removing non-adherent bacteria, the biofilms were treated with various concentrations of aureolysin. As expected, 10833 (Fig. 4) displayed adherent biofilms when grown in either TSBG or 12.5 mM EGTA in the absence of any added aureolysin and 10833aur : : Tn displayed a similar trend (Fig. S4a). However, increasing concentrations of aureolysin promoted dispersion of the biofilms established in EGTA and ≥5 mM aureolysin prevented any detectable biofilm from being retained (P<0.0001). In contrast, the biofilms pre-established within TSBG were not affected by aureolysin treatment. Results obtained with Newmanaur : : Tn (Fig. S4b) were similar to 10833 with dispersion of biofilms pre-established in EGTA following treatment with ≥5 mM aureolysin (P<0.0001). Together these results demonstrate that aureolysin disrupts biofilms established under chelating conditions and suggest that it mediates this effect through the degradation of ClfB. They also suggest that in strain 10833, endogenous aureolysin activity is too low to inhibit or disrupt biofilms formed in EGTA.
Fig. 3.
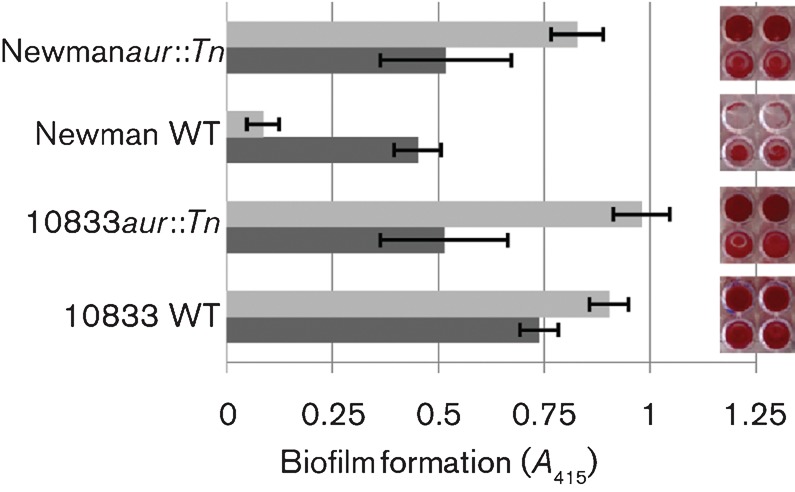
Insertional inactivation of the aureolysin gene (aur) augments biofilm formation by Newman in EGTA. Strains 10833 and Newman and their respective aur transposon mutants were tested for biofilm formation in TSBG (dark grey) and TSBG+12.5 mM EGTA (light grey). Safranin-stained biofilms are shown to the right and the quantified A415 results obtained from three biological replicates, each with four technical replicates, are shown on the left. Bars represent mean±sd.
Fig. 4.
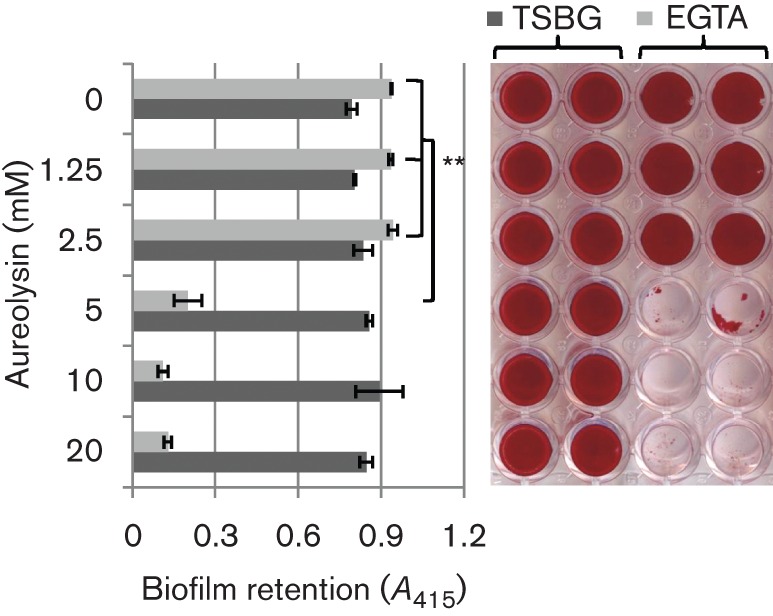
Biofilms pre-established in EGTA are dispersed by exogenous aureolysin. Biofilms pre-established by S. aureus strain 10883 were treated with varying concentrations of purified aureolysin for 18 h at 37 °C. The remaining biofilms were stained with safranin and imaged (right). For quantitative data, the safranin was solubilized and the A415 was recorded. Three biological replicates, each with four technical replicates, were used to generate the statistical analysis. **P<0.0001 as determined by Student’s t-test. Bars represent mean±sd.
The Newman aur gene encodes functional aureolysin whereas 10833 aur is non-functional
We hypothesize that 10833 produces either very little aureolysin or none at all. To test this, we cloned aur genes from both 10833 and Newman into an inducible expression vector so that gene expression would not be background-dependent. We then expressed 10833 aur in both 10833aur : : Tn and Newmanaur : : Tn and Newman aur in both 10833aur : : Tn and Newmanaur : : Tn. When 10833aur : : Tn and Newmanaur : : Tn were complemented with a plasmid-borne copy of 10833 aur (p1aur), the biofilm phenotypes of the complemented strains were indistinguishable from the deletion mutants and we observed strong biofilm formation in EGTA (Fig. 5a). However, when 10833aur : : Tn and Newmanaur : : Tn were complemented with a plasmid-borne copy of Newman aur (pNaur), biofilm formation in EGTA was inhibited in both Newmanaur : : Tn and 10833aur : : Tn. We found similar results for clumping activity as for biofilm forming activity (Fig. S5). These results suggest that the 10833 aur gene itself, rather than expression of the gene, differed from the Newman aur gene and that in 10833, aureolysin activity was low enough to allow for expression of ClfB on the bacterial cell surface. To prove that ClfB was able to accumulate on the surface of 10833 and contribute to biofilm formation, we performed Western blot analysis of surface-associated proteins. As shown previously, ClfB (150 kDa) was not detected on the surface of Newman in stationary cultures (Ní Eidhin et al., 1998); however, a slight band was visible at 120 kDa (Fig. 5b, lane 5). This 120 kDa band has been previously reported and appears to be ClfB that has been partially degraded by aureolysin (McAleese et al., 2001). In contrast, ClfB was detected on the surface of 10833 (lane 1). Deletion of aur in Newman resulted in ClfB expression (lane 6) but did not have an effect on 10833 (lane 2) and expression of 10833 aur had no effect on ClfB expression (lanes 3 and 7), whereas expression of Newman aur resulted in loss of the 150 kDa ClfB protein (lanes 4 and 8).
Fig. 5.
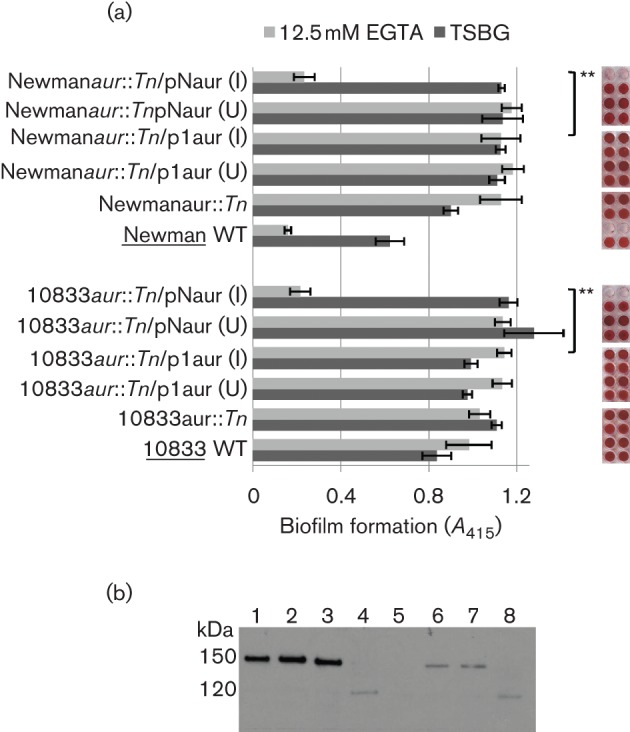
Strain 10833 aur fails to complement the aur deletion in Newman. (a) Strains 10833aur : : Tn and Newmanaur : : Tn were complemented with either 10833 (p1aur) or Newman aur (pNaur) and tested for biofilm formation in the presence or absence of EGTA. Complementation was induced (I) in the presence of 1 mM IPTG; uninduced samples (U) had no IPTG. Statistical analysis was performed using three independent biological replicates, each containing four technical replicates. **P<0.0001 as determined by Student’s t-test. Bars represent mean±sd. (b) Western blot analysis of surface-expressed ClfB in EGTA. Lanes: 1, 10833; 2, 10833aur : : Tn; 3, 10833aur : : Tn+p1aur; 4, 10833aur : : Tn+pNaur; 5, Newman; 6, Newmanaur : : Tn; 7, Newmanaur : : Tn+p1aur; 8, Newmanaur : : Tn+pNaur.
In order to determine the basis for the lack of activity associated with 10833 aureolysin, we compared the aur gene sequences from Newman and 10833. The 10833 aur coding sequence contained 4 nt changes, one of which, a deletion of thymine 565, resulted in a frame shift of the protein coding sequence and a TAG stop codon. Therefore, translation of 10833 aur message would stop prematurely and full-length, active protein would not be expected to be produced. Thus, the molecular basis for the phenotypic disparity in biofilm-forming activity between strains 10833 and Newman is the lack of a functional aur gene in 10833, which leads to the accumulation of ClfB on the surface and biofilm formation in the presence of chelating agents such as EGTA.
Calcium induces dispersal of EGTA-induced biofilms
Our data suggested that a mutation in 10833 aur explained the strain-dependent difference in the abilities of 10833 and Newman to form biofilms in the presence of chelators but we also wanted to determine why 10833 formed ClfB-dependent biofilms under chelating conditions but formed ClfB-independent biofilms in TSBG alone. We hypothesized that in TSBG, Ca2+ inhibited the autoaggregative activity mediated by the helix–loop EF-Hand domain of ClfB and that EGTA alleviated this inhibition by removing free Ca2+. To test this hypothesis, we pre-established ClfB-dependent 10833 biofilms in TSBG+12.5 mM EGTA and treated them with varying concentrations of CaCl2, MgCl2 and MnCl2 to determine their effect on biofilm dispersal (Fig. 6a). Ca2+ induced biofilm dispersal in a concentration-dependent manner and 3.125 mM Ca2+ prevented any visible biofilm from being retained (P<0.0001). Neither Mg2+, Mn2+ nor water (vehicle control) affected the biofilms.
Fig. 6.
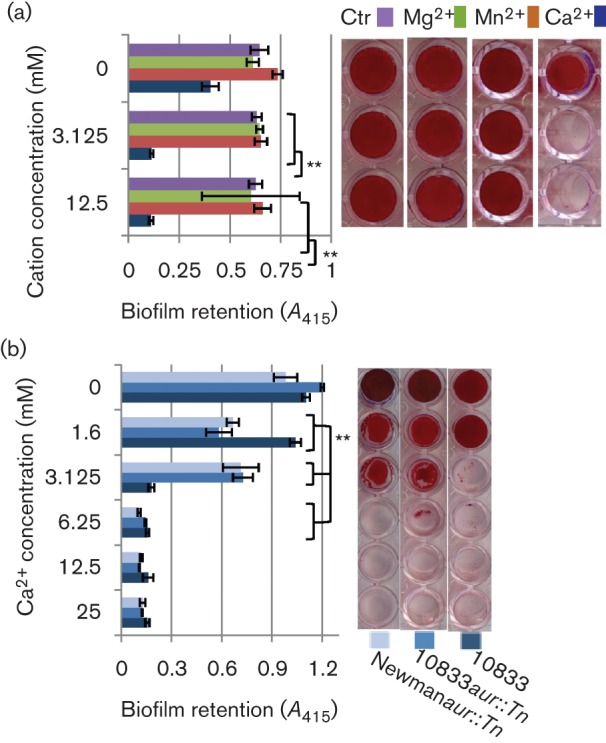
Ca2+ ions disperse S. aureus biofilms pre-established in EGTA. (a) Strain 10833 was grown to pre-establish biofilms within 12.5 mM EGTA and thereafter treated with varying concentrations of divalent cations (Mg2+, Mn2+ or Ca2+) or deionized water (Ctr). Once the biofilms were treated with the cations or water they were washed to remove non-adherent bacteria, stained (right) and quantified at 415 nm (left). (b) The effect of the Ca2+ ions was independent of aureolysin as determined by pre-establishing biofilms of strains 10833 wild-type or 10833 and Newman aur mutants in 12.5 mM EGTA which were thereafter treated with varying concentrations of Ca2+. The experiments were performed in biological triplicate with each condition being tested in technical quadruplets. **P<0.0001 as determined by Student’s t-test. Bars represent mean±sd.
Aureolysin reportedly requires Ca2+ for maximum activity. Therefore, the possibility remained that Ca2+-induced dispersal of the biofilms was caused by activation of aureolysin. To support our hypothesis that Ca2+ mediated biofilm dispersal by inhibiting ClfB activity and not by inducing aureolysin activity, we tested the effect of Ca2+on 10833aur : : Tn and Newmanaur : : Tn biofilms pre-established in TSBG containing EGTA. As illustrated in Fig. 6(b), Ca2+ induced the dispersal of biofilms produced by both aur mutants. Thus, these results indicate that Ca2+ induces dispersal of pre-established ClfB-mediated biofilms and suggest that EGTA mediates the formation of ClfB-mediated biofilms by removing free Ca2+. They also suggest that the effect of Ca2+ removal is mediated mainly through ClfB activity rather than through aureolysin activity.
Discussion
Biofilm formation is an important virulence mechanism associated with S. aureus and is involved in catheter-related infections, a significant and growing worldwide concern. Catheter lock solutions containing chemical chelators like trisodium citrate and EDTA are anti-thrombotic and have antimicrobial and antibiofilm effects. In rare cases, however, rapid infusion of citrate has resulted in toxicity and it is therefore not approved by the FDA for locking catheters in the USA (O’Grady et al., 2011). Further supporting the potential dangers of citrate in catheter lock solutions, we found (Abraham et al., 2012), a subgroup of strains including 10833, a close relative of sequenced strain Newman, that actually form thicker biofilms in the presence of Ca2+-chelating agents. In the current study, we confirmed the suspected requirement for the MSCRAMM protein ClfB in these chelator-induced biofilms.
ClfB is a large surface-associated protein, which has been shown to bind fibrinogen. However, we have found that ClfB mediates aggregation even in the absence of fibrinogen and we have confirmed in this study that it is required for the formation of biofilm under chelating conditions. We also sought to answer two additional questions: ‘How is it that 10833 can form ClfB-dependent biofilms in chelating conditions, but the closely related strain Newman fails to form biofilms under these conditions?’ and ‘Why is it that biofilms formed in the presence of chelators are ClfB-dependent but biofilms formed in the absence of chelators are ClfB-independent?’.
To address the first question, we tested our hypothesis that ClfB activity was differentially regulated in the two strains. Previous work accomplished by McAleese and colleagues demonstrated that aureolysin (aur) regulates ClfB stability by cleaving it, resulting in decreased binding to fibrinogen (McAleese et al., 2001). We found that aureolysin was, in fact, able to negatively regulate ClfB-dependent biofilm formation and that the striking disparity between the ability of 10833 and Newman to form biofilms in the presence of chelators was due to the fact that Newman encodes functional aureolysin, which presumably cleaves ClfB and prevents ClfB-mediated biofilm formation whereas 10833 has a mutation in aur that results in premature termination of translation and a lack of functional aureolysin. Thus, ClfB remains stable and mediates biofilm formation in this strain. Our study does not rule out the presence of additional differences between 10833 and Newman that could contribute to the difference in chelator-induced biofilm-forming activity, but the data suggest that the sequence differences in aur play a major role. To investigate the possibility that aur mutations may contribute to ClfB-mediated biofilm formation in other strains, we compared the aur genes of 80 previously sequenced S. aureus strains. Out of these, we found only two, TCH60 and ST38, with mutations leading to premature stop codons. Because we found in our earlier study (Abraham et al., 2012) that nearly 30 % of S. aureus isolates form strong biofilms in EGTA, and we confirmed in a number of these strains that biofilm formation in EGTA was ClfB-dependent, we hypothesize that additional genetic differences must lead to the observed phenotype in other strains. For example, clfB transcript levels could be higher in some strains or aur transcript levels could be lower.
In order to answer the question of how EGTA induces ClfB-mediated biofilms, we hypothesized that Ca2+ was the key. EGTA has greater affinity for Ca2+ than for other divalent cations. Plus, there is precedent in the literature for a role of Ca2+ in regulating ClfB activity (Ní Eidhin et al., 1998). EF-Hand domains in S. aureus proteins like Bap have been observed to bind Ca2+ and this prevents aggregation and biofilm formation (Arrizubieta et al., 2004). Similar EF-Hand domains within the ClfB protein suggested a role for Ca2+-mediated regulation of biofilm formation. Dispersion of biofilms with high concentrations of Ca2+ took place even when aur was deleted, confirming this hypothesis (Fig. 7). This suggested an added level of regulation whereby binding of Ca2+ to these helix–loop domains prevented the ClfB protein from promoting intercellular aggregation and biofilm formation.
Fig. 7.
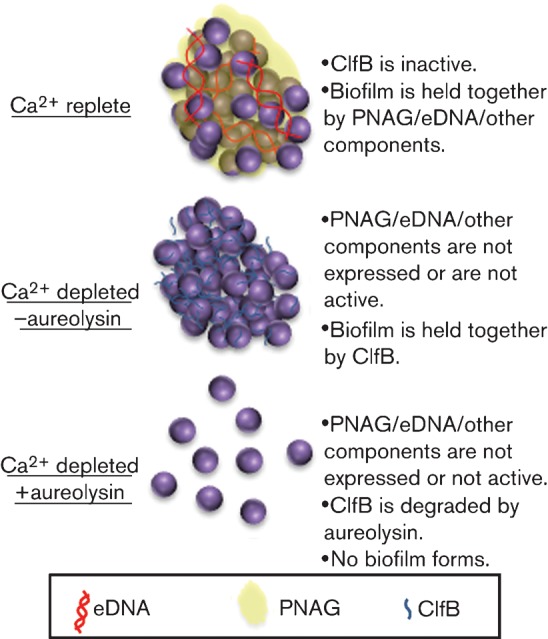
Schematic representation of proposed model based on data obtained in this study. eDNA, extracellular DNA.
In summary, two distinct types of biofilm are formed in the presence and absence of Ca2+. Under Ca2+-replete conditions, PNAG and other components play a role in the formation of the biofilm, whereas under Ca2+-depleted conditions, biofilms depend upon ClfB. ClfB appears to accumulate on the bacterial surface in the absence of functional aureolysin and mediates biofilm formation in the absence of Ca2+. This is unique in that other biofilm mediators are inhibited by Ca2+ depletion. The role for Ca2+ has important clinical implications as Ca2+ is one of the most abundant metal cations in the human body but its accessibility is tightly regulated by a number of calcium-binding proteins. Furthermore, it suggests that certain strains of S. aureus may vary in their response to the chelating agents present in certain catheter lock solutions.
Acknowledgements
We appreciate the donation of ClfB-specific antiserum from Drs Clarissa Pozzi and Gerald Pier (Brigham and Women’s Hospital, Harvard Medical School, Boston, MA). This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (R01AI068892).
Abbreviations:
- cm
chloramphenicol
- erm
erythromycin
- MSCRAMM
microbial surface component recognizing adhesive matrix molecule
- PNAG
poly-N-acetylglucosamine
Footnotes
Five supplementary figures are available with the online version of this paper.
References
- Abraham N. M., Lamlertthon S., Fowler V. G., Jr, Jefferson K. K. (2012). Chelating agents exert distinct effects on biofilm formation in Staphylococcus aureus depending on strain background: role for Clumping factor B. J Med Microbiol [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud M., Chastanet A., Débarbouillé M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70, 6887–6891. 10.1128/AEM.70.11.6887-6891.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrizubieta M. J., Toledo-Arana A., Amorena B., Penadés J. R., Lasa I. (2004). Calcium inhibits Bap-dependent multicellular behavior in Staphylococcus aureus. J Bacteriol 186, 7490–7498. 10.1128/JB.186.22.7490-7498.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen G. D., Simpson W. A., Younger J. J., Baddour L. M., Barrett F. F., Melton D. M., Beachey E. H. (1985). Adherence of coagulase-negative staphylococci to plastic tissue culture plates: a quantitative model for the adherence of staphylococci to medical devices. J Clin Microbiol 22, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramton S. E., Gerke C., Schnell N. F., Nichols W. W., Götz F. (1999). The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67, 5427–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crnich C., Maki D. (2005). Infections caused by intravascular devices: epidemiology, pathogenesis, diagnosis, prevention, and treatment. In APIC Text of Infection Control and Epidemiology, 2nd edn., vol 1, pp. 24.21–24.26. Washington, DC: Association for Professionals in Infection Control and Epidemiology. [Google Scholar]
- Entenza J. M., Foster T. J., Ní Eidhin D., Vaudaux P., Francioli P., Moreillon P. (2000). Contribution of clumping factor B to pathogenesis of experimental endocarditis due to Staphylococcus aureus. Infect Immun 68, 5443–5446. 10.1128/IAI.68.9.5443-5446.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- François P., Schrenzel J., Stoerman-Chopard C., Favre H., Herrmann M., Foster T. J., Lew D. P., Vaudaux P. (2000). Identification of plasma proteins adsorbed on hemodialysis tubing that promote Staphylococcus aureus adhesion. J Lab Clin Med 135, 32–42. 10.1016/S0022-2143(00)70018-7 [DOI] [PubMed] [Google Scholar]
- Grundmeier M., Hussain M., Becker P., Heilmann C., Peters G., Sinha B. (2004). Truncation of fibronectin-binding proteins in Staphylococcus aureus strain Newman leads to deficient adherence and host cell invasion due to loss of the cell wall anchor function. Infect Immun 72, 7155–7163. 10.1128/IAI.72.12.7155-7163.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M., Herrmann M., von Eiff C., Perdreau-Remington F., Peters G. (1997). A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect Immun 65, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasatiya S. S., Baldwin J. N. (1967). Nature of the determinant of tetracycline resistance in Staphylococcus aureus. Can J Microbiol 13, 1079–1086. 10.1139/m67-144 [DOI] [PubMed] [Google Scholar]
- Kennedy A. D., Otto M., Braughton K. R., Whitney A. R., Chen L., Mathema B., Mediavilla J. R., Byrne K. A., Parkins L. D., et al. (2008). Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc Natl Acad Sci U S A 105, 1327–1332. 10.1073/pnas.0710217105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiswirth B. N., Löfdahl S., Betley M. J., O’Reilly M., Schlievert P. M., Bergdoll M. S., Novick R. P. (1983). The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712. 10.1038/305709a0 [DOI] [PubMed] [Google Scholar]
- Lee J. C. (1993). Electrotransformation of Staphylococci. Totowa, NJ: Humana Press. [Google Scholar]
- Luong T. T., Lee C. Y. (2006). The arl locus positively regulates Staphylococcus aureus type 5 capsule via an mgrA-dependent pathway. Microbiology 152, 3123–3131. 10.1099/mic.0.29177-0 [DOI] [PubMed] [Google Scholar]
- Maki D. G., Kluger D. M., Crnich C. J. (2006). The risk of bloodstream infection in adults with different intravascular devices: a systematic review of 200 published prospective studies. Mayo Clin Proc 81, 1159–1171. 10.4065/81.9.1159 [DOI] [PubMed] [Google Scholar]
- McAleese F. M., Walsh E. J., Sieprawska M., Potempa J., Foster T. J. (2001). Loss of clumping factor B fibrinogen binding activity by Staphylococcus aureus involves cessation of transcription, shedding and cleavage by metalloprotease. J Biol Chem 276, 29969–29978. 10.1074/jbc.M102389200 [DOI] [PubMed] [Google Scholar]
- McDevitt D., François P., Vaudaux P., Foster T. J. (1994). Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol Microbiol 11, 237–248. 10.1111/j.1365-2958.1994.tb00304.x [DOI] [PubMed] [Google Scholar]
- Miajlovic H., Loughman A., Brennan M., Cox D., Foster T. J. (2007). Both complement- and fibrinogen-dependent mechanisms contribute to platelet aggregation mediated by Staphylococcus aureus clumping factor B. Infect Immun 75, 3335–3343. 10.1128/IAI.01993-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels J., Xi C., Verhaert J., Vanderleyden J. (2002). The functions of Ca2+ in bacteria: a role for EF-Hand proteins? Trends Microbiol 10, 87–93. 10.1016/S0966-842X(01)02284-3 [DOI] [PubMed] [Google Scholar]
- Ní Eidhin D., Perkins S., François P., Vaudaux P., Höök M., Foster T. J. (1998). Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol 30, 245–257. 10.1046/j.1365-2958.1998.01050.x [DOI] [PubMed] [Google Scholar]
- O’Brien L. M., Walsh E. J., Massey R. C., Peacock S. J., Foster T. J. (2002). Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell Microbiol 4, 759–770. 10.1046/j.1462-5822.2002.00231.x [DOI] [PubMed] [Google Scholar]
- O’Connell D. P., Nanavaty T., McDevitt D., Gurusiddappa S., Höök M., Foster T. J. (1998). The fibrinogen-binding MSCRAMM (clumping factor) of Staphylococcus aureus has a Ca2+-dependent inhibitory site. J Biol Chem 273, 6821–6829. 10.1074/jbc.273.12.6821 [DOI] [PubMed] [Google Scholar]
- O’Grady N. P., Alexander M., Burns L. A., Dellinger E. P., Garland J., Heard S. O., Lipsett P. A., Masur H., Mermel L. A., et al. (2011). Guidelines for the prevention of intravascular catheter-related infections. Am J Infect Control 39 (Suppl. 1), S1–S34. 10.1016/j.ajic.2011.01.003 [DOI] [PubMed] [Google Scholar]
- Perkins S., Walsh E. J., Deivanayagam C. C., Narayana S. V., Foster T. J., Höök M. (2001). Structural organization of the fibrinogen-binding region of the clumping factor B MSCRAMM of Staphylococcus aureus. J Biol Chem 276, 44721–44728. 10.1074/jbc.M106741200 [DOI] [PubMed] [Google Scholar]
- Ramos E. R., Reitzel R., Jiang Y., Hachem R. Y., Chaftari A. M., Chemaly R. F., Hackett B., Pravinkumar S. E., Nates J., et al. (2011). Clinical effectiveness and risk of emerging resistance associated with prolonged use of antibiotic-impregnated catheters: more than 0.5 million catheter days and 7 years of clinical experience. Crit Care Med 39, 245–251. 10.1097/CCM.0b013e3181feb83e [DOI] [PubMed] [Google Scholar]
- Schneewind O., Mihaylova-Petkov D., Model P. (1993). Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J 12, 4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudaux P. E., François P., Proctor R. A., McDevitt D., Foster T. J., Albrecht R. M., Lew D. P., Wabers H., Cooper S. L. (1995). Use of adhesion-defective mutants of Staphylococcus aureus to define the role of specific plasma proteins in promoting bacterial adhesion to canine arteriovenous shunts. Infect Immun 63, 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz J. M., Memtsoudis S. G., Heard S. O. (2010). Prevention of central venous catheter bloodstream infections. J Intensive Care Med 25, 131–138. 10.1177/0885066609358952 [DOI] [PubMed] [Google Scholar]