

Abstract
Sweet to the Core Enantioselective formal total syntheses of the Stemona alkaloids didehydrostemofoline and isodidehydrostemofoline were accomplished in 24 steps from commercially available 2-deoxy-d-ribose. The synthesis features a novel cascade of reactions culminating in an intramolecular dipolar cycloaddition to form the cage-like, tricyclic core of the stemofoline alkaloids from an acyclic diazo imine intermediate.
Keywords: cascade reaction, stemofoline alkaloids, enantioselective, total synthesis, azomethine ylide, dipolar cycloaddition
Plants of the Stemonacea family, which are indigenous to a number of areas in Southeast Asia, have long been used in traditional oriental medicine for treating a variety of ailments.[1] Extraction of the roots and leaves of these plants have yielded a number of biologically active alkaloids that have been targets of many synthetic investigations.[2] Arguably the most complex members of the Stemona alkaloids are those belonging to the stemofoline family, which are characterized by a densely-functionalized, caged hexacyclic architecture and differ in the geometry of the C11–C12 double bond and the oxidation state of the butyl side chain at C3 (Figure 1). These alkaloids, which were first reported by Irie and coworkers in 1970[ 3 ] and later isolated from other Stemona species,[4,5,6] exhibit strong insecticidal activity because they act as insect acetylcholine receptor antagonists.[7] Didehydrostemofoline (1) is not only the most potent acetylcholine receptor antagonist,[8] but it also exhibits in vivo anti-oxytocin activity as well as antitumor activity against gastric carcinoma.[4,9] A recent study has shown that stemofoline (2) increases the sensitivity of anticancer drugs such as vinblastine, paclitaxel, and doxorubicin by reversal of P-glycoprotein mediated multi-drug resistance.[ 10 ] A number of semisynthetic analogs of these alkaloids have been prepared and found to exhibit acetylcholinesterase inhibitory activity.[8,11]
Figure 1.
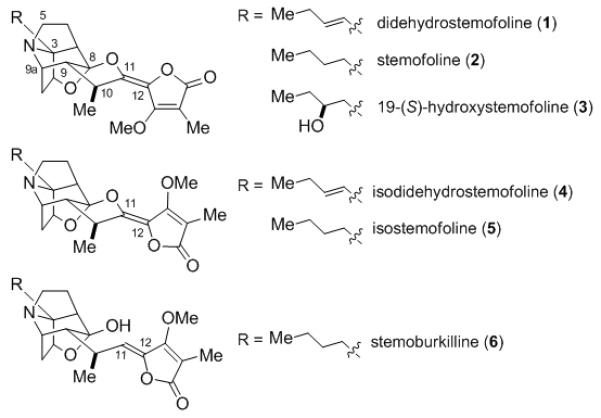
The Stemofoline Family of Natural Products
Because of their complex polycyclic structures and their biological activities, there has been considerable interest in the synthesis of the stemofoline alkaloids. Despite these efforts,[12,13] the only total syntheses are Kende’s synthesis of (±)-isostemofoline (5) in 1999[14] and Overman’s syntheses of (±)-1 and (±)-4 in 2003.[15] Each of the Kende and the Overman strategies relied upon clever cascade processes to construct the bridged polycyclic core of these alkaloids. However, because neither of these approaches is enantioselective, we queried whether we might be able to achieve an enantioselective synthesis of selected members of the stemofoline alkaloids using a single, enantiomerically pure starting material as the sole source of chirality.
Examination of the structures of didehydrostemofoline (1) and isodidehydrostemofoline (4) reveals a bicyclic γ-ylidenetetronate moiety that is fused to a tetracyclic, cage-like framework. The problem of creating this array was solved by Overman by coupling 7 with 8 (Scheme 1), followed by two additional operations to furnish an intermediate diol that was subjected to a Corey–Winter olefination to give a mixture of (±)-1 and (±)-4.[15] Given that the synthesis of 8 would thus represent formal syntheses of 1 and 4, we were attracted by the prospect that we might be able to prepare 8 via an approach that featured an intramolecular [3+2] cycloaddition of an azomethine ylide. Indeed, we had previously demonstrated that such reactions can be used to create the tricyclic core of the stemofoline alkaloids, but we had encountered difficulties with either poor regioselectivity in the cycloaddition or with refunctionalizing the cycloadduct in ways that might lead to 8.[13] These studies, however, did suggest that the presence of an electron-withdrawing group at C5 and a protected alcohol at C8 (didehydrostemofoline numbering) on an azomethine ylide such as 10 would preferentially afford the cycloadduct 9. We envisioned that 10 might be generated in situ from the diazo imine 11, which in turn would be accessible from commercially available 2-deoxy-d-ribose (12). Herein, we report the enantioselective formal syntheses of 1 and 4 using an intramolecular dipolar cycloaddition cascade as a key step to rapidly assemble the tricyclic core.
Scheme 1.
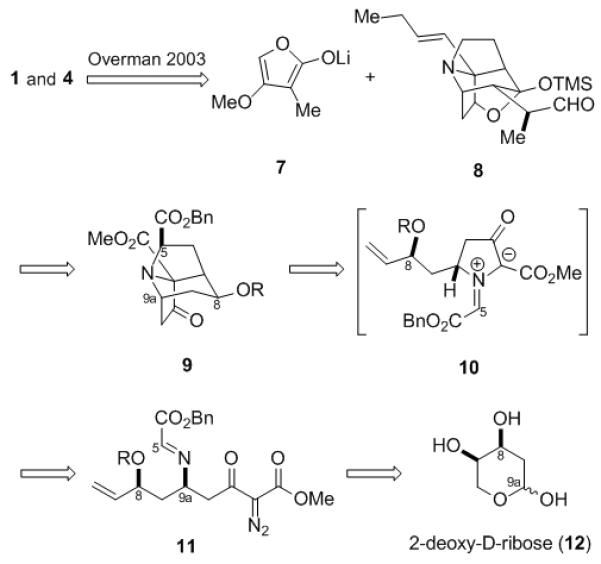
Retrosynthesis of Didehydrostemofoline and Isodidehydrostemofoline
The synthesis commenced with transforming 12 via a one-pot Wittig olefination and iodination sequence to give an intermediate iodo diol that was not isolated but was acetylated directly to give an iodo bis-acetate. This compound was then treated immediately with zinc granules in refluxing methanol to effect both a Boord elimination and a transesterification to furnish the allylic alcohol 13 in 54% overall yield from 12 (Scheme 2).[16] The physical nature of zinc metal used to induce the Boord elimination was important, as zinc dust led to a lower yield (46% vs. 62%). The optimized process for synthesizing 13 did not require chromatographic purification of any intermediate and was easily executed on >100 g scale. The allylic alcohol 13 was then converted into carbamate 14 in quantitative yield, thereby setting the stage for a key Hirama–Ito cyclization,[ 17 ] which transferred the chirality at C8 to C9a preferentially via the transition state 15 to give the key intermediate 16 as the major product. When this reaction was performed in THF using NaH as the base at room temperature, 16 was isolated in 75% yield and moderate diastereoselectivity (dr ≈ 4:1). However, when the reaction was conducted in CH2Cl2 at −10 °C, 16 was isolated in 80% yield and improved diastereoselectivity (dr ≈ 8:1).
Scheme 2.
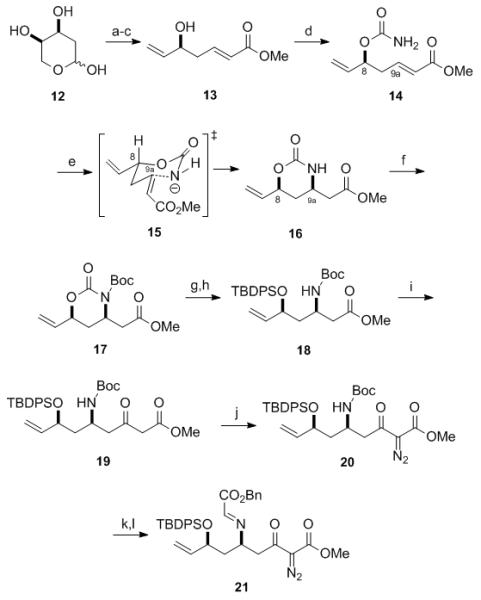
Synthesis of Cycloaddition Precursor 21. Reagents and Conditions: a) Ph3PCHCO2Me, THF, reflux; I2, Ph3P, imidazole; b) Ac2O, pyridine, DMAP, CH2Cl2, 87% (two steps); c) Zn granules, MeOH, reflux, 62%; d) ClSO2NCO, CH2Cl2; H2O, 99%; e) NaH, CH2Cl2, −10 °C, 80% (dr = 8:1 cis:trans); f) Boc2O, NEt3, DMAP, CH2Cl2, 87%; g) Cs2CO3, MeOH; h) TBDPS-Cl, imidazole, DMAP, CH2Cl2, 80% (two steps); i) MeCO2Me, NaHMDS, THF, −78 °C to −10 °C, 75% (+ 17% recovered 18); j) p-ABSA, NEt3, MeCN, 92%; k) TFA, CH2Cl2; l) BnO2CCHO, NEt3, 4 Å MS, CH2Cl2, 99% (two steps).
The cyclic carbamate 16 was transformed into the imide 17, which underwent regioselective, methoxide-induced cleavage to give an intermediate alcohol that was protected as the silyl ether 18. The subsequent Claisen condensation of 18 with the sodium enolate of methyl acetate provided the β-ketoester 19 in 75% yield together with recovered 18 (ca 17%). When the corresponding lithium enolate was employed, the Claisen reaction proceeded to give 19 in significantly lower yields (35–60%). β-Ketoester 19 was then converted into the diazo-β-ketoester 20 using p-acetamidobenzenesulfonyl azide (p-ABSA) as the diazo transfer reagent. Acid-induced deprotection of 20 with trifluoroacetic acid gave an ammonium salt that was condensed in situ with benzyl glyoxylate; subsequent removal of the solvent under reduced pressure gave the crude diazo imine 21, which was contaminated with an approximately equimolar amount of TFA•NEt3 (1H NMR spectrum) and several minor impurities.
Having thus prepared 21, we were anxious to determine whether it might be induced to undergo the much anticipated cascade of reactions to deliver the tricyclic core of the stemofoline alkaloids as depicted in Scheme 1. Some support for the possible efficacy of this conversion is found in previous work of Padwa,[18 ] although a sequence in which all reactions proceed via an intramolecular manifold is unprecedented. Owing to our zeal to quickly put our plan to the test, we used the crude diazo imine 21 in initial experiments. As will become evident, this was a most fortuitous decision. In the event, crude 21 was heated in refluxing xylenes in the presence of Rh2(OAc)4 (3 mol %) to give the desired azatricycle 22 in 75% yield as a single regioisomer and stereoisomer (Scheme 3). We quickly learned, however, that this reaction sequence was considerably more complex than we had initially envisioned. Namely, in an effort to improve the yield of this pivotal cascade process, the imine 21 was purified by column chromatography using basic alumina to remove TFA•NEt3 and the other minor impurities. Surprisingly, when pure 21 was heated in refluxing xylenes in the presence of Rh2(OAc)4 (3 mol %), a mixture (1.5:1) of the regioisomers 22 and 29 was obtained in 66% combined yield.
Scheme 3.
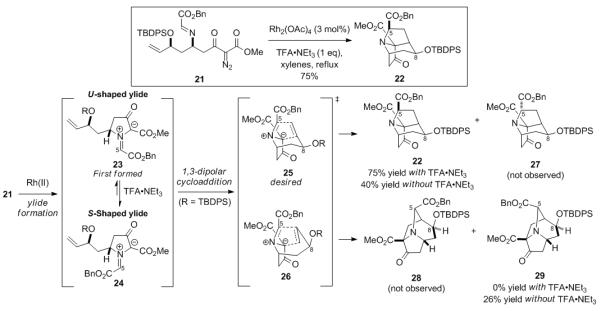
Rhodium Catalyzed Diastereoselective Dipolar Cycloaddition Cascade and Mechanistic Rationale for the Observed Selectivity
The divergent results obtained with crude and purified 21 beg an explanation. We envision that the rhodium carbene formed upon diazo decomposition of 21, which presumably has the imine stereochemistry shown, undergoes cyclization to generate the U-shaped azomethine ylide 23. This intermediate may then either undergo dipolar cycloaddition or isomerization, which appears to be accelerated by the presence of a weak acid, to form the more stable S-shaped azomethine ylide 24. The two regioisomeric transition states for the cycloaddition of 23 and 24 are depicted by 25 and 26, wherein the stereochemistry at C5 in 25 and 26 is dictated by the stereochemistry at C5 in 23 or 24. The dipolar cycloaddition of 23 via transition state 25 will furnish 27, which was not detected, whereas cycloaddition of 23 via transition state 26 will provide 29, which was only isolated when the reaction was conducted in the absence of acid. The cycloaddition of the diastereomeric azomethine ylide 24 via transition state 25 generates the desired adduct 22, whereas cycloaddition of 24 by the regioisomeric transition state 26 will give 28, which was not observed. Examination of transition state models reveals that those leading to 27 and 28 are much more hindered than those leading to 22 and 29. In the presence of acid, it thus appears that isomerization of 23 to 24 is more facile than the cyclization of 23 via the slightly less favorable transition state shown in 26 to give 29. This remarkably efficient cascade reaction is notable because the tricyclic core of the stemofoline alkaloids is generated with high stereoselectivity and regioselectivity in a single chemical operation from an acyclic precursor.
Having thus generated the key tricyclic intermediate 22, the stage was set to complete the formal syntheses of 1 and 4 (Scheme 4). Stereoselective hydride reduction of the ketone moiety in 22 delivered the requisite endo-alcohol, which was subsequently protected as MOM ether 30. Hydrogenolysis of the benzyl ester 31 afforded the amino acid, which was subjected directly to a modified Barton decarboxylation protocol using CHCl3 as the solvent to give 31 in 63% yield from 30.[19 ] The butenyl side chain was then introduced by DIBAL-H reduction of 31, followed by a Julia–Kocienski olefination[20] of the intermediate aldehyde 32 to furnish 33. TBAF mediated removal of the TBDPS group from 33 and subsequent Parikh–Doering oxidation[21] of the secondary alcohol thus formed afforded ketone 34. The alkylation of the enolate derived from 34 with ICH2CO2Et provided the axial-alkylated product 35. Base-mediated epimerization of 35 provided the equatorially-substituted product, which was treated with TFA to effect deprotection of the MOM group and furnish the hemiketal 36 in 29% overall yield from 31. The spectral data of synthetic 36 thus obtained are consistent with those reported by Overman for racemic 36 that was subsequently elaborated into didehydrostemofoline (1) and isodidehydrostemofoline (4).[15] Accordingly, preparation of enantiomerically pure 36 completes the formal enantioselective syntheses of 1 and 4. It also completes enantioselective syntheses of stemofoline (2) and stemoburkilline (6) because these two compounds were recently prepared from 1 by semisynthesis.[6b]
Scheme 4.
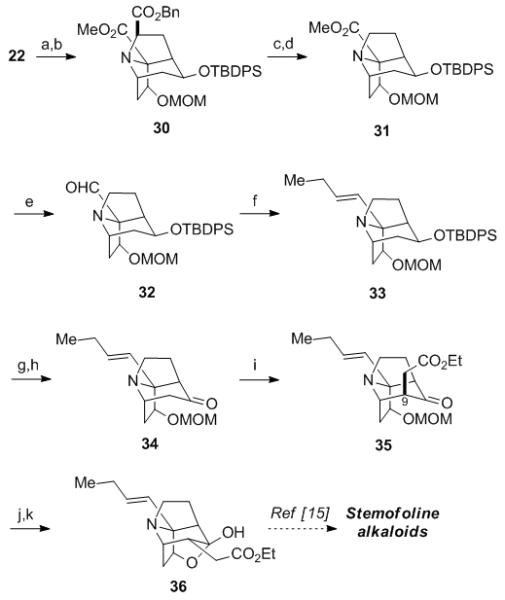
Completion of the Formal Total Synthesis. Reagents and Conditions: a) NaBH4, MeOH, −30 °C; b) MOM-Cl, NEtiPr2, DMF, 50 °C, 75% (two steps); c) Pd/C, H2, EtOH; d) 2-mercaptopyridine-N-oxide, DCC, DMAP, tBuSH, CHCl3, hν (250 W tungsten), 63% (two steps); e) DIBAL-H, CH2Cl2, −78 °C, 90%; f) Ph-N4CSO2-nPr, KHMDS, DME, −55 °C, 89%; g) TBAF, THF, 50 °C, 95%; h) SO3•py, NEt3, DMSO, CH2Cl2, 77%; i) LDA, ICH2CO2Et, THF, −10 °C, 62%; j) DBU, toluene, 130 °C; k) TFA, CH2Cl2, 81% (two steps).
In summary, the tricyclic compound 36, a key intermediate in Overman’s elegant synthesis of didehydrostemofoline (1) and isodidehydrostemofoline (4), has been prepared in enantiomerically pure form, thereby completing the first enantioselective approach to these alkaloids. Inasmuch as 1 has also been transformed into other stemofoline alkaloids,[6b] this accomplishment also constitutes a formal synthesis of many other members of the stemofoline family of natural products. The synthesis begins with commercially available 2-deoxy-d-ribose and features a novel cascade of reactions that culminates in the intramolecular dipolar cycloaddition of an acyclic diazo imine intermediate to form the cage-like, tricyclic core of the stemofoline alkaloids. Further applications of similar cascade reactions to complex molecule synthesis are in progress as is the use of 22 as an intermediate in even shorter routes to the stemofoline alkaloids. The results of these investigations will be reported in due course.
Supplementary Material
Footnotes
We thank the National Institutes of Health (GM 25439 and GM 31077) and The Robert A. Welch Foundation (F-0652) for their generous support. DHP thanks NIH for postdoc fellowship (GM096557).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1] a).Pilli RA, Ferreira de Oliveira MC. Nat. Prod. Rep. 2000;17:117–127. doi: 10.1039/a902437i. [DOI] [PubMed] [Google Scholar]; b) Greger H. Planta Med. 2006;72:99–113. doi: 10.1055/s-2005-916258. [DOI] [PubMed] [Google Scholar]
- [2] a).Pilli RA, Rosso GB, Ferreira de Oliveira MC. Nat. Prod. Rep. 2010;27:1908–1937. doi: 10.1039/c005018k. [DOI] [PubMed] [Google Scholar]; b) Alibes R, Figueredo M. Euro. J. Org. Chem. 2009:2421–2435. [Google Scholar]; c) Pilli RA, Rosso GB, Ferreira de Oliveira MC. Alkaloids. 2005;62:77–173. doi: 10.1016/s1099-4831(05)62002-0. [DOI] [PubMed] [Google Scholar]
- [3].Irie H, Masaki N, Ohno K, Osaki K, Taga T, Uyeo S. J. Chem. Soc. D; Chem. Commun. 1970:1066–1066. [Google Scholar]
- [4] a).Sekine T, Fukasawa N, Kashiwagi Y, Ruangrungsi N, Murakoshi I. Chem. Pharm. Bull. 1994;42:1360–1362. [Google Scholar]; b) Sekine T, Ikegami F, Fukasawa N, Kashiwagi Y, Aizawa T, Fujii Y, Ruangrungsi N, Murakashi I. J. Chem. Soc., Perkin Trans. 1995;1:391–393. [Google Scholar]; c) Jiwajinda S, Hirai N, Watanabe K, Santisopasri V, Chuengsamarnyart N, Koshimizu K, Ohigashi H. Phytochemistry. 2001;56:693–695. doi: 10.1016/s0031-9422(00)00443-x. [DOI] [PubMed] [Google Scholar]; d) Sastraruji T, Jatisatienr A, Pyne SG, Ung AT, Lie W, Williams MC. J. Nat. Prod. 2005;68:1763–1767. doi: 10.1021/np050361y. [DOI] [PubMed] [Google Scholar]; e) Mungkornasawakul P, Chaiyong S, Sastraruji T, Jatisatienr A, Jatisatienr C, Pyne SG, Ung AT, Korth J, Lie W. J. Nat. Prod. 2009;72:848–851. doi: 10.1021/np900030y. [DOI] [PubMed] [Google Scholar]
- [5].Brem B, Seger C, Pacher T, Hofer O, Vajrodaya S, Greger H. J. Agric. Food Chem. 2002;50:6383–6388. doi: 10.1021/jf0205615. [DOI] [PubMed] [Google Scholar]
- [6] a).Mungkornasawakul P, Pyne SG, Jatisatienr A, Lie W, Ung AT, Issakul K, Sawatwanich A, Supyen D, Jatisatienr C. J. Nat. Prod. 2004;67:1740–1743. doi: 10.1021/np049791z. [DOI] [PubMed] [Google Scholar]; b) Sastraruji K, Pyne SG, Ung AT, Mungkornasawakul P, Lie W, Jatisatienr A. J. Nat. Prod. 2009;72:316–318. doi: 10.1021/np800755p. [DOI] [PubMed] [Google Scholar]
- [7].Kaltenegger E, Brem B, Mereiter K, Kalchhauser H, Kahlig H, Hofer O, Vajrodaya S, Greger H. Phytochemistry. 2003;63:803–816. doi: 10.1016/s0031-9422(03)00332-7. [DOI] [PubMed] [Google Scholar]
- [8].Baird MC, Pyne SG, Ung AT, Lie W, Sastraruji T, Jatisatienr A, Jatisatienr C, Dheeranupattana S, Lowlam J, Boonchalermkit S. J. Nat. Prod. 2009;72:679–684. doi: 10.1021/np800806b. [DOI] [PubMed] [Google Scholar]
- [9].Tip-Pyang S, Tangpraprutgul P, Wiboopun N, Veerachato G, Phuwapraisirisan P, Sup-Udomphol B. ACGC Chem. Res. Commun. 2000;12:31–35. [Google Scholar]
- [10] a).Chanmahasathien W, Ohnuma S, Ambudkar SV, Limtrakul P. Planta Medica. 2011;77:1990–1995. doi: 10.1055/s-0031-1280054. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chanmahasathien W, Ampasavate C, Greger H, Limtrakul P. Phytomedicine. 2011;18:199–204. doi: 10.1016/j.phymed.2010.07.014. [DOI] [PubMed] [Google Scholar]
- [11] a).Sastraruji K, Sastraruji T, Pyne SG, Ung AT, Jatisatienr A, Lie W. J. Nat. Prod. 2010;73:935–941. doi: 10.1021/np100137h. [DOI] [PubMed] [Google Scholar]; b) Sastraruji K, Sastraruji T, Ung AT, Griffith R, Jatisatienr A, Pyne SG. Tetrahedron. 2012;68:7103–7115. [Google Scholar]
- [12] a).Epperson MT, Gin DY. Angew. Chem. 2002;114:1856–1858. doi: 10.1002/1521-3773(20020517)41:10<1778::aid-anie1778>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. 2002;41:1778–1780. doi: 10.1002/1521-3773(20020517)41:10<1778::aid-anie1778>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; b) Ye Y, Velten RF. Tetrahedron Lett. 2003;44:7171–7173. [Google Scholar]; c) Baylis AM, Davies MPH, Thomas EJ. Org. Biomol. Chem. 2007;5:3139–3155. doi: 10.1039/b708910d. [DOI] [PubMed] [Google Scholar]; d) Carra RJ, Epperson MT, Gin DY. Tetrahedron. 2008;64:3629–3641. doi: 10.1016/j.tet.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Thomas EJ, Vickers CF. Tetrahedron: Asymmetry. 2009. 20:970–979. [Google Scholar]
- [13] a).Dietz J, Martin SF. Tetrahedron Lett. 2011;52:2048–2050. doi: 10.1016/j.tetlet.2010.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shanahan CS, Fuller NO, Ludolph B, Martin SF. Tetrahedron Lett. 2011;52:4076–4079. doi: 10.1016/j.tetlet.2011.05.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kende AS, Smalley TL, Jr., Huang H. J. Am. Chem. Soc. 1999;121:7431–7432. [Google Scholar]
- [15].Brueggemann M, McDonald AI, Overman LE, Rosen MD, Schwink L, Scott JP. J. Am. Chem. Soc. 2003;125:15284–15285. doi: 10.1021/ja0388820. [DOI] [PubMed] [Google Scholar]
- [16].Swallen LC, Boord CE. J. Am. Chem. Soc. 1930;52:651–660. [Google Scholar]
- [17].Hirama M, Shigemoto T, Ito S. Tetrahedron Lett. 1985;26:4137–4140. [Google Scholar]
- [18].Padwa A, Dean DC, Osterhout MH, Precedo L, Semones MA. J. Org. Chem. 1994;59:5347–5357. [Google Scholar]
- [19].Ko EJ, Savagem GP, Williams CM. Org. Lett. 2011;13:1944–1947. doi: 10.1021/ol200290m. [DOI] [PubMed] [Google Scholar]
- [20].Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]
- [21].Parikh JR, Doering W. v. E. J. Am. Chem. Soc. 1967;89:5505–5507. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.