

Abstract
In common with other bacterial taxa, members of the genus Neisseria are classified using a range of phenotypic and biochemical approaches, which are not entirely satisfactory in assigning isolates to species groups. Recently, there has been increasing interest in using nucleotide sequences for bacterial typing and taxonomy, but to date, no broadly accepted alternative to conventional methods is available. Here, the taxonomic relationships of 55 representative members of the genus Neisseria have been analysed using whole-genome sequence data. As genetic material belonging to the accessory genome is widely shared among different taxa but not present in all isolates, this analysis indexed nucleotide sequence variation within sets of genes, specifically protein-coding genes that were present and directly comparable in all isolates. Variation in these genes identified seven species groups, which were robust to the choice of genes and phylogenetic clustering methods used. The groupings were largely, but not completely, congruent with current species designations, with some minor changes in nomenclature and the reassignment of a few isolates necessary. In particular, these data showed that isolates classified as Neisseria polysaccharea are polyphyletic and probably include more than one taxonomically distinct organism. The seven groups could be reliably and rapidly generated with sequence variation within the 53 ribosomal protein subunit (rps) genes, further demonstrating that ribosomal multilocus sequence typing (rMLST) is a practicable and powerful means of characterizing bacteria at all levels, from domain to strain.
Introduction
The genus Neisseria comprises Gram-negative oxidase-positive diplococci, many of which are harmless commensal inhabitants of the mucosal and dental surfaces of humans (Zaura et al., 2009). The genus contains two human pathogens that cause very different diseases, both of global significance: Neisseria meningitidis, the meningococcus, which causes meningitis and septicaemia; and Neisseria gonorrhoeae, the gonococcus, which causes gonorrhoea and, occasionally, disseminated infections. Conventionally, species of the genus Neisseria are distinguished based on their phenotypic properties, using assays such as carbohydrate utilization and enzyme substrate tests. While these techniques are generally satisfactory for the identification of the meningococcus, gonococcus and the lactose-fermenting organism Neisseria lactamica, misclassification is not uncommon using these methods and can have important medical consequences (Dossett et al., 1985).
A number of approaches have been used to explore the relationships and species assignment of the genus Neisseria, including DNA–DNA hybridization (Tønjum et al., 1989), numerical taxonomy (Barrett & Sneath, 1994), 16S rRNA gene sequencing (Harmsen et al., 2001) and analysis of the seven housekeeping gene fragments (Bennett et al., 2007) used in MLST (Maiden et al., 1998). DNA–DNA relatedness studies have shown that four members of the genus, N. meningitidis, N. gonorrhoeae, N. lactamica and Neisseria polysaccharea, are closely related (Guibourdenche et al., 1986), although two cause distinct human diseases. Phylogenies constructed from 16S rRNA gene sequences provide sufficient resolution to distinguish the genus Neisseria from its close relatives; however, Neisseria isolates classified as distinct species may have identical or very similar 16S rRNA gene sequences to other species within the genus (Harmsen et al., 2001).
The genus Neisseria is an instructive model system for examining the relationships of epidemiology, population genetics and evolution with the emergence of distinct phenotypes, especially those associated with invasive disease (Maiden, 2008). Members of the genus are naturally competent for the uptake of DNA by transformation, which is mediated by a specific uptake mechanism involving DNA uptake sequences (DUS) (Treangen et al., 2008). For over 20 years, the genus has played a central part in establishing the importance of horizontal genetic exchange in bacterial population structure and evolution (Maynard Smith et al., 1991; Spratt, 1988). Genomic studies of individual isolates have been combined with population analyses using MLST data (Bennett et al., 2010). These studies suggest first that the accessory genome, which includes genes thought to be associated with the ability to cause invasive disease, is widely shared among pathogenic and non-pathogenic members of the genus (Marri et al., 2010), and second, that sequence polymorphism in core genes, those present in all isolates, is important in defining the groups of genetically related isolates currently assigned species status (Bennett et al., 2010).
The present study analysed Neisseria species described in Bergey’s Manual of Systematic Bacteriology (Tønjum, 2005) to determine the phylogenetic relationships among these species and specifically their relationship to N. meningitidis. Species structure within the genus was investigated using whole-genome sequence data from 15 Neisseria species: N. meningitidis, N. gonorrhoeae, N. lactamica, N. polysaccharea, Neisseria cinerea, Neisseria flavescens, the Neisseria subflava biovars Neisseria subflava, Neisseria perflava and Neisseria flava, Neisseria mucosa and the Neisseria mucosa variant Neisseria mucosa var. heidelbergensis, Neisseria sicca, Neisseria elongata subsp. glycolytica, Neisseria bacilliformis, Neisseria macacae, Neisseria canis, Neisseria dentiae and Neisseria weaveri. Type strains from 12 of the commensal Neisseria were included as reference species (see Table S1 available with the online version of this paper). The type strains of N. meningitidis, N. gonorrhoeae and N. lactamica were not included, as fully annotated genomes were already available for these species and their species status is not in doubt.
The database platform Bacterial Isolate Genome Sequence Database (BIGSdb) (Jolley & Maiden, 2010), which is able to store genomic sequence data and has the capacity to define and identify any number of loci and genetic variants at these loci, was employed to identify nucleotide variation in genes present in all taxa. A reference gene approach using previously annotated Neisseria genomes for initial locus designation (Bennett et al., 2010) identified successive sets of genes that generated distinct groups of isolates, with the set of 53 ribosomal protein subunit (rps) genes, used in the ribosomal MLST (rMLST) typing scheme (Jolley et al., 2012), providing a minimal set of genes that clustered the isolates into groups, broadly consistent with current species assignments. These data demonstrate that some isolates currently in culture collections have been misnamed and that some minor changes in nomenclature are required.
Methods
Isolates.
A total of 36 Neisseria isolates were sequenced de novo, four N. lactamica isolates obtained from asymptomatic carriage in children in Oxfordshire (Bennett et al., 2005) and 32 isolates from the Culture Collection of the University of Göteborg (CCUG), Sweden (Tables S1 and S2). The CCUG isolates comprised 28 isolates designated human commensal Neisseria: five N. polysaccharea, four N. cinerea, three N. flavescens, one N. mucosa, one N. mucosa var. heidelbergensis, three N. sicca, one N. bacilliformis and 10 N. subflava, which comprised the biovars N. perflava (three), N. subflava (five) and N. flava (two). In addition, the CCUG isolates included four Neisseria (N. canis, N. dentiae, N. weaveri and N. macacae) not isolated from humans.
Microbiology and sequencing.
Freeze-dried bacterial isolates were inoculated onto Columbia horse-blood agar (Oxoid) and incubated for 24 h at 37 °C in a 5 % CO2 atmosphere. Genomic DNA was prepared using the Wizard Genomic DNA Purification kit (Promega), according to the manufacturer’s instructions. Standard Illumina multiplex libraries were generated according to the manufacturer’s instructions, using 1 µg genomic DNA sheared to between 200 and 300 bp using a Covaris E210 acoustic shearing device. Up to 12 libraries were pooled together in an equimolar ratio for sequencing in one flow cell lane on the Illumina Genome Analyzer II platform; 54 bp paired end reads were generated. Genomes were assembled using Velvet 1.0.10 (Zerbino & Birney, 2008); the assembly process was optimized using default parameters for the VelvetOptimizer script provided with the Velvet software package. Assembly data are available as Table S2.
Public sequence data.
Whole-genome data from 19 isolates were downloaded from either the Integrated Microbial Genomes (IMG) database found at http://img.jgi.doe.gov/cgi-bin/w/main.cgi (Markowitz et al., 2010) or GenBank (http://www.ncbi.nlm.nih.gov/genbank/). These data included genome sequences of five N. meningitidis isolates (Bentley et al., 2007; Parkhill et al., 2000; Peng et al., 2008; Schoen et al., 2008; Tettelin et al., 2000), six N. gonorrhoeae, including one published genome (Chung et al., 2008), one N. lactamica (Bennett et al., 2010), and one each of N. cinerea, N. flavescens, N. mucosa, N. sicca, N. polysaccharea, N. subflava and N. elongata subsp. glycolytica (Marri et al., 2010) (Table S1).
Uploading and annotation of sequence data with BIGSdb.
All genome data were uploaded to BIGSdb, along with available taxonomic and provenance data and links to the appropriate PubMed record; these data are accessible through the PubMLST database (http://pubmlst.org). The identifiers used for the isolates were usually those provided with the isolates, but all other known names associated with these isolates were included as aliases. Where isolates were obtained from culture collections, the culture collection name was accorded priority and the species designation provided with the isolate was used. Genes within the sequences were annotated with the tagging functionality included in BIGSdb (Jolley & Maiden, 2010; Jolley et al., 2012). Briefly, known genes were used as query strings for iterative searches with progressively decreasing stringency of the whole-genome data by means of the blastn and tblastx algorithms (Altschul et al., 1997). This process identified likely genes, which were tagged in the database, enabling them to be extracted and exported in formats suitable for various analyses. For a given locus, each unique complete sequence identified was assigned an arbitrary allele number. Allele sequences were manually checked to ensure that only in-frame sequences without internal stop codons were included and that the sequences began at common start codons where possible. For a small number of the gene sequences analysed, some of the data were missing from the ends of the contigs assembled from the short-read data, and in a few cases, apparent frameshift mutations were present, resulting in internal stop codons. These data were included in the analysis but were not assigned allele designations. Gene sequences from the isolate database were exported as XMFA files containing each locus as an aligned block, and then converted to fasta format for importing into mega version 5.0 (Tamura et al., 2007).
Analyses.
The BIGSdb genome comparator tool, which identifies loci shared among genomes and their allelic diversity, was used to detect genes present among all taxa. The annotated gene sequences from the published FAM18 genome (Bentley et al., 2007) were compared to whole-genome sequence data from 54 isolates using the following parameters: minimum percentage identity of 50, minimum percentage alignment of 30 and blastn word size of 11. As the search used nucleotide sequences, it would be expected to retrieve only conserved protein-coding genes. This level of stringency was chosen to ensure that only homologous genes were analysed.
Neighbor-joining phylogenies (Saitou & Nei, 1987) and a neighbor-net phylogeny (Bryant & Moulton, 2004) using nucleotide p-distances were constructed in mega version 5.0 and SplitsTree version 4 (Huson & Bryant, 2006), respectively. Genetic distances were calculated using mega version 4.0, DnaSP version 5 (Librado & Rozas, 2009) was used to calculate shared polymorphisms and fixed differences, and Arlequin version 3.11 (Excoffier et al., 2005) was used to calculate fixation index (FST) values.
Results and Discussion
While bacterial nomenclature is covered by the International Code of Nomenclature of Bacteria (Lapage et al., 1992), the bacterial species concept remains contentious at both a conceptual (Doolittle, 2008) and practical level (Stackebrandt et al., 2002). As increasing volumes of nucleotide sequence data become available from across the bacterial domain, the need for the systematic organization of bacterial groups becomes increasingly important (Achtman & Wagner, 2008). The long-established gold standard of DNA–DNA relatedness (Wayne et al., 1987) is not easily applied to all specimens and cannot resolve closely related members of certain groups, even though these may have distinct phenotypic properties deserving of distinct species status (Achtman & Wagner, 2008). There is general agreement that taxonomic schemes should be backwards-compatible, phylogenetically consistent and reflect genetic relatedness (Stackebrandt et al., 2002; Wayne et al., 1987); however, there is no consensus as to how this is best achieved (Achtman & Wagner, 2008). Approaches based on sequencing of multiple chromosomal loci, first envisioned in the late 1980s (Wayne et al., 1987), have been proposed (Gevers et al., 2005), but no practical method is yet in universal use. Here, we explore the use of genomic sequence data from members of the genus Neisseria to define species groups, concentrating on sequence variation in comparable subsets of genes present among all isolates examined.
16S rRNA and MLST gene phylogenies
A 456 bp gene fragment was extracted from one 16S rRNA gene from each of the 55 genomes examined, resulting in 36 unique alleles with an overall mean p-distance among alleles of 0.053. A neighbor-joining phylogeny generated with these data was poorly congruent with species designations of the isolates and only one group contained isolates assigned to a single species (N. gonorrhoeae) (Fig. 1). Consistent with previous findings (Tønjum, 2005), some isolates assigned the same species names occupied very different positions in the tree. For example, while four N. lactamica sequences formed a distinct group, the 16S rRNA sequence from N. lactamica isolate 020-06 was highly divergent. Furthermore, one cluster included species described as N. meningitidis, N. polysaccharea, N. cinerea and N. flavescens, and isolates thought to be N. polysaccharea and N. flavescens had 16S rRNA gene sequences identical to the type strain of N. cinerea (ATCC 14685). Other strains described as particular species did not cluster with the type strains of their designated species, indicating that further taxonomic investigation is required to clarify the species identity of these strains. These data confirmed that the 16S phylogeny was not useful for species assignment within the genus, due to a combination of low and unevenly distributed sequence diversity – a consequence of shared ancestry, inter-species horizontal genetic exchange (Smith et al., 1999) or both. The 16S rRNA phylogeny was not used further in this analysis.
Fig. 1.
Evolutionary relationships among Neisseria based on 16S rRNA fragments. The evolutionary history was inferred using the neighbor-joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown next to the branches. The analysis involved 55 nt sequences consisting of 456 nt. ‘T’ denotes type strain.
Gene fragments corresponding to the loci used for MLST were extracted from the database, concatenated and used to generate a neighbor-joining phylogeny, effectively the multilocus sequence analysis (MLSA) approach (Gevers et al., 2005). This phylogeny (Fig. S1), generated groups that were consistent with microbiological designations for isolates characterized as N. meningitidis, N. gonorrhoeae and N. lactamica, as has been described previously (Bennett et al., 2007). All of the isolates microbiologically assigned to N. cinerea clustered with the type strain of N. cinerea (ATCC 14685), along with one isolate previously identified as N. flavescens (CCUG 28662). The phylogeny indicated that this isolate could be a misidentified N. cinerea. Three N. polysaccharea isolates (CCUG 24845, CCUG 24846 and CCUG 18031) grouped with the N. polysaccharea type strain ATCC 43768 (Riou et al., 1983), but N. polysaccharea isolates 15883 and CCUG 27182 did not, with 15883 more distantly related. The other isolates did not cluster clearly into species-specific groups, indicating that variation at the MLST loci provides insufficient power to resolve all Neisseria into distinct species groups.
Examination of common genes sets
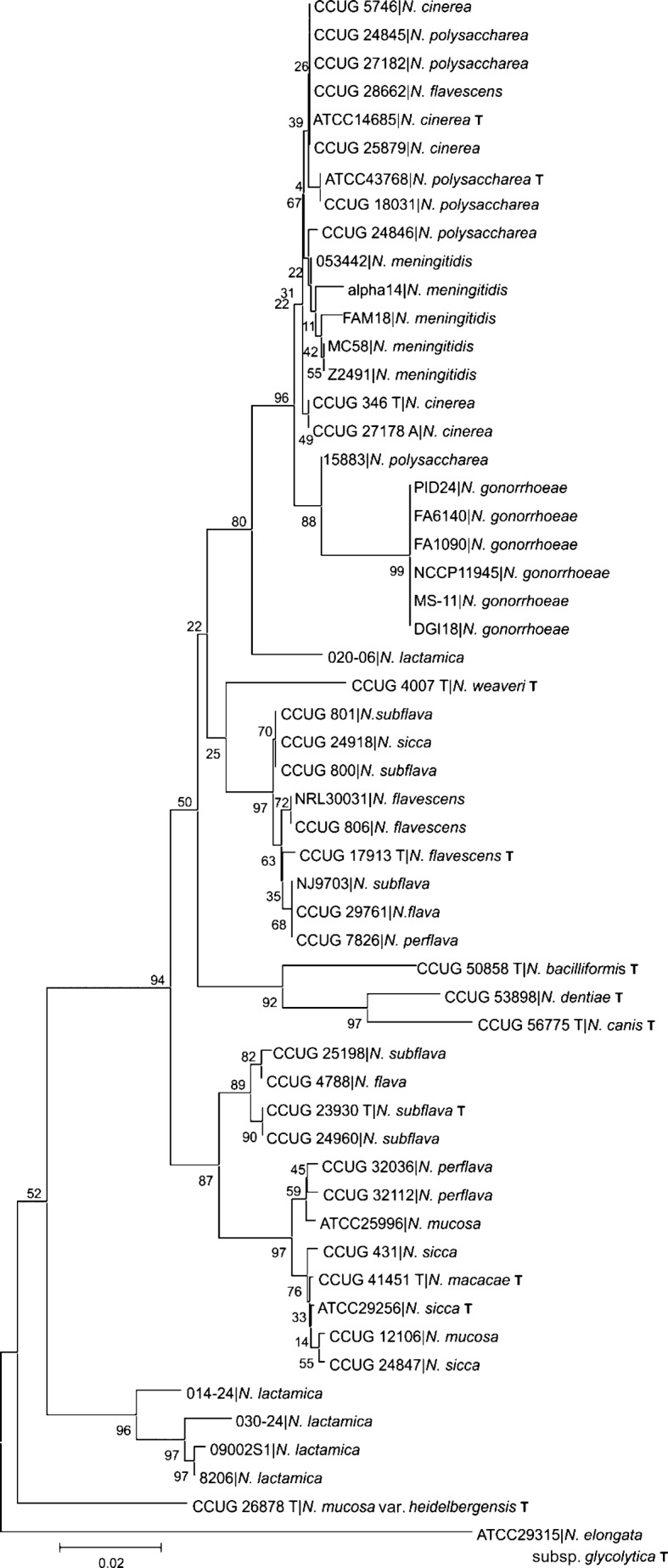
The genome comparator module of BIGSdb was employed to identify comparable coding sequences shared among the Neisseria genomes, with N. meningitidis FAM18 used as the reference genome. Using blastn, 246 genes, totalling 190 534 nt and amounting to 8.68 % of the query genome, were identified in all genomes (Table S3) using the blastn criteria described. A neighbor-joining phylogeny reconstructed from concatenated sequences of these genes generated seven groups (Fig. 2). Four of the groups comprised isolates belonging to single species: N. meningitidis, N. gonorrhoeae, N. lactamica and N. cinerea, with N. flavescens CCUG 28662 grouped again with N. cinerea, confirming the suggestion from MLST data that this isolate was a misidentified N. cinerea.
Fig. 2.

Evolutionary relationships among Neisseria based on concatenated sequences from 246 genes. The evolutionary history was inferred using the neighbor-joining method. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches. The analysis involved 55 nt sequences consisting of 190 534 nt. ‘T’ denotes type strain.
A further group, which contained the N. subflava type strain (CCUG 23930 T), consisted mainly of species defined in Bergey’s Manual of Systematic Bacteriology (Tønjum, 2005) as N. subflava and N. subflava biovars. There was little distinction among the biovars N. subflava biovar subflava, N. subflava biovar perflava and N. subflava biovar flava, confirming that they are variants of the same species. The three N. flavescens isolates were also included in this group, and were almost identical, consistent with their evolution from a single clone (Branham, 1930). The similarity between N. flavescens and the N. subflava biovars suggests that this species may also require reclassification as an N. subflava biovar. The N. sicca isolate (CCUG 24918) which clustered in this group is likely to be a misidentified N. subflava species.
The sixth group, distinct from the other five, consisted of isolates described as the following species: N. mucosa, N. sicca, N. perflava and the non-human isolate N. macacae. Here, the term ‘N. mucosa group’ has been used to define these organisms, as N. mucosa (originally given the name Diplococcus mucosus by Von Lingelsheim in 1906) was the first of these species to be identified (Tønjum, 2005). N. mucosa var. heidelbergensis (Berger, 1971) was shown to be distinct from the other N. mucosa isolates, as described by Tønjum (2005). Phenotypically and biochemically, N. sicca and N. mucosa are very similar, except that N. mucosa reduces nitrates and forms mucoid colonies, whereas N. sicca does not and forms dry, wrinkled colonies (Tønjum, 2005). This, taken together with the genetic data, suggests that these two species are variants of one species group. An examination of the alleles of the two isolates named N. perflava (CCUG 32036 and CCUG 32112) that clustered with this group indicated that they were misidentified N. mucosa variants.
The non-human N. macacae isolate, CCUG 41451, is closely related to the other isolates in the N. mucosa group, whereas the other non-human isolates in this study, N. canis (CCUG 56775 T), N. dentiae (CCUG 53898) and N. weaveri (CCUG 4007 T), are only distantly related to the human isolates. The N. macacae type strain was isolated from the oropharnyx of a captive primate (Rhesus monkey), and the close sequence identity of isolates from primate hosts suggests that some Neisseria are likely to colonize more than one host species. The rod-shaped human isolates (N. elongata subsp. glycolytica ATCC 29315 and N. bacilliformis CCUG 50858 T) were not closely related to the other human isolates or to each other.
All of the isolates previously defined as N. polysaccharea were closely related to the N. meningitidis, N. gonorrhoeae and N. lactamica isolates, but did not represent a monophyletic group. Isolate 15883 was less closely related to the type strain ATCC 43768. This bacterium was isolated along with strain 25862 (CCUG 18031), which were the first examples of this species to be described in Germany (Berger, 1985). At the time of discovery, it was observed that both isolates were different from the type strain in that they did not grow on Thayer–Martin medium (TMM) and that isolate 15883 differed from the others in its degradation of sugar, but otherwise appeared identical to N. polysaccharea. Another study of N. polysaccharea, which included these isolates, found two distinct subsets among the isolates, with some resistant to colistin, an antibiotic used in TMM, and some susceptible, indicating further variability within this taxon and that the documentation of this species is incomplete (Anand et al., 1991). Analysis of the set of genes employed here confirmed that isolate 15883 is distinct from other isolates of N. polysaccharea and could be either an N. polysaccharea variant or perhaps a separate species.
A subset of 98 genes (Table S3), which excluded the 53 ribosomal genes and consisted of 84 685 nt (amounting to 3.86 % of the query genome), were concatenated and used to reconstruct a neighbor-joining phylogeny. The same group structure as seen with the 246 gene analysis was evident (Fig. S2). Measures of FST (Table 1), fixed differences and shared polymorphisms (Table 2) calculated on the basis of the species groups revealed in this report showed that with the exception of the N. polysaccharea isolates, there was high differentiation between species groups. N. polysaccharea was most closely related to N. meningitidis, with an FST value of 0.35, the lowest number of fixed differences between species (511) and a high number of shared polymorphisms (2209).
Table 1. Gene flow between a set of 98 genes from seven species groups of Neisseria.
The divergent strains for which there is only one example (N. mucosa var. heidelbergensis CCUG 26878 T, N. polysaccharea 15883, N. elongata subsp. glycolytica ATCC 29315, N. bacilliformis CCUG 50858 T, N. dentiae CCUG 53898, N. weaveri CCUG 4007 T and N. canis CCUG 56775 T) have been excluded from this analysis. Figures above the diagonal are FST values, those below are P values (significance level = 0.05). Nmu, N. mucosa; Nsu, N. subflava; Npo, N. polysaccharea; Nme, N. meningitidis; Ngo, N. gonorrhoeae; Nla, N. lactamica; Nci, N. cinerea. Numbers of isolates are shown in parentheses.
| Species group | Nmu | Nsu | Npo | Nme | Ngo | Nla | Nci |
| Nmu (8)* | 0.67 | 0.65 | 0.69 | 0.79 | 0.71 | 0.68 | |
| Nsu (13)† | 0.00 | 0.67 | 0.71 | 0.78 | 0.72 | 0.68 | |
| Npo (5) | 0.01 | 0.00 | 0.35 | 0.66 | 0.47 | 0.51 | |
| Nme (5) | 0.00 | 0.00 | 0.01 | 0.71 | 0.59 | 0.64 | |
| Ngo (6) | 0.00 | 0.00 | 0.00 | 0.00 | 0.81 | 0.80 | |
| Nla (5) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.65 | |
| Nci (6) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
This group includes N. mucosa, N. sicca and N. macacae.
This group includes isolates defined as N. subflava, N. perflava, N. flava and N. flavescens.
Table 2. Fixed difference and shared polymorphisms between a set of 98 genes from seven species groups of Neisseria.
Figures above the diagonal are fixed differences, those below are shared polymorphisms. Numbers of isolates are shown in parentheses. Nmu, N. mucosa; Nsu, N. subflava; Npo, N. polysaccharea; Nme, N. meningitidis; Ngo, N. gonorrhoeae; Nla, N. lactamica; Nci, N. cinerea. The divergent strains for which there is only one example (N. mucosa var. heidelbergensis CCUG 26878 T, N. polysaccharea 15883, N. elongata subsp. glycolytica ATCC 29315, N. bacilliformis CCUG 50858 T, N. dentiae CCUG 53898, N. weaveri CCUG 4007 T and N. canis CCUG 56775 T) have been excluded from this analysis.
| Species group | Nmu | Nsu | Npo | Nme | Ngo | Nla | Nci |
| Nmu (8)* | 4493 | 4218 | 5015 | 6249 | 5169 | 4418 | |
| Nsu (13)† | 2585 | 4907 | 5864 | 7112 | 5871 | 4893 | |
| Npo (5) | 1642 | 2202 | 511 | 1738 | 1231 | 1237 | |
| Nme (5) | 1227 | 1611 | 2209 | 1745 | 2065 | 2578 | |
| Ngo (6) | 67 | 67 | 115 | 103 | 3439 | 4059 | |
| Nla (5) | 1028 | 1383 | 1956 | 1321 | 82 | 2706 | |
| Nci (6) | 1664 | 2303 | 2314 | 1555 | 96 | 1321 |
This group includes N. mucosa, N. sicca and N. macacae.
This group includes isolates defined as N. subflava, N. perflava, N. flava and N. flavescens.
Examination of ribosomal genes
The 53 rps genes represent ideal candidates for a bacterial classification scheme, as they are universally present, conserved and distributed around the bacterial chromosome. Concatenated gene sequences from the rps loci used in the rMLST scheme (Jolley et al., 2012) have been shown to produce phylogenies that cluster species in groups substantiated by current nomenclature. The groups generated using rMLST data were also consistent and not dependent on the clustering algorithm used.
A Neisseria phylogeny was reconstructed from the concatenated rps gene sequences using the neighbor-joining method (Fig. 3), which showed the same groups as the phylogenies produced using either 246 or 98 concatenated gene sequence sets. These groups were also generated using neighbor-net (Fig. S3). The 53 genes used in these analyses consisted of 21 398 nt in total, amounting to 0.97 % of the genome of the reference meningococcal genome sequence FAM18.
Fig. 3.

Evolutionary relationships among Neisseria based on concatenated sequences of 53 ribosomal protein genes. The evolutionary history was inferred using the neighbor-joining method. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches. The analysis involved 55 nt sequences consisting of 21 398 nt. ‘T’ denotes type strain.
Measures of gene flow, genetic differentiation and divergence among species supported the groups defined. An FST value of 0.54 between N. polysaccharea and N. meningitidis (Table 3) indicated that N. polysaccharea is more closely related to N. meningitidis than the other species examined here. This is supported by the 239 fixed differences between N. polysaccharea and N. meningitidis (Table 4), which is the lowest number of fixed differences between species, and the number of shared polymorphisms, which is the highest number of shared polymorphisms between species (194). FST values ranging from 0.74 (N. gonorrhoeae vs N. polysaccharea) to 0.95 (N. lactamica vs N. gonorrhoeae) indicate the higher level of differentiation between these other species. N. gonorrhoeae shared very few polymorphisms with other Neisseria, as N. gonorrhoeae isolates are likely to have descended from a single clone, have very low diversity and normally inhabit a different niche to other Neisseria. These data suggest that N. meningitidis and N. gonorrhoeae evolved from a common ancestor shared with isolates currently designated N. polysaccharea.
Table 3. Gene flow between 53 ribosomal genes from seven species groups of Neisseria.
Figures above the diagonal are FST values, those below are P values (significance level = 0.05). Nmu, N. mucosa; Nsu, N. subflava; Npo, N. polysaccharea; Nme, N. meningitidis; Ngo, N. gonorrhoeae; Nla, N. lactamica; Nci, N. cinerea. Numbers of isolates are shown in parentheses. The divergent strains for which there is only one example (N. mucosa var. heidelbergensis CCUG 26878 T, N. polysaccharea 15883, N. elongata subsp. glycolytica ATCC 29315, N. bacilliformis CCUG 50858 T, N. dentiae CCUG 53898, N. weaveri CCUG 4007 T and N. canis CCUG 56775 T) have been excluded from this analysis.
| Species group | Nmu | Nsu | Npo | Nme | Ngo | Nla | Nci |
| Nmu (8)* | 0.79 | 0.76 | 0.80 | 0.85 | 0.80 | 0.75 | |
| Nsu (13)† | 0.00 | 0.85 | 0.88 | 0.91 | 0.87 | 0.81 | |
| Npo (5) | 0.00 | 0.00 | 0.54 | 0.74 | 0.82 | 0.78 | |
| Nme (5) | 0.00 | 0.00 | 0.00 | 0.79 | 0.88 | 0.84 | |
| Ngo (6) | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.90 | |
| Nla (5) | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.82 | |
| Nci (6) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
This group includes N. mucosa, N. sicca and N. macacae.
This group includes isolates defined as N. subflava, N. perflava, N. flava and N. flavescens.
Table 4. Fixed difference and shared polymorphisms between 53 ribosomal genes from seven species groups of Neisseria.
Figures above the diagonal are fixed differences, those below are shared polymorphisms. Nmu, N. mucosa; Nsu, N. subflava; Npo, N. polysaccharea; Nme, N. meningitidis; Ngo, N. gonorrhoeae; Nla, N. lactamica; Nci, N. cinerea. Numbers of isolates are shown in parentheses. The divergent strains for which there is only one example (N. mucosa var. heidelbergensis CCUG 26878 T, N. polysaccharea 15883, N. elongata subsp. glycolytica ATCC 29315, N. bacilliformis CCUG 50858 T, N. dentiae CCUG 53898, N. weaveri CCUG 4007 T and N. canis CCUG 56775 T) have been excluded from this analysis.
| Species group | Nmu | Nsu | Npo | Nme | Ngo | Nla | Nci |
| Nmu (8)* | 916 | 1147 | 1259 | 1431 | 1162 | 834 | |
| Nsu (13)† | 162 | 1247 | 1450 | 1677 | 1298 | 863 | |
| Npo (5) | 133 | 142 | 239 | 394 | 1075 | 950 | |
| Nme (5) | 71 | 66 | 194 | 314 | 1103 | 1047 | |
| Ngo (6) | 12 | 4 | 11 | 3 | 1307 | 1240 | |
| Nla (5) | 48 | 46 | 115 | 57 | 5 | 829 | |
| Nci (6) | 162 | 136 | 187 | 84 | 3 | 80 |
This group includes N. mucosa, N. sicca and N. macacae.
This group includes isolates defined as N. subflava, N. perflava, N. flava and N. flavescens.
An analysis of the individual ribosomal allele sequences used for rMLST showed that some identical allele sequences were shared among different species groups, consistent with a common ancestry for these genes that encode proteins that are under stabilizing selection for functional conservation. Another explanation is that there is frequent genetic recombination between species, with recombination acting as a mechanism for repairing core genes rather than as a method of diversification (Treangen et al., 2008). Identical ribosomal alleles shared between species were more frequent between N. polysaccharea and other Neisseria than between any other species group and the rest of the genus examined here. This suggests that if recombination is frequent among the ribosomal genes of Neisseria, then carriage of N. polysaccharea is more common than currently recognized.
In contrast, there was little support for frequent inter-species recombination among the MLST alleles identified using BIGSdb, as these were unique to each species group defined here. This was also true when the whole genes from which the MLST fragments were extracted were examined (data not shown). This indicates that metabolic housekeeping genes evolve and diverge as they adapt to a particular niche, and as they diverge, distinct alleles become evident that are specific to a particular species, consistent with previous findings (Bennett et al., 2010).
Further analyses of the individual ribosomal allele sequences for these isolates provided further evidence for isolate or species misclassification. For example, the N. flavescens isolate CCUG 28662, which grouped with N. cinerea, had 19 ribosomal gene sequences identical to other N. cinerea isolates, including 13 identical to the type strain, but none identical to any other N. flavescens isolate examined, confirming that it is N. cinerea. It was isolated in Sweden in 1991, and is not closely related to the N. flavescens isolates originally identified during an outbreak of meningitis in Chicago in 1928 (Branham, 1930).
All the isolates within the N. mucosa group shared similar ribosomal gene sequences and can be considered variants of one species, as is the case for the N. subflava variants. The close relationship between N. macacae and other Neisseria within the N. mucosa group was supported by the observation that 13 N. macacae ribosomal gene sequences were identical to sequences present in other N. mucosa group genomes. Also in the N. mucosa group were two isolates identified originally as N. subflava biovar N. perflava (CCUG 32112 and CCUG 32036). These shared no ribosomal alleles with other isolates in the N. subflava group, but shared a large number with isolates clustering in the N. mucosa group, confirming their identity as N. mucosa variants. Another isolate, which was originally identified as N. sicca (CCUG 24918), but clustered with the N. subflava group, shared ribosomal alleles with all subspecies from the N. subflava group, including N. flavescens, but none with any isolates clustering in the N. mucosa group, confirming that it is an N. subflava variant, along with the N. flavescens isolates in this group.
Neisseria classification
The availability of genomic data and the development of the BIGSdb platform have facilitated a classification method (rMLST) which has sufficient power to classify species within the genus Neisseria rapidly and reliably. Species assignments for the human isolates N. meningitidis, N. gonorrhoeae and N. lactamica are well established, but a number of other species require some reclassification. These data indicate that the species N. sicca and N. macacae should be classed as variants of N. mucosa, and that N. mucosa var. heidelbergensis is sufficiently diverse to be assigned species status (Neisseria heidelbergensis). N. flavescens is closely related genetically to the N. subflava variants and could be considered a variant of this species (Neisseria subflava var. flavescens). The N. cinerea isolates formed a distinct group, although isolate CCUG 5746 was more dissimilar to the other representatives of this species. This isolate may form a distinct variant of this species, but more data from other N. cinerea isolates would be required to confirm this.
The isolates currently designated N. polysaccharea examined here form a polyphyletic group. Data from historical studies indicate that N. polysaccharea is diverse (Anand et al., 1991), is more closely related to N. meningitidis than other Neisseria species (Guibourdenche et al., 1986; Zhu et al., 2003), is carried by children of primary school age (Cann & Rogers, 1989; Saez-Nieto et al., 1985) and may act as a reservoir for antibiotic resistance (Saez-Nieto et al., 1990). Taken together these data indicate that further examination of the N. polysaccharea variants is required to define this species group accurately, and that additional research is needed to determine its genetic relationship to N. meningitidis, its epidemiology and its rate of carriage in young children. The most diverse of the N. polysaccharea isolates (15883) requires reclassification, as it differs from the other N. polysaccharea variants both phenotypically (Berger, 1985) and genotypically, and is less closely related to N. meningitidis than the other isolates designated N. polysaccharea. A suggested name for the reclassification of this strain is Neisseria bergeri.
Conclusions
Reliable identification and classification of bacteria is important in all areas of microbiology, but is essential in clinical applications. It is important that commensal Neisseria are accurately distinguished, as some may be misidentified as pathogenic species, and occasionally some are isolated from unusual sites and must be correctly identified for clinical purposes (Knapp, 1988). Accurately identified bacterial species are an essential starting point to investigate the genetic determination of phenotypes by the comparison of related isolates that exhibit diverse properties. The availability of whole-genome sequences has greatly increased the number of possible comparative studies, but it is essential that the isolates used in such investigations are well characterized to realize the opportunities presented by association studies of diverse phenotypes with particular genotypes. As in many micro-organisms, the accessory genome is widely shared among Neisseria that have distinct pathologies; for example, many ‘virulence-associated’ genes identified for N. meningitidis and N. gonorrhoea are also present in the non-pathogen N. lactamica. Consequently, it is necessary to examine sequence divergence in core genes to accurately characterize bacterial isolates. This analysis demonstrates that in the genus Neisseria, reproducible species groups can be generated from various sets of genes including a ‘minimal core genome’, the 53 rps genes. These groups are largely congruent with previous nomenclatures, and therefore this approach represents an effective and rapid method for taxonomic classification that can be readily applied to other bacterial groups. The method has the potential to replace approaches such as DNA association studies as a reproducible and generally applicable basis for bacterial identification and classification.
Acknowledgements
This project was funded by the Wellcome Trust. M. C. J. M. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Science.
Abbreviations:
- FST
fixation index
- rMLST
ribosomal MLST
Footnotes
Three supplementary figures and three supplementary tables are available with the online version of this paper.
References
- Achtman M., Wagner M. (2008). Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6, 431–440. [DOI] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997). Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand C. M., Ashton F., Shaw H., Gordon R. (1991). Variability in growth of Neisseria polysaccharea on colistin-containing selective media for Neisseria spp. J Clin Microbiol 29, 2434–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett S. J., Sneath P. H. A. (1994). A numerical phenotypic taxonomic study of the genus Neisseria. Microbiology 140, 2867–2891. 10.1099/00221287-140-10-2867 [DOI] [PubMed] [Google Scholar]
- Bennett J. S., Griffiths D. T., McCarthy N. D., Sleeman K. L., Jolley K. A., Crook D. W., Maiden M. C. (2005). Genetic diversity and carriage dynamics of Neisseria lactamica in infants. Infect Immun 73, 2424–2432. 10.1128/IAI.73.4.2424-2432.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J. S., Jolley K. A., Sparling P. F., Saunders N. J., Hart C. A., Feavers I. M., Maiden M. C. (2007). Species status of Neisseria gonorrhoeae: evolutionary and epidemiological inferences from multilocus sequence typing. BMC Biol 5, 35. 10.1186/1741-7007-5-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J. S., Bentley S. D., Vernikos G. S., Quail M. A., Cherevach I., White B., Parkhill J., Maiden M. C. J. (2010). Independent evolution of the core and accessory gene sets in the genus Neisseria: insights gained from the genome of Neisseria lactamica isolate 020-06. BMC Genomics 11, 652. 10.1186/1471-2164-11-652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley S. D., Vernikos G. S., Snyder L. A., Churcher C., Arrowsmith C., Chillingworth T., Cronin A., Davis P. H., Holroyd N. E., et al. (2007). Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet 3, e23. 10.1371/journal.pgen.0030023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger U. (1971). [Neisseria mucosa var. heidelbergensis]. Z Med Mikrobiol Immunol 156, 154–158 (in German). 10.1007/BF02124646 [DOI] [PubMed] [Google Scholar]
- Berger U. (1985). First isolation of Neisseria polysacchareae species nova in the Federal Republic of Germany. Eur J Clin Microbiol 4, 431–433. 10.1007/BF02148705 [DOI] [PubMed] [Google Scholar]
- Branham S. E. (1930). A new meningococcus-like organism (Neisseria flavescens n. sp.) from epidemic meningitis. Public Health Rep 45, 845–849. 10.2307/4579618 [DOI] [Google Scholar]
- Bryant D., Moulton V. (2004). Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21, 255–265. 10.1093/molbev/msh018 [DOI] [PubMed] [Google Scholar]
- Cann K. J., Rogers T. R. (1989). The phenotypic relationship of Neisseria polysaccharea to commensal and pathogenic Neisseria spp. J Med Microbiol 29, 251–254. 10.1099/00222615-29-4-251 [DOI] [PubMed] [Google Scholar]
- Chung G. T., Yoo J. S., Oh H. B., Lee Y. S., Cha S. H., Kim S. J., Yoo C. K. (2008). Complete genome sequence of Neisseria gonorrhoeae NCCP11945. J Bacteriol 190, 6035–6036. 10.1128/JB.00566-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle W. F. (2008). Microbial evolution: stalking the wild bacterial species. Curr Biol 18, R565–R567. 10.1016/j.cub.2008.05.029 [DOI] [PubMed] [Google Scholar]
- Dossett J. H., Appelbaum P. C., Knapp J. S., Totten P. A. (1985). Proctitis associated with Neisseria cinerea misidentified as Neisseria gonorrhoeae in a child. J Clin Microbiol 21, 575–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L., Laval G., Schneider S. (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1, 47–50. [PMC free article] [PubMed] [Google Scholar]
- Gevers D., Cohan F. M., Lawrence J. G., Spratt B. G., Coenye T., Feil E. J., Stackebrandt E., Van de Peer Y., Vandamme P., et al. (2005). Opinion: re-evaluating prokaryotic species. Nat Rev Microbiol 3, 733–739. 10.1038/nrmicro1236 [DOI] [PubMed] [Google Scholar]
- Guibourdenche M., Popoff M. Y., Riou J. Y. (1986). Deoxyribonucleic acid relatedness among Neisseria gonorrhoeae, N. meningitidis, N. lactamica, N. cinerea and “Neisseria polysaccharea”. Ann Inst Pasteur Microbiol 137B, 177–185. 10.1016/S0769-2609(86)80106-5 [DOI] [PubMed] [Google Scholar]
- Harmsen D., Singer C., Rothgänger J., Tønjum T., de Hoog G. S., Shah H., Albert J., Frosch M. (2001). Diagnostics of Neisseriaceae and Moraxellaceae by ribosomal DNA sequencing: ribosomal differentiation of medical microorganisms. J Clin Microbiol 39, 936–942. 10.1128/JCM.39.3.936-942.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D. H., Bryant D. (2006). Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23, 254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Jolley K. A., Maiden M. C. (2010). BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595. 10.1186/1471-2105-11-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley K. A., Bliss C. M., Bennett J. S., Bratcher H. B., Brehony C. M., Colles F. M., Wimalarathna H. M., Harrison O. B., Sheppard S. K., et al. (2012). Ribosomal multi-locus sequence typing: universal characterisation of bacteria from domain to strain. Microbiology 4, 1005–1015. 10.1099/mic.0.055459-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp J. S. (1988). Historical perspectives and identification of Neisseria and related species. Clin Microbiol Rev 1, 415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapage S. P., Sneath P. H. A., Lessel E. F., Skerman V. B. D., Seeliger H. P. R., Clark W. A. (editors) (1992). International Code of Nomenclature of Bacteria (1990 Revision). Bacteriological Code. Washington, DC: American Society for Microbiology. [PubMed] [Google Scholar]
- Librado P., Rozas J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Maiden M. C. J., Bygraves J. A., Feil E., Morelli G., Russell J. E., Urwin R., Zhang Q., Zhou J., Zurth K., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95, 3140–3145. 10.1073/pnas.95.6.3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden M. C. (2008). Population genomics: diversity and virulence in the Neisseria. Curr Opin Microbiol 11, 467–471. 10.1016/j.mib.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz V. M., Chen I. M., Palaniappan K., Chu K., Szeto E., Grechkin Y., Ratner A., Anderson I., Lykidis A., et al. (2010). The integrated microbial genomes system: an expanding comparative analysis resource. Nucleic Acids Res 38 (Database issue), D382–D390. 10.1093/nar/gkp887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marri P. R., Paniscus M., Weyand N. J., Rendón M. A., Calton C. M., Hernández D. R., Higashi D. L., Sodergren E., Weinstock G. M., et al. (2010). Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS ONE 5, e11835. 10.1371/journal.pone.0011835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J. M., Dowson C. G., Spratt B. G. (1991). Localized sex in bacteria. Nature 349, 29–31. 10.1038/349029a0 [DOI] [PubMed] [Google Scholar]
- Parkhill J., Achtman M., James K. D., Bentley S. D., Churcher C., Klee S. R., Morelli G., Basham D., Brown D., et al. (2000). Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature 404, 502–506. 10.1038/35006655 [DOI] [PubMed] [Google Scholar]
- Peng J., Yang L., Yang F., Yang J., Yan Y., Nie H., Zhang X., Xiong Z., Jiang Y., et al. (2008). Characterization of ST-4821 complex, a unique Neisseria meningitidis clone. Genomics 91, 78–87. 10.1016/j.ygeno.2007.10.004 [DOI] [PubMed] [Google Scholar]
- Riou J. Y., Guibourdenche M., Popoff M. Y. (1983). A new taxon in the genus Neisseria. Ann Microbiol (Paris) 134B, 257–267. [DOI] [PubMed] [Google Scholar]
- Saez-Nieto J. A., Dominguez J. R., Monton J. L., Cristobal P., Fenoll A., Vazquez J., Casal J., Taracena B. (1985). Carriage of Neisseria meningitidis and Neisseria lactamica in a school population during an epidemic period in Spain. J Hyg (Lond) 94, 279–288. 10.1017/S0022172400061507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Nieto J. A., Lujan R., Martinez-Suarez J. V., Berron S., Vazquez J. A., Viñas M., Campos J. (1990). Neisseria lactamica and Neisseria polysaccharea as possible sources of meningococcal β-lactam resistance by genetic transformation. Antimicrob Agents Chemother 34, 2269–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Schoen C., Blom J., Claus H., Schramm-Glück A., Brandt P., Müller T., Goesmann A., Joseph B., Konietzny S., et al. (2008). Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proc Natl Acad Sci U S A 105, 3473–3478. 10.1073/pnas.0800151105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. H., Holmes E. C., Donovan G. M., Carpenter G. A., Spratt B. G. (1999). Networks and groups within the genus Neisseria: analysis of argF, recA, rho, and 16S rRNA sequences from human Neisseria species. Mol Biol Evol 16, 773–783. [DOI] [PubMed] [Google Scholar]
- Spratt B. G. (1988). Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature 332, 173–176. 10.1038/332173a0 [DOI] [PubMed] [Google Scholar]
- Stackebrandt E., Frederiksen W., Garrity G. M., Grimont P. A., Kämpfer P., Maiden M. C., Nesme X., Rosselló-Mora R., Swings J., et al. (2002). Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52, 1043–1047. 10.1099/ijs.0.02360-0 [DOI] [PubMed] [Google Scholar]
- Tamura K., Dudley J., Nei M., Kumar S. (2007). mega4: Molecular Evolutionary Genetics Analysis (mega) software version 4.0. Mol Biol Evol 24, 1596–1599. 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- Tettelin H., Saunders N. J., Heidelberg J., Jeffries A. C., Nelson K. E., Eisen J. A., Ketchum K. A., Hood D. W., Peden J. F., et al. (2000). Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287, 1809–1815. 10.1126/science.287.5459.1809 [DOI] [PubMed] [Google Scholar]
- Tønjum T. (2005). Genus I. Neisseria. In Bergey’s Manual of Systematic Bacteriology, pp. 777–798. Edited by Garrity G. M., Brenner D. J., Krieg N. R., Staley J. R. New York: Springer-Verlag. [Google Scholar]
- Tønjum T., Bukholm G., Bøvre K. (1989). Differentiation of some species of Neisseriaceae and other bacterial groups by DNA–DNA hybridization. APMIS 97, 395–405. 10.1111/j.1699-0463.1989.tb00806.x [DOI] [PubMed] [Google Scholar]
- Treangen T. J., Ambur O. H., Tonjum T., Rocha E. P. (2008). The impact of the neisserial DNA uptake sequences on genome evolution and stability. Genome Biol 9, R60. 10.1186/gb-2008-9-3-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne L. G., Brenner D. J., Colwell R. R., Grimont P. A. D., Kandler O., Krichevsky M. I., Moore L. H., Moore W. E. C., Murray R. G. E., et al. (1987). Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37, 463–464. 10.1099/00207713-37-4-463 [DOI] [Google Scholar]
- Zaura E., Keijser B. J., Huse S. M., Crielaard W. (2009). Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol 9, 259. 10.1186/1471-2180-9-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D. R., Birney E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P., Tsang R. S., Tsai C. M. (2003). Nonencapsulated Neisseria meningitidis strain produces amylopectin from sucrose: altering the concept for differentiation between N. meningitidis and N. polysaccharea. J Clin Microbiol 41, 273–278. 10.1128/JCM.41.1.273-278.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]