

Abstract
Vaccinia virus (VACV) strain Western Reserve protein C4 has been characterized and its function and contribution to virus virulence assessed. Bioinformatic analysis showed that C4 is conserved in six orthopoxvirus species and shares 43 % amino acid identity with VACV protein C16, a known virulence factor. A recombinant VACV expressing a C-terminally tagged version of C4 showed that, like C16, this 37 kDa protein is expressed early during infection and localizes to both the cytoplasm and the nucleus. Functional assays using a firefly luciferase reporter plasmid under the control of a nuclear factor kappa B (NF-κB)-dependent promoter demonstrated that C4 inhibits NF-κB activation at, or downstream of, the inhibitor of kappa kinase (IKK) complex. Consistent with this, C4 inhibited interleukin-1β-induced translocation of p65 into the nucleus. A VACV lacking the C4L gene (vΔC4) showed no significant differences from wild-type virus in growth kinetics or spread in cell culture, but had reduced virulence in a murine intranasal model of infection. vΔC4-infected mice exhibited fewer symptoms, lost less weight and recovered 7 days earlier than animals infected with control viruses expressing C4. Furthermore, bronchoalveolar lavage fluid from vΔC4-infected mice had increased cell numbers at day 5 post-infection, which correlated with reduced lung virus titres from this time onward. C4 represents the ninth VACV protein to inhibit NF-κB activation and remarkably, in every case examined, loss of each protein individually caused an alteration in virus virulence, despite the presence of other NF-κB inhibitors.
Introduction
Vaccinia virus (VACV) is the prototypical member of the genus Orthopoxvirus (OPV) of the Poxviridae, a family of large, complex viruses with dsDNA genomes of 135 kb or more (Moss, 2007). Like other poxviruses, VACV replicates in cytoplasmic factories and encodes many proteins needed for virus transcription and DNA replication (Moss, 2007). VACV is the live vaccine used to immunize against smallpox, an eradicated human disease caused by the antigenically related OPV variola virus (Fenner et al., 1988). After smallpox eradication, interest in VACV continued due to the development of recombinant VACVs as candidate vaccines for other micro-organisms, for instance hepatitis B virus (Smith et al., 1983; Moss et al., 1984; Paoletti et al., 1984), and because VACV is a useful expression vector (Moss, 1996), a tool for immunologists (Bennink et al., 1984, 1986: Yewdell et al., 1985; McMichael et al., 1986) and a good model system for studying virus–host interactions (Cudmore et al., 1995; Alcamí & Smith, 1996; Doceul et al., 2010).
The VACV genome contains approximately 200 ORFs (Goebel et al., 1990) with a highly conserved central region encoding proteins required for VACV transcription, replication and assembly (Upton et al., 2003; Gubser et al., 2004). In contrast, the more variable terminal regions encode proteins that are non-essential for virus replication in cell culture, but which affect virus host range, virulence and immunomodulation in vivo. The latter group includes proteins that are secreted from infected cells to bind cytokines, chemokines, interferons (IFNs) or complement factors, and intracellular proteins that inhibit apoptosis, synthesize steroid hormones or block signalling cascades leading to activation of transcription factors that promote expression of IFNs and pro-inflammatory molecules (Smith, 1994; Alcami, 2003; Seet et al., 2003).
A subset of these VACV immunomodulatory proteins inhibit activation of nuclear factor kappa B (NF-κB), a transcription factor that is retained in the cytosol of resting cells bound to the inhibitor of kappa B alpha (IκBα). When upstream signalling pathways are activated by, for instance, engagement of the interleukin (IL)-1 receptor with IL-1β, tumour necrosis factor (TNF) receptor with TNF-α, or Toll-like receptors (TLRs) with their respective ligands, IκBα is phosphorylated, ubiquitinated and degraded. Consequently, NF-κB is released and translocates to the nucleus, where it activates NF-κB-responsive genes (Hayden & Ghosh, 2008). NF-κB is an important transcription factor for the expression of many pro-inflammatory molecules and therefore it is not surprising that viruses encode proteins that inhibit activation of NF-κB. VACV is no exception and encodes many proteins that target this pathway, including A46, A52, N1, B14, M2, K1, K7 and E3.
Protein A52 binds TNF receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 2 (IRAK2) and inhibits NF-κB activation downstream of TLRs and the IL-1 receptor (Bowie et al., 2000; Harte et al., 2003). Protein A46 targets the host Toll–IL-1 receptor (TIR) adaptors myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like, TIR domain-containing adaptor inducing IFN-β (TRIF) and the TRIF-related adaptor molecule, and thereby interferes with NF-κB activation (Bowie et al., 2000; Stack et al., 2005). Protein B14 binds to the inhibitor of kappa B kinase (IKK)β and thereby inhibits its activation and the consequential phosphorylation of IκBα (Chen et al., 2006, 2008). Protein N1 was reported to bind the IKK complex (DiPerna et al., 2004), but this has been disputed (Chen et al., 2008) and so the mechanism by which N1 inhibits NF-κB activation remains uncertain. Protein M2 reduced ERK2 phosphorylation induced by phorbol myristate acetate and prevented p65 nuclear translocation (Gedey et al., 2006). Proteins K7 and K1 inhibit NF-κB activation either by inhibiting TLR-induced signalling (K7) (Schröder et al., 2008) or by preventing IκBα degradation (K1) (Shisler & Jin, 2004). Finally, protein E3 inhibits NF-κB activity in both PKR-dependent and -independent manners (Myskiw et al., 2009), and also by antagonizing the RNA polymerase III–dsDNA-sensing pathway (Valentine & Smith, 2010).
This study reports a characterization of VACV strain Western Reserve (WR) protein C4, encoded by gene C4L. Protein C4 has a predicted size of 37.2 kDa and is conserved within other VACV strains and OPVs (http://www.poxvirus.org), suggesting an important function. Protein C4 also shows 43 % amino acid identity to VACV protein C16, which is an immunomodulator and affects virus virulence (Fahy et al., 2008). However, C4 and C16 are functionally distinct because viruses lacking C16 exhibit an attenuated phenotype despite the presence of C4. Data presented here show that C4 is expressed early, is present in both the cytoplasm and the nucleus and inhibits activation of NF-κB at, or downstream of, the IKK complex. Despite being the ninth VACV protein assigned this function, deletion of C4 from the VACV genome caused attenuation of virus virulence. Possible explanations for this observation are discussed.
Results
Computational analysis of the C4L gene
VACV WR gene C4L (GenBank accession no. YP_232906) encodes a 37.2 kDa protein without a transmembrane domain or signal peptide (http://www.poxvirus.org) and without obvious cellular orthologues. The C-terminal sequence VTKYYI is very similar to VTKFYF present in the same position of the IL-1 receptor antagonist (IL1-ra) protein. This peptide is also conserved in the related VACV protein C16 (VTRFYF) (Fahy et al., 2008) and peptides containing this sequence were reported to have immunosuppressive activity (Kluczyk et al., 2002). C4 is conserved in seven of 15 VACV strains, including WR, Copenhagen, Lister (and derivatives such as LC16m0), which all contain the C-terminal VTKYYI peptide, but is absent from VACV strains chorioallantois vaccinia virus Ankara (CVA), modified vaccinia Ankara (MVA) and its derivatives, Acambis 2000 and its derivatives, 3737 and DUKE. C4 is also present in six of eight OPV species (95–99 % amino acid identity), including VACV, cowpox virus, camelpox virus (CMLV), taterapoxvirus (TATV), variola virus and monkeypox virus (MPXV), but not in ectromelia virus (ECTV) or horsepox virus. C16 is conserved (97–100 % amino acid identity) in all sequenced strains of VACV, but only in five OPV species (Fahy et al., 2008). Notably, the three OPVs lacking C16 (CMLV, TATV and MPXV) encode C4. Outside OPVs, C4 showed limited conservation (23–32 % amino acid identity) in eight chordopoxviruses, including mule deer poxvirus, sheeppox virus and lumpy skin disease virus.
Construction of C4L deletion, revertant and TAP VACVs
Several recombinant VACVs (strain WR) were constructed (Methods) to study the C4 protein within VACV-infected cells. These included a plaque-purified wild-type virus (vC4), a virus lacking the C4L gene (vΔC4) and a revertant virus in which the C4L gene was reinserted at its natural locus (vC4-Rev). To characterize the C4 protein during VACV infection in the absence of a C4 antibody, a virus expressing C4 from its natural promoter and TAP-tagged at the C terminus (vC4-TAP) was constructed. PCR utilizing C4L primers confirmed the presence of C4L in vC4, vC4-Rev and vC4-TAP and its absence in vΔC4 (Fig. S1, available in JGV Online). Analysis of genomic DNA by restriction endonuclease digestion showed that the only discernible difference between these viruses was at the C4L locus (data not shown).
Analysis of C4 expression during VACV infection
To determine when C4 is expressed, BSC-1 cells were infected with vC4-TAP in the presence or absence of cytosine arabinoside (AraC), an inhibitor of viral DNA replication and late protein expression, and extracts of cells were analysed by immunoblotting at different times post-infection (p.i.) (Fig. 1). C4–TAP was detected as a 37 kDa protein, consistent with its predicted size. Like protein C16 (Fahy et al., 2008), the C4 protein was detected from 2 h p.i. onwards and in the presence of AraC, indicating early expression. In contrast, AraC inhibited expression of D8, a virion structural protein expressed late during infection (Niles & Seto, 1988). Immunoblotting with anti-α-tubulin confirmed equivalent loading of samples.
Fig. 1.
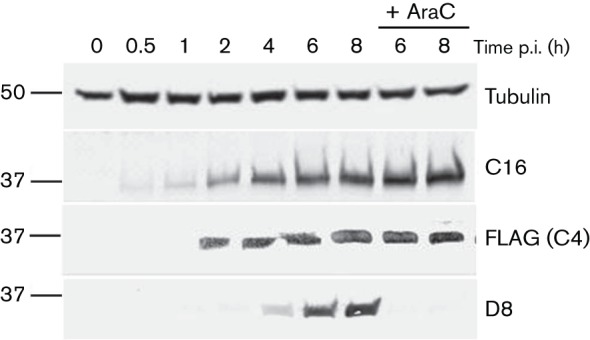
C4 is expressed early during infection. BSC-1 cells were infected with vC4-TAP at 10 p.f.u. per cell in the presence or absence of 40 µg AraC ml−1. At the indicated times p.i., the cells were washed with PBS and lysed. Samples were resolved by SDS-PAGE and analysed by immunoblotting with antibodies against the indicated proteins. The positions of molecular mass markers are indicated (kDa).
The subcellular localization of C4 was investigated by infecting HeLa cells with vC4-TAP and then either examining fixed cells by immunofluorescence (Fig. 2a) or performing immunoblotting of biochemically fractionated cellular lysates (Fig. 2b). Consistent with temporal expression data, immunofluorescence detected the C4 protein from 2 h p.i. in the cytoplasm, but C4 subsequently became increasingly nuclear. By 6 h p.i., C4 was detectable within both cytoplasm and nucleus, but by 24 h it was predominantly nuclear. Immunoblotting of fractionated cells corroborated data obtained by immunofluorescence and showed that C16 had a distribution similar to that of C4 (Fig. 2b). Successful separation of cytoplasmic and nuclear compartments was confirmed by the presence of α-tubulin and lamin A/C in only the cytoplasmic and nuclear fractions, respectively.
Fig. 2.

Subcellular localization of C4. (a) Immunofluorescence. HeLa cells were infected with vC4-TAP at 0.5 p.f.u. per cell for the indicated times. Cells were washed with PBS, fixed and stained with anti-FLAG mAb. The localization of C4/FLAG (red; middle panels), DNA stained with DAPI (blue; left panels) and phase-contrast/merged images (right panels) are shown. Bars, 20 µm (0–6 h); 10 µm (24 h). (b) Immunoblotting. BSC-1 cells were infected at 10 p.f.u. per cell with vC4-TAP for the indicated times, harvested and fractionated into cytoplasmic and nuclear fractions. Protein fractions were resolved by SDS-PAGE and analysed by immunoblotting with antibodies against the indicated proteins. The positions of molecular mass markers are shown (kDa).
C4 does not affect virus replication in cell culture
The fact that not all VACV strains contain gene C4L indicated that it was non-essential for virus replication, and this was confirmed for VACV strain WR by the isolation of the C4 deletion mutant, vΔC4. To ascertain whether C4 affected virus replication or spread, the size of plaque formed by vΔC4 was compared with that of control viruses in RK-13 and BSC-1 cells; no significant differences were observed (Fig. 3a). Next, the replication of vΔC4 in BSC-1 cells was investigated after infection at low (0.01) or high (10) m.o.i. and viruses in the intra- and extracellular fractions were titrated by plaque assay. Again, no differences were observed between vΔC4 and control viruses (Figs 3b, c and S2). Collectively, these data indicate that C4 is non-essential for virus replication and spread.
Fig. 3.
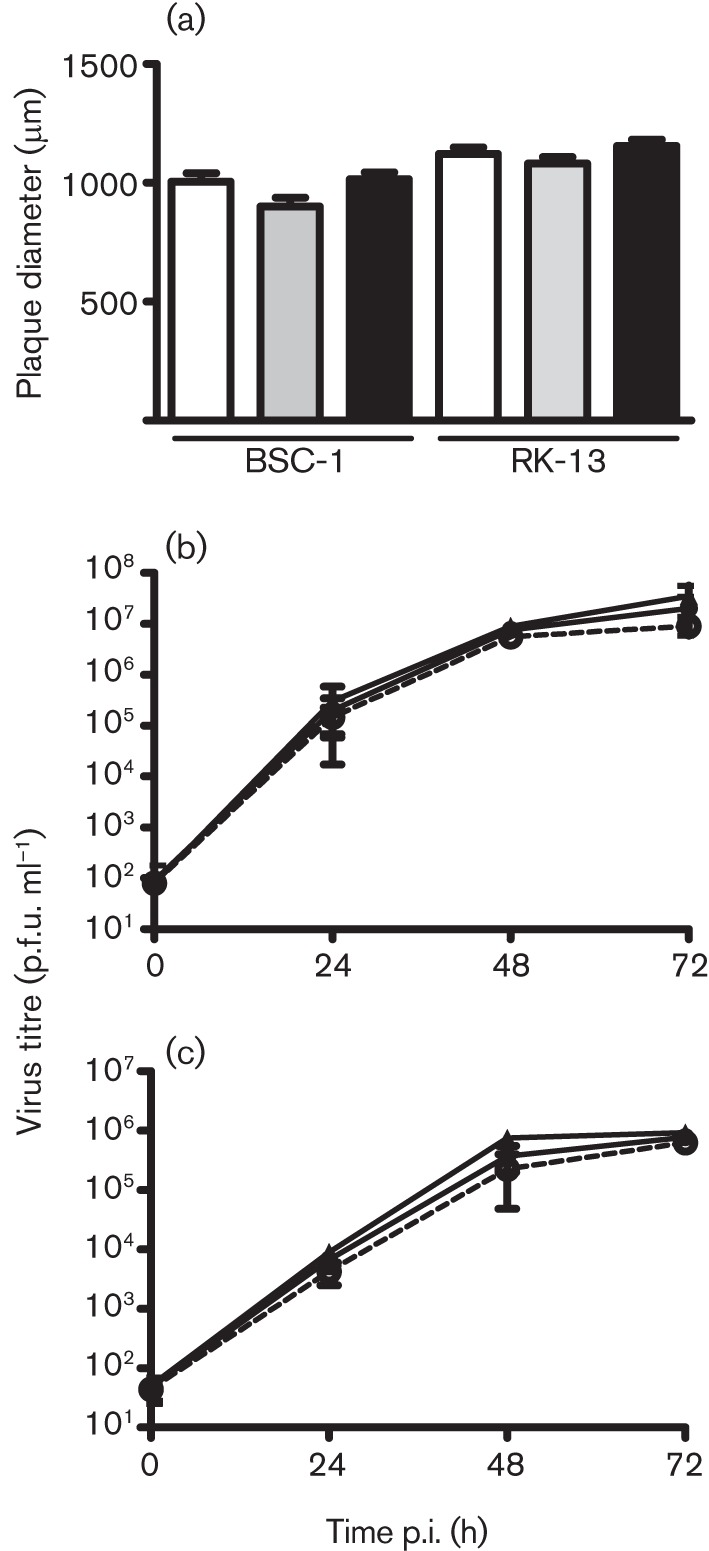
C4 is non-essential for virus replication and spread. (a) Plaque size. Monolayers of BSC-1 or RK-13 cells were infected with viruses (empty bars, vC4; shaded bars, vΔC4; filled bars, vC4-Rev) for 72 h. The sizes of 30 plaques were measured for each virus. Data are expressed as the mean±sd plaque diameter (μm). (b, c) Growth curves. BSC-1 cells were infected at 0.01 p.f.u. per cell and (b) intracellular and (c) extracellular virus were collected at the indicated times and titrated by plaque assay on BSC-1 cells. •, vC4; ○, vΔC4; ▴, vC4-Rev. Data are presented as the mean±sd log10(p.f.u.).
C4 inhibits NF-κB activation
Given that C4 was intracellular, its proposed possible function as an extracellular IL-1ra-like protein seemed improbable. Therefore, we investigated whether C4 inhibited intracellular signalling pathways, using a reporter plasmid with the IFN-β promoter driving expression of firefly luciferase. This was transfected into HEK293T cells that were stimulated subsequently by transfection with poly(dA : dT), a ligand for intracellular DNA sensors, or poly(I : C), a ligand of retinoic acid-inducible gene (RIG)-I-like receptors. These stimuli each induced luciferase activity, which was inhibited by C4 but not by a GFP control (Fig. 4a, b). Inhibition was also achieved by VACV protein B14, which inhibits NF-κB activation (Chen et al., 2008), and by C6, which inhibits interferon regulatory factor (IRF) 3 activation (Unterholzner et al., 2011), because the IFN-β promoter contains binding sites for both IRF3 and NF-κB.
Fig. 4.

C4 inhibits NF-κB activation. HEK293T cells were transfected with a firefly luciferase reporter plasmid under the control of an (a, b) IFN-β-, (c, d) ISG56.1- or (e, f) NF-κB-dependent promoter, a Renilla luciferase transfection control, and a C4, B14, C6 or GFP expression plasmid or empty vector control (EV). Cells were (a–d) transfected 24 h later with (a, c) poly(dA : dT) or (b, d) poly(I : C) at 800 ng per well for 24 h or (e, f) stimulated directly with (e) IL-1β or (f) TNF-α at 40 ng ml−1 for 8 h and then harvested. Firefly luciferase activity was normalized to Renilla luciferase activity. Data are from one experiment representative of at least three, each performed in triplicate and presented as means±sd. *P<0.05; **P<0.005; ***P<0.0005, compared with EV.
To dissect the mechanism of action of C4, its ability to inhibit activation of IRF3, NF-κB and activator protein 1 (AP-1) was investigated with reporter plasmids specific for these transcription factors. To measure IRF3 activity, an ISG56.1-dependent promoter was stimulated by the transfection of poly(dA : dT) and poly(I : C). ISG56.1 promoter activity was inhibited by C6, a recently characterized antagonist of IRF3 activation (Unterholzner et al., 2011), but not by C4 or B14 (Fig. 4c, d). Next, the effect of C4 on NF-κB-specific promoter activity was assessed using an NF-κB-dependent promoter (Fig. 4e, f). Stimulation of HEK293T cells with IL-1β and TNF-α resulted in a 70- and 18-fold induction of NF-κB promoter activity, respectively, and both were inhibited by C4. As expected B14, but not C6, was able to inhibit NF-κB activation (Chen et al., 2008; Unterholzner et al., 2011). Satisfactory stimulation of an AP-1 reporter plasmid was not achieved under several conditions tried and so the effect of C4 on this transcription factor could not be determined. Collectively, these data indicate that C4 inhibits IFN-β promoter activity by inhibiting NF-κB, but not IRF3, activation.
The mechanism by which C4 inhibited NF-κB activation was investigated by overexpression of proteins acting at different stages in the signalling cascade. Expression of TRAF2, which acts downstream of the TNF receptor, or TRAF6, which acts downstream of the IL-1 receptor, both stimulated NF-κB expression and both pathways were inhibited by C4 and B14 (Fig. 5a, b). C4 was therefore inhibiting both IL-1- and TNF-mediated signalling and so was likely to act at, or downstream of, the position where these pathways converge. To test this hypothesis, NF-κB was activated by overexpression of IKKβ, part of the IKK complex (Fig. 5c); once again both C4 and B14, but not C6, inhibited this activation. Taken together, these data demonstrate that C4 inhibits the activation of NF-κB at, or downstream of, the IKK complex. This was confirmed by measuring the translocation of p65 into the nucleus in response to IL-1β stimulation (Fig. 6). In resting cells p65 was cytoplasmic but, in response to IL-1β, it became nuclear and this was inhibited by C4 expression (Fig. 6a, b).
Fig. 5.
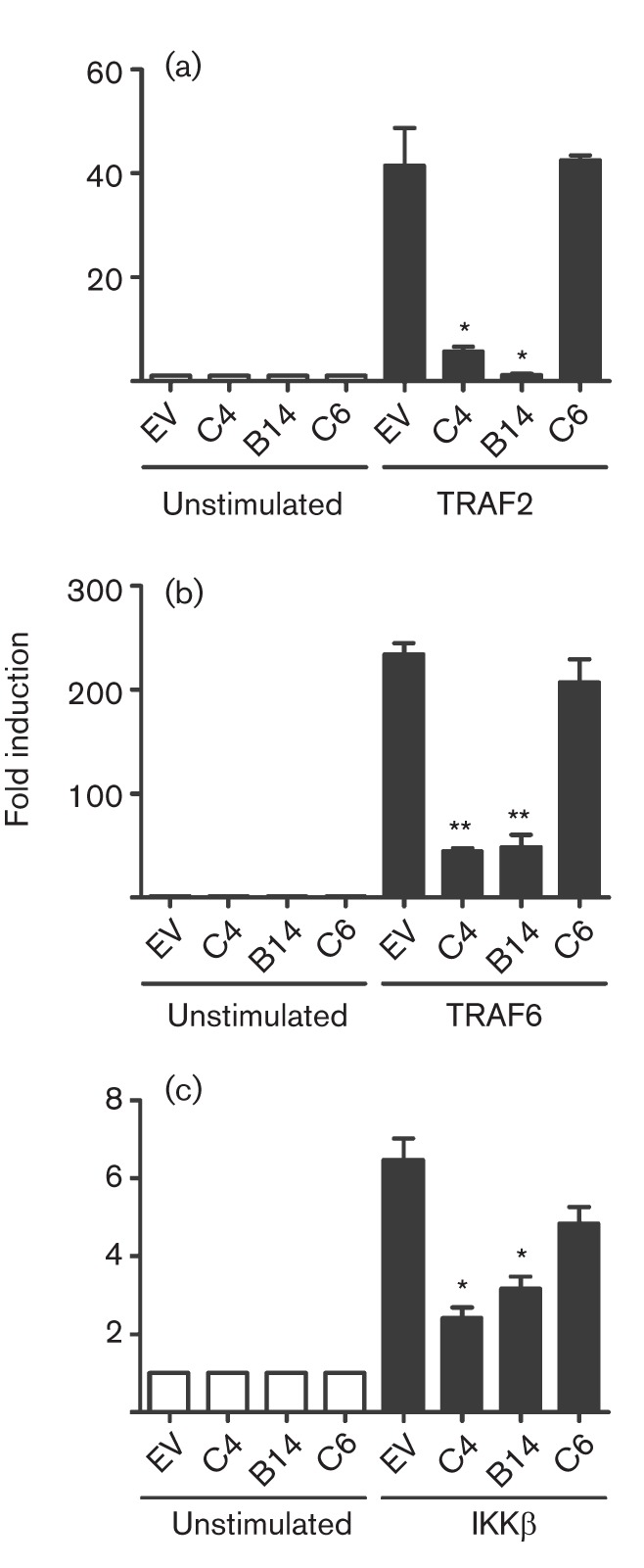
C4 inhibits NF-κB activation at or downstream of IKKβ. HEK293T cells were transfected with a firefly luciferase reporter plasmid under the control of an NF-κB-dependent promoter and (a) TRAF2, (b) TRAF6 or (c) IKK-β expression plasmids. Cells were co-transfected with a Renilla luciferase control and a C4, B14 or C6 expression plasmid or empty vector control (EV) for 24 h. Firefly luciferase activity was normalized to Renilla luciferase activity. Data are from one experiment representative of at least three, each performed in triplicate and presented as mean±sd. *P<0.05; **P<0.005, compared with EV.
Fig. 6.
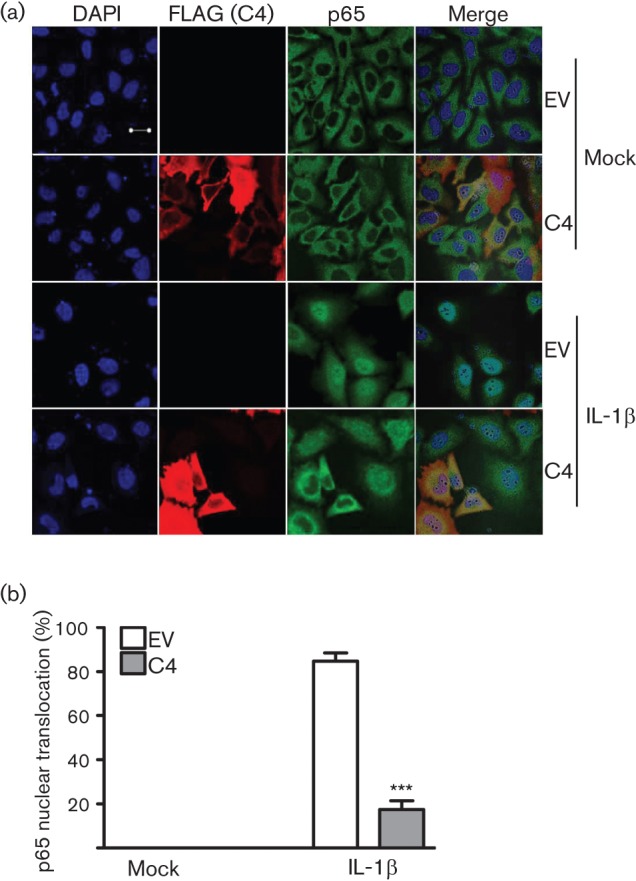
C4 blocks p65 translocation. (a) Immunofluorescence. HEK293T cells were transfected with a C4 expression plasmid or empty vector control (EV). After 24 h, cells were mock-stimulated or stimulated with 20 ng IL-1β ml−1 for 20 min. Cells were washed with PBS, fixed and stained with anti-FLAG (red) and anti-p65 (green) mAbs. DNA was stained with DAPI (blue) and phase-contrast/merged images (right panels) are shown. Bar, 20 µm. (b) Scoring: 90 cells per condition were quantified for nuclear translocation of p65. Data shown are from one experiment representative of three. ***P<0.0005, compared with EV.
C4 affects virus virulence and cell recruitment
As C4 inhibited NF-κB activation, it was possible that C4 might affect virus virulence, and this was tested utilizing intradermal and intranasal murine models of VACV infection (Williamson et al., 1990; Tscharke & Smith, 1999). In the intradermal model, there was no difference in lesion size in mice infected with vΔC4 compared with control viruses (Fig. S3). However in the intranasal model, mice infected with vΔC4 lost less weight, exhibited fewer signs of illness and recovered faster than mice infected with control viruses (Fig. 7a, b). These differences were statistically significant (P<0.05) from days 6 and 5 p.i., respectively. Measurement of virus titres in lungs at day 2 p.i. showed that all viruses had replicated to equivalent levels, demonstrating that, as in vitro, the loss of C4 did not impair virus replication in vivo. However, by days 5 and 8 p.i., vΔC4-infected mice had significantly lower virus titres than controls, suggesting more rapid clearance of virus in the absence of C4 (Fig. 8a). To investigate the basis for this phenotype, the number of viable cells in bronchoalveolar lavage (BAL) fluids was investigated on days 2, 5 and 8 p.i. (Fig. 8b). This revealed that, as early as day 2 p.i., there was an increase in the number of cells in BALs from vΔC4-infected mice, although this difference was not statistically significant. However, by day 5 a larger and statistically significant difference was seen. By day 8 p.i., there were fewer cells in the BAL fluid of mice infected with vΔC4 compared with controls. The recruitment of cells into BALs therefore showed quantitative and kinetic differences following infection by vΔC4, with earlier recruitment and higher numbers of cells, and more rapid resolution. Note that, at day 8 p.i., the number of cells present in BALs of mice infected with control viruses was equivalent to that at day 5, whereas in vΔC4-infected animals, the cell number had already declined from that at day 5, indicating recovery from infection. The more rapid and greater recruitment of cells into BAL fluids of vΔC4-infected mice correlated well with the reduced virus titres at days 5 and 8 p.i. C4 is therefore both an immunomodulator and a virulence factor.
Fig. 7.
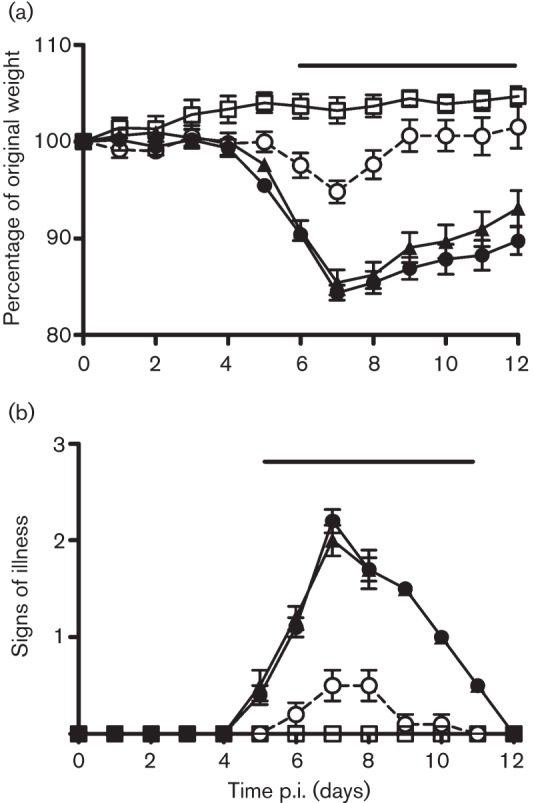
C4 is a virulence factor. BALB/c mice (n = 5) were infected intranasally with the indicated viruses and their weights and signs of illness were measured daily (Methods). (a) Weights. Data (means±sd) are expressed as a percentage of the mean weight of the same group of animals on day 0. (b) Signs of illness. The mean ±sem score of each group of animals is shown. □, Mock; •, vC4; ○, vΔC4; ▴, vC4-Rev. The horizontal bars indicate days on which the weight loss or signs of illness induced by vΔC4 was statistically different (P<0.05) from both vC4 and vC4-Rev.
Fig. 8.
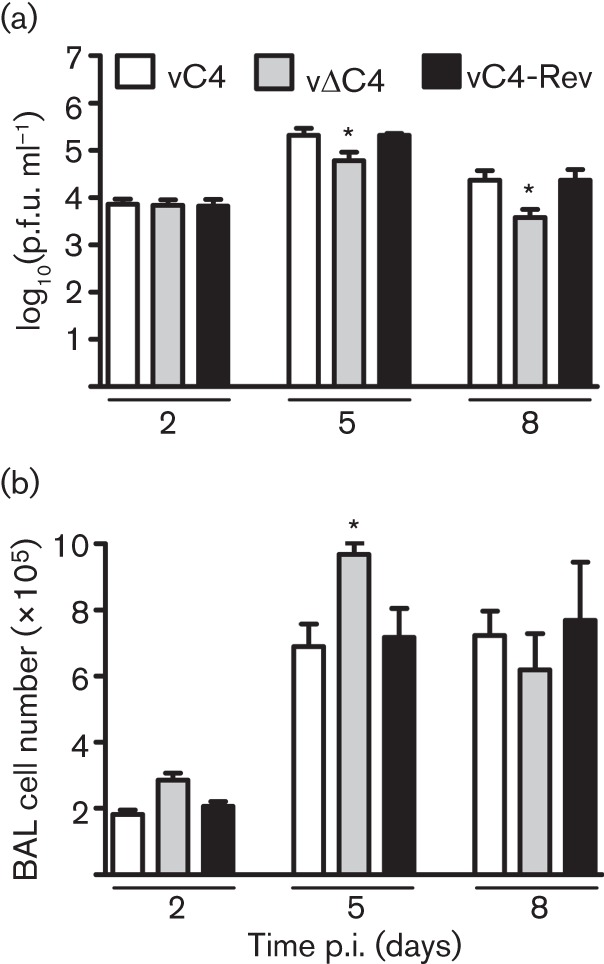
Lower virus titres and increased cell infiltration are associated with vΔC4 infection. BALB/c mice (n = 5) were infected intranasally with the indicated viruses. (a) Virus titres in lungs. At the indicated times, groups (n = 5) were sacrificed, lungs were removed and infectious virus (ml lung homogenate)−1 was determined by plaque assay. Data are presented as mean±sd titre. (b) Total number of viable cells in BAL fluid. At the indicated times, groups (n = 5) were sacrificed and cells were extracted from BAL fluid. Data are presented as mean±sd cell counts. *P<0.05 for vΔC4, compared with both vC4 and vC4Rev.
Discussion
Characterization of the C4 protein from VACV strain WR is reported. Data presented show that C4 is an intracellular, 37 kDa protein that is expressed early after infection and is non-essential for virus replication. Functional assays demonstrated C4 to inhibit NF-κB activation, acting at or downstream of the IKK complex, and C4 was shown to be a virulence factor in a murine intranasal model of infection.
The C4L gene is conserved (95–99 % amino acid identity) in six of eight OPV species and seven of 15 sequenced VACV strains. It is also related closely to VACV protein C16 and a family of OPV proteins (Fahy et al., 2008). It was noteworthy that C4 is encoded by the three OPVs that do not encode C16 (CMLV, TATV and MPXV). The close relatedness of C4 and C16 (43 % amino acid identity) and the fact that C4 is present in those OPVs lacking C16 suggest that these proteins have an important role in the virus life cycle and may have partially overlapping functions. However, it is clear that C4 and C16 are not redundant because removal of either gene causes an alteration in virus virulence, as shown in this study and by Fahy et al. (2008).
The detection of C4 early during infection is consistent with the results of transcriptional analysis which reported that C4 mRNA was detectable within 30 min p.i. (Assarsson et al., 2008). The expression of C4 also parallels that of C16, which is also made early during infection (Fahy et al., 2008), and this timing is consistent with the immunomodulatory function of these proteins. VACV proteins that inhibit the innate immune response to infection are generally expressed rapidly after infection to prevent activation of signalling cascades leading to production of IFNs and pro-inflammatory cytokines and chemokines. One exception to this principle is the case of the VACV IL-1β-binding protein encoded by VACV strain WR gene B15R, which is expressed late during infection and controls the body temperature of infected animals (Smith & Chan, 1991; Alcamí & Smith, 1992, 1996).
The localization of C4 during infection is unusual and shows a predominant cytoplasmic expression early (2 h) p.i., but this changes with time and by 6 h it is in both nuclear and cytoplasmic fractions, and by 24 h is predominantly nuclear (Fig. 2). Similarly, the related protein C16 is also predominantly cytoplasmic early after infection and accumulates in the nucleus as infection progresses (Fahy et al., 2008). C4 does not contain an identifiable nuclear-localization sequence, suggesting that it might bind to another viral or cellular protein to mediate this translocation, or that it can move between cytoplasmic and nuclear compartments by passive diffusion (which is possible for proteins ≤50 kDa). However, the strong nuclear predominance later during infection argues against simple diffusion for this localization. Proteins C6 and E3 are other examples of VACV immunomodulatory proteins that have a nucleocytoplasmic localization during the virus life cycle (Yuwen et al., 1993; Unterholzner et al., 2011).
The absence of C4 in several VACV strains and in ECTV indicated that the protein was non-essential for OPV replication and this was consistent with isolation of a VACV strain WR deletion mutant vΔC4. Despite this, a minor reduction in plaque size was noted, which, although not statistically significant with the sample size studied, was observed consistently in multiple cell types. Similarly, vΔC16 also produced a smaller plaque, but in that instance, the difference was large enough to be statistically significant (Fahy et al., 2008). In the future, it will be interesting to investigate the plaque size of a virus lacking both genes. An analysis of virus replication kinetics in vitro and in vivo showed no difference between vΔC4 and control viruses.
Functional screens revealed C4 to be an inhibitor of IFN-β promoter activation in response to poly(dA : dT) and poly(I : C). Mechanistically, this was attributable to inhibition of activation of NF-κB rather than IRF3. C4 is therefore distinct from VACV protein C6, which inhibits IRF3 activation by binding to the adaptor proteins TANK, SINTBAD and NAP1 (Unterholzner et al., 2011), but similar to B14, which inhibits NF-κB activation by binding to IKKβ (Chen et al., 2008; Graham et al., 2008; Benfield et al., 2011). The position in the pathway leading to NF-κB activation at which C4 functions was deduced to be at, or downstream of, the IKK complex, because C4 inhibited activation mediated by overexpression of TRAF2, TRAF6 and IKKβ. This was consistent with its ability to block NF-κB activation by both IL-1β and TNF-α. C4 is therefore comparable to B14, which binds to IKKβ and prevents the phosphorylation of the IKKβ activation loop (Chen et al., 2008; McCoy et al., 2010; Benfield et al., 2011).
Given the similar function of C4 and B14 and the fact that C4 represents the ninth VACV protein to inhibit NF-κB (see Introduction), it was surprising that loss of C4 caused an attenuated phenotype (Fig. 7). Furthermore, in all cases where the loss of an individual NF-κB inhibitor has been studied in vivo – A46 (Stack et al., 2005), A52 (Harte et al., 2003), B14 (Chen et al., 2006), N1 (Bartlett et al., 2002) and C4 (this paper) – there is also an attenuated phenotype, despite the presence of other inhibitors. These observations indicate that these proteins are non-redundant and that this non-redundancy might be explained in different ways. First, the proteins may have multiple functions, as has been demonstrated for N1, which is an inhibitor of both apoptosis and NF-κB activation (DiPerna et al., 2004; Cooray et al., 2007; Maluquer de Motes et al., 2011). Second, the position in the NF-κB activation pathway at which an inhibitor functions could affect outcome. For instance, if an inhibitor blocked only IL-1β or TNF-α-induced NF-κB activation, the influence in vivo could be different from that of an inhibitor that acts on both pathways. Furthermore, crosstalk between NF-κB and other signalling pathways could also allow an inhibitor to influence outcomes in vivo differently, depending on where it acts.
A further surprise is that losses of C4 or B14 give different in vivo phenotypes, despite blocking the same pathway. With vΔB14, the attenuation was evident in the intradermal but not the intranasal model (Chen et al., 2006), whereas with vΔC4, the opposite is observed. C4 represents another example where a phenotype is seen in only one model and illustrates the value of using both models, as noted previously (Tscharke et al., 2002). Mechanistically, a propensity for an immunomodulator to show a phenotype in only one in vivo model has been attributed to its mechanism of action and the different inflammatory responses elicited by infection via either route (Tscharke et al., 2002; Reading & Smith, 2003). In this case, given that B14 and C4 target the same pathway and at a similar position, the different in vivo phenotype is more surprising and suggests, perhaps, that one or both of these proteins have additional functions.
In summary, VACV protein C4 is an intracellular inhibitor of NF-κB and promotes virus virulence. C4 represents the ninth VACV inhibitor of NF-κB, and the fact that VACV has evolved so many inhibitors of this pathway highlights the importance of NF-κB signalling for the host response to virus infection. A notable feature of these inhibitors is that, in all cases where the virulence of viruses lacking individual proteins has been studied, reduced virulence is observed, despite the presence of the other inhibitors. This indicates additional complexity of these proteins and signalling pathways that is worthy of further investigation and may enhance our understanding of virus pathogenesis and innate immunity.
Methods
Cell culture.
BSC-1, CV-1 and HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10 % FBS (Harlan Laboratories) that was heated at 56 °C for 1 h to inactivate complement, and penicillin/streptomycin (50 µg ml−1; Gibco). HeLa and RK-13 cells were grown in minimum essential medium (Gibco) supplemented as above and with the addition of non-essential amino acids (Sigma). All cell lines were maintained at 37 °C in a 5 % CO2 atmosphere.
Construction of plasmids.
For construction of a VACV lacking the C4L gene, 300–350 bp flanking regions of the C4L gene were amplified by overlapping PCR from VACV WR genomic DNA, and then cloned into the Z11 mammalian expression vector containing Escherichia coli guanine phosphoribosyltransferase (Ecogpt) and enhanced green fluorescent protein (EGFP) genes, generating plasmid Z11ΔC4, as described previously for another VACV gene (Unterholzner et al., 2011). The 5′ DNA fragment was generated with oligonucleotides 5′-GCCACGCGTTCCAATTATCTTTACCG-3′ (L-FA) containing an MluI restriction site (underlined) and 5′-CCCGGGTCCGGAGAGCTCTAGTATTGGTTAAAAATGAAAATGG-3′ containing nucleotides from the 3′ fragment (italics) at the 5′ end. The 3′ DNA fragment was generated with oligonucleotides 5′-GAGCTCTCCGGACCCGGGTTTATATCACTACGG-3′ containing complementary sequence to the 5′ fragment (italics) and 5′-TAGCGGCCGCGGTGTCCTGTGTTCAGG-3′ (L-FB) containing a NotI restriction site (underlined). Left and right amplicons were joined by PCR using the L-FA and L-FB oligonucleotides to generate an FA-FB fragment. For construction of a C4 revertant virus in which the C4L gene was reinserted into its natural locus within the C4 deletion mutant, C4L together with the C4L flanking regions was amplified from VACV WR genomic DNA using oligonucleotides L-FA and L-FB and then cloned into Z11, generating plasmid Z11C4Rev.
The sequence of C4L was codon-optimized for expression in mammalian cells (GeneArt, Invitrogen Life Technologies). C4L was then subcloned into the mammalian expression vector pcDNA4.0/TO (Invitrogen) containing a C-terminal tandem affinity purification (TAP) tag consisting of two FLAG and two StrepII epitopes (Gloeckner et al., 2007), generating plasmid C4-TAP. For construction of the TAP-tagged C4 revertant virus (vC4-TAPRev), C4L and the C4L left flanking region were cloned into the pcDNA4.0/TO vector and the C4L right flanking region was then cloned downstream of the TAP tag. The complete C4L locus and TAP tag was subsequently subcloned into the pUC13 mammalian expression vector modified to contain the Ecogpt and EGFP genes (Maluquer de Motes et al., 2011), generating the plasmid pUC13-C4TAPRev. The fidelity of all plasmids constructed using PCR was confirmed by DNA sequencing.
Construction of recombinant VACVs.
C4 recombinant viruses were constructed by transfecting plasmid Z11ΔC4 into VACV-WR-infected CV-1 cells (to make vΔC4) or transfecting Z11C4Rev or pUC13-C4TAPRev into vΔC4-infected cells (to make vC4-Rev and vC4-TAPRev, respectively). Recombinant VACVs were subsequently isolated by transient dominant selection (Falkner & Moss, 1990) as described for the construction of other VACV deletion mutants (Chen et al., 2006; Fahy et al., 2008), using three cycles of plaque purification. The genotype of viruses was determined by C4L locus-specific PCR, and a plaque-purified wild-type virus (vC4) and a C4L deletion mutant virus (vΔC4) were identified and amplified. A revertant virus expressing C4 and a recombinant virus expressing C4 with a C-terminal TAP tag were isolated from vΔC4 by the same methodology. The genomes of all viruses were confirmed to have the predicted structure by PCR and restriction endonuclease digestion of genomic DNA.
Immunoblotting.
BSC-1 cells were infected with the indicated viruses and cell lysates prepared as described previously (Bartlett et al., 2002). Antibodies were from the following sources: mouse anti-FLAG (Sigma), mouse anti-α-tubulin (Upstate Biotech) and mouse anti-lamin A/C (Abcam). The rabbit C16 polyclonal antiserum and mouse anti-D8 mAb AB1.1 were described previously (Parkinson & Smith, 1994; Fahy et al., 2008). Secondary antibodies and protein detection were performed as described previously (Bartlett et al., 2002).
Cell fractionation.
BSC-1 cells were infected with vC4-TAP at 10 p.f.u. per cell for the indicated times and processed as described previously (Fahy et al., 2008). Proteins were resolved by SDS-PAGE and detected by immunoblotting.
Immunofluorescence.
HeLa cells were seeded into six-well plates containing sterile coverslips (borosilicate glass; BDH) 24 h before infection at 0.5 p.f.u. per cell. At various times p.i., the cells were washed twice with ice-cold PBS and processed as described previously (Unterholzner et al., 2011). Antibodies used for staining were rabbit anti-FLAG primary antibody (1 : 500 in blocking buffer; Sigma), mouse anti-p65 primary antibody (1 : 500 in blocking buffer; Santa Cruz Biotechnology), Alexa Fluor 488–donkey anti-mouse secondary antibody [1 : 500 in blocking buffer containing 10 % donkey serum (Sigma); Invitrogen] and Alexa Fluor 546–goat anti-rabbit secondary antibody (1 : 500 in blocking buffer containing 10 % donkey serum; Invitrogen).
Plaque-size assay.
Monolayers of BSC-1 or RK-13 cells were infected in duplicate at 50 p.f.u. per well and incubated for 72 h in medium containing 1.5 % carboxymethylcellulose. The cells were washed once with PBS and then stained with crystal violet. The diameter of 30 plaques per virus was measured using Axiovision 4.6 on a Zeiss Axiovert 200 M microscope (Zeiss).
Virus growth curves.
Intracellular and extracellular virus production after high or low m.o.i. was determined as described previously (Chen et al., 2006) and the clarified culture supernatant was used for extracellular virus.
Reporter-gene assays.
Luciferase reporter-gene assays were performed by seeding HEK293T cells at a density of 10 000 cells per well before transfection with polyethyleneimine (PEI; Polysciences). Transfection reactions contained 10 ng GL3-Renilla control plasmid per well, 60 ng IFN-β–, AP-1– or NF-κB–firefly reporter plasmid per well (gifts from A. Bowie, Trinity College Dublin, Ireland, and G. Sen, Lerner Research Institute, Cleveland, OH, USA) and 70 ng expression vector or pcDNA4.0/TO empty vector control per well. After 24 h incubation, transfected cells were stimulated with 40 ng IL-1β or TNF-α ml−1 (Peprotech) for 8 h, or transfected with 800 ng poly(dA : dT) or poly(I : C) ml−1 (Sigma) for 24 h. The cells were harvested in Passive Lysis Buffer (Promega) and data were analysed using mars data analysis software on a FLUOstar Omega instrument (BMG Labtech). For data analysis, firefly luciferase activity values were normalized against Renilla luciferase activity values. Experiments were performed in biological triplicate and repeated a minimum of three times. Data are expressed as means±sd.
Virulence assay.
Female BALB/c mice (n = 5; 6–8 weeks old) were anaesthetized and infected intranasally with 5×103 p.f.u. and monitored as described previously (Alcamí & Smith, 1992). Female C57BL/6 mice (n = 5; 6–8 weeks old) were anaesthetized and infected intradermally with 104 p.f.u. and the lesion size was measured daily with a micrometer as described previously (Tscharke & Smith, 1999).
Analysis of cell populations.
Mice were sacrificed and the BAL fluid and lung tissue were prepared and processed as described previously (Clark et al., 2006; Jacobs et al., 2006). Viable cells in BAL fluid were stained with 0.4 % trypan blue (Invitrogen) and counted.
Statistical analysis.
Data were analysed using an unpaired Student’s t-test. Statistical significance is expressed as follows: *P<0.05, **P<0.005 and ***P<0.0005.
Acknowledgements
This work was supported by grants from the Medical Research Council and Wellcome Trust. B. J. F. is an Imperial College Junior Research Fellow and G. L. S. is a Wellcome Trust Principal Research Fellow.
Footnotes
Three supplementary figures are available with the online version of this paper.
References
- Alcami A. (2003). Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3, 36–50 10.1038/nri980 [DOI] [PubMed] [Google Scholar]
- Alcamí A., Smith G. L. (1992). A soluble receptor for interleukin-1β encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell 71, 153–167 10.1016/0092-8674(92)90274-G [DOI] [PubMed] [Google Scholar]
- Alcamí A., Smith G. L. (1996). A mechanism for the inhibition of fever by a virus. Proc Natl Acad Sci U S A 93, 11029–11034 10.1073/pnas.93.20.11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assarsson E., Greenbaum J. A., Sundström M., Schaffer L., Hammond J. A., Pasquetto V., Oseroff C., Hendrickson R. C., Lefkowitz E. J. & other authors (2008). Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc Natl Acad Sci U S A 105, 2140–2145 10.1073/pnas.0711573105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett N., Symons J. A., Tscharke D. C., Smith G. L. (2002). The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence. J Gen Virol 83, 1965–1976 [DOI] [PubMed] [Google Scholar]
- Benfield C. T., Mansur D. S., McCoy L. E., Ferguson B. J., Bahar M. W., Oldring A. P., Grimes J. M., Stuart D. I., Graham S. C., Smith G. L. (2011). Mapping the IκB kinase β (IKKβ)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKβ-mediated activation of nuclear factor κB. J Biol Chem 286, 20727–20735 10.1074/jbc.M111.231381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennink J. R., Yewdell J. W., Smith G. L., Moller C., Moss B. (1984). Recombinant vaccinia virus primes and stimulates influenza haemagglutinin-specific cytotoxic T cells. Nature 311, 578–579 10.1038/311578a0 [DOI] [PubMed] [Google Scholar]
- Bennink J. R., Yewdell J. W., Smith G. L., Moss B. (1986). Recognition of cloned influenza virus hemagglutinin gene products by cytotoxic T lymphocytes. J Virol 57, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie A., Kiss-Toth E., Symons J. A., Smith G. L., Dower S. K., O’Neill L. A. (2000). A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc Natl Acad Sci U S A 97, 10162–10167 10.1073/pnas.160027697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. A., Jacobs N., Smith G. L. (2006). Vaccinia virus strain Western Reserve protein B14 is an intracellular virulence factor. J Gen Virol 87, 1451–1458 10.1099/vir.0.81736-0 [DOI] [PubMed] [Google Scholar]
- Chen R. A., Ryzhakov G., Cooray S., Randow F., Smith G. L. (2008). Inhibition of IκB kinase by vaccinia virus virulence factor B14. PLoS Pathog 4, e22 10.1371/journal.ppat.0040022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R. H., Kenyon J. C., Bartlett N. W., Tscharke D. C., Smith G. L. (2006). Deletion of gene A41L enhances vaccinia virus immunogenicity and vaccine efficacy. J Gen Virol 87, 29–38 10.1099/vir.0.81417-0 [DOI] [PubMed] [Google Scholar]
- Cooray S., Bahar M. W., Abrescia N. G., McVey C. E., Bartlett N. W., Chen R. A., Stuart D. I., Grimes J. M., Smith G. L. (2007). Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gen Virol 88, 1656–1666 10.1099/vir.0.82772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore S., Cossart P., Griffiths G., Way M. (1995). Actin-based motility of vaccinia virus. Nature 378, 636–638 10.1038/378636a0 [DOI] [PubMed] [Google Scholar]
- DiPerna G., Stack J., Bowie A. G., Boyd A., Kotwal G., Zhang Z., Arvikar S., Latz E., Fitzgerald K. A., Marshall W. L. (2004). Poxvirus protein N1L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by Toll-like receptors. J Biol Chem 279, 36570–36578 10.1074/jbc.M400567200 [DOI] [PubMed] [Google Scholar]
- Doceul V., Hollinshead M., van der Linden L., Smith G. L. (2010). Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327, 873–876 10.1126/science.1183173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy A. S., Clark R. H., Glyde E. F., Smith G. L. (2008). Vaccinia virus protein C16 acts intracellularly to modulate the host response and promote virulence. J Gen Virol 89, 2377–2387 10.1099/vir.0.2008/004895-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkner F. G., Moss B. (1990). Transient dominant selection of recombinant vaccinia viruses. J Virol 64, 3108–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner F., Anderson D. A., Arita I., Jezek Z., Ladnyi I. D. (1988). Smallpox and its Eradication. Geneva: World Health Organization [Google Scholar]
- Gedey R., Jin X. L., Hinthong O., Shisler J. L. (2006). Poxviral regulation of the host NF-κB response: the vaccinia virus M2L protein inhibits induction of NF-κB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J Virol 80, 8676–8685 10.1128/JVI.00935-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner C. J., Boldt K., Schumacher A., Roepman R., Ueffing M. (2007). A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7, 4228–4234 10.1002/pmic.200700038 [DOI] [PubMed] [Google Scholar]
- Goebel S. J., Johnson G. P., Perkus M. E., Davis S. W., Winslow J. P., Paoletti E. (1990). The complete DNA sequence of vaccinia virus. Virology 179, 247–266, 517–563 10.1016/0042-6822(90)90294-2 [DOI] [PubMed] [Google Scholar]
- Graham S. C., Bahar M. W., Cooray S., Chen R. A., Whalen D. M., Abrescia N. G., Alderton D., Owens R. J., Stuart D. I. & other authors (2008). Vaccinia virus proteins A52 and B14 share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog 4, e1000128 10.1371/journal.ppat.1000128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubser C., Hué S., Kellam P., Smith G. L. (2004). Poxvirus genomes: a phylogenetic analysis. J Gen Virol 85, 105–117 10.1099/vir.0.19565-0 [DOI] [PubMed] [Google Scholar]
- Harte M. T., Haga I. R., Maloney G., Gray P., Reading P. C., Bartlett N. W., Smith G. L., Bowie A., O’Neill L. A. (2003). The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med 197, 343–351 10.1084/jem.20021652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M. S., Ghosh S. (2008). Shared principles in NF-κB signaling. Cell 132, 344–362 10.1016/j.cell.2008.01.020 [DOI] [PubMed] [Google Scholar]
- Jacobs N., Chen R. A., Gubser C., Najarro P., Smith G. L. (2006). Intradermal immune response after infection with Vaccinia virus. J Gen Virol 87, 1157–1161 10.1099/vir.0.81556-0 [DOI] [PubMed] [Google Scholar]
- Kluczyk A., Siemion I. Z., Szewczuk Z., Wieczorek Z. (2002). The immunosuppressive activity of peptide fragments of vaccinia virus C10L protein and a hypothesis on the role of this protein in the viral invasion. Peptides 23, 823–834 10.1016/S0196-9781(02)00006-2 [DOI] [PubMed] [Google Scholar]
- Maluquer de Motes C., Cooray S., Ren H., Almeida G. M., McGourty K., Bahar M. W., Stuart D. I., Grimes J. M., Graham S. C., Smith G. L. (2011). Inhibition of apoptosis and NF-κB activation by vaccinia protein N1 occur via distinct binding surfaces and make different contributions to virulence. PLoS Pathog 7, e1002430 10.1371/journal.ppat.1002430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy L. E., Fahy A. S., Chen R. A., Smith G. L. (2010). Mutations in modified virus Ankara protein 183 render it a non-functional counterpart of B14, an inhibitor of nuclear factor κB activation. J Gen Virol 91, 2216–2220 10.1099/vir.0.022343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael A. J., Michie C. A., Gotch F. M., Smith G. L., Moss B. (1986). Recognition of influenza A virus nucleoprotein by human cytotoxic T lymphocytes. J Gen Virol 67, 719–726 10.1099/0022-1317-67-4-719 [DOI] [PubMed] [Google Scholar]
- Moss B. (1996). Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci U S A 93, 11341–11348 10.1073/pnas.93.21.11341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. (2007). Poxviridae: the viruses and their replicaton. In Fields Virology, 5th edn, pp. 2905–2946 Edited by Knipe D. M. Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- Moss B., Smith G. L., Gerin J. L., Purcell R. H. (1984). Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature 311, 67–69 10.1038/311067a0 [DOI] [PubMed] [Google Scholar]
- Myskiw C., Arsenio J., van Bruggen R., Deschambault Y., Cao J. (2009). Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-κB, and IRF3 pathways. J Virol 83, 6757–6768 10.1128/JVI.02570-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niles E. G., Seto J. (1988). Vaccinia virus gene D8 encodes a virion transmembrane protein. J Virol 62, 3772–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti E., Lipinskas B. R., Samsonoff C., Mercer S., Panicali D. (1984). Construction of live vaccines using genetically engineered poxviruses: biological activity of vaccinia virus recombinants expressing the hepatitis B virus surface antigen and the herpes simplex virus glycoprotein D. Proc Natl Acad Sci U S A 81, 193–197 10.1073/pnas.81.1.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J. E., Smith G. L. (1994). Vaccinia virus gene A36R encodes a Mr 43–50 K protein on the surface of extracellular enveloped virus. Virology 204, 376–390 10.1006/viro.1994.1542 [DOI] [PubMed] [Google Scholar]
- Reading P. C., Smith G. L. (2003). A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol 84, 1973–1983 10.1099/vir.0.19285-0 [DOI] [PubMed] [Google Scholar]
- Schröder M., Baran M., Bowie A. G. (2008). Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKϵ-mediated IRF activation. EMBO J 27, 2147–2157 10.1038/emboj.2008.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seet B. T., Johnston J. B., Brunetti C. R., Barrett J. W., Everett H., Cameron C., Sypula J., Nazarian S. H., Lucas A., McFadden G. (2003). Poxviruses and immune evasion. Annu Rev Immunol 21, 377–423 10.1146/annurev.immunol.21.120601.141049 [DOI] [PubMed] [Google Scholar]
- Shisler J. L., Jin X. L. (2004). The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J Virol 78, 3553–3560 10.1128/JVI.78.7.3553-3560.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. L. (1994). Virus strategies for evasion of the host response to infection. Trends Microbiol 2, 81–88 10.1016/0966-842X(94)90539-8 [DOI] [PubMed] [Google Scholar]
- Smith G. L., Chan Y. S. (1991). Two vaccinia virus proteins structurally related to the interleukin-1 receptor and the immunoglobulin superfamily. J Gen Virol 72, 511–518 10.1099/0022-1317-72-3-511 [DOI] [PubMed] [Google Scholar]
- Smith G. L., Mackett M., Moss B. (1983). Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature 302, 490–495 10.1038/302490a0 [DOI] [PubMed] [Google Scholar]
- Stack J., Haga I. R., Schröder M., Bartlett N. W., Maloney G., Reading P. C., Fitzgerald K. A., Smith G. L., Bowie A. G. (2005). Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med 201, 1007–1018 10.1084/jem.20041442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscharke D. C., Smith G. L. (1999). A model for vaccinia virus pathogenesis and immunity based on intradermal injection of mouse ear pinnae. J Gen Virol 80, 2751–2755 [DOI] [PubMed] [Google Scholar]
- Tscharke D. C., Reading P. C., Smith G. L. (2002). Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J Gen Virol 83, 1977–1986 [DOI] [PubMed] [Google Scholar]
- Unterholzner L., Sumner R. P., Baran M., Ren H., Mansur D. S., Bourke N. M., Randow F., Smith G. L., Bowie A. G. (2011). Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog 7, e1002247 10.1371/journal.ppat.1002247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton C., Slack S., Hunter A. L., Ehlers A., Roper R. L. (2003). Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77, 7590–7600 10.1128/JVI.77.13.7590-7600.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine R., Smith G. L. (2010). Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J Gen Virol 91, 2221–2229 10.1099/vir.0.021998-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. D., Reith R. W., Jeffrey L. J., Arrand J. R., Mackett M. (1990). Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol 71, 2761–2767 10.1099/0022-1317-71-11-2761 [DOI] [PubMed] [Google Scholar]
- Yewdell J. W., Bennink J. R., Smith G. L., Moss B. (1985). Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc Natl Acad Sci U S A 82, 1785–1789 10.1073/pnas.82.6.1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuwen H., Cox J. H., Yewdell J. W., Bennink J. R., Moss B. (1993). Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195, 732–744 10.1006/viro.1993.1424 [DOI] [PubMed] [Google Scholar]