

Abstract
Until recently, transmissible spongiform encephalopathy (TSE) disease in cattle was thought to be caused by a single agent strain, bovine spongiform encephalopathy (BSE) (classical BSE or BSE-C). However, due to the initiation of a large-scale surveillance programme throughout Europe, two atypical BSE strains, bovine amyloidotic spongiform encephalopathy (BASE, also named BSE-L) and BSE-H have since been discovered. These atypical BSE isolates have been previously transmitted to a range of transgenic mouse models overexpressing PrP from different species at different levels, on a variety of genetic backgrounds. To control for genetic background and expression level in the analysis of these isolates, we performed here a comprehensive comparison of the neuropathological and molecular properties of all three BSE agents (BASE, BSE-C and BSE-H) upon transmission into the same gene-targeted transgenic mouse line expressing the bovine prion protein (Bov6) and a wild-type control of the same genetic background. Significantly, upon challenge with these BSE agents, we found that BASE did not produce shorter survival times in these mice compared with BSE-C, contrary to previous studies using overexpressing bovine transgenic mice. Amyloid plaques were only present in mice challenged with atypical BSE and neuropathological features, including intensity of PrP deposition in the brain and severity of vacuolar degeneration were less pronounced in BASE compared with BSE-C-challenged mice.
Introduction
Bovine spongiform encephalopathy (BSE) is a fatal neurodegenerative disorder of cattle, and belongs to a group of diseases known as transmissible spongiform encephalopathies (TSEs) or prion diseases. BSE was first reported in 1987 (Bruce et al., 1997; Wells et al., 1987) and its transmission to humans through contaminated food is thought to be the cause of the variant form of Creutzfeldt–Jakob disease (vCJD) (Bruce et al., 1997; Hill et al., 1997). BSE is characterized by the accumulation in the brain of PrPTSE, which is a protease-resistant conformational variant of the host encoded cellular prion protein (PrPc). Although BSE has been extensively characterized, the true nature and origin of the infectious agent remains unknown.
Until 2004, TSE disease in cattle was believed to be caused by a single prion strain, classical BSE (BSE-C). This conclusion was based on classical strain typing in mice (incubation time, lesion profiles, patterns of PrP staining in the brain) (Bruce, 1996) and biochemical features of the proteinase K (PK)-resistant PrP (PrPTSE) in natural and experimental BSE (Collinge et al., 1996; Kuczius & Groschup, 1999; Kuczius et al., 1998), which showed consistent results from all cattle isolates. However, two atypical BSE agents have recently been reported (Biacabe et al., 2004; Casalone et al., 2004; Jacobs et al., 2007; Stack et al., 2009), and are identified as H-type BSE (BSE-H) and bovine amyloidotic spongiform encephalopathy (BASE, also named BSE-L). These atypical bovine TSEs mainly affect older cattle and can be distinguished by the electrophoretic positions of their protease-resistant PrPTSE isoforms (Biacabe et al., 2004; Buschmann et al., 2006; Casalone et al., 2004; Jacobs et al., 2007). Indeed, PrP is a glycoprotein and has two sites for the attachment of N-linked glycans, which depending on their utilization will produce di-, mono- and unglycosylated PrP (Stimson et al., 1999). BSE-H PrPTSE shows a significantly higher molecular mass unglycosylated PrP isoform by immunoblot when compared with BSE-C PrPTSE. Similarly, BASE has a slightly lower molecular size than BSE-C PrPTSE and a clearly different glycoform pattern. Furthermore, following transmission into transgenic mice that overexpress the bovine prion protein, both BASE and BSE-H show neuropathological and molecular phenotypes, which are distinct from BSE-C (Béringue et al., 2006, 2007; Buschmann et al., 2006; Capobianco et al., 2007; Okada et al., 2011a). However, interestingly a recent study has found that survival times are similar in transgenic mice that overexpress the bovine prion protein challenged with either BSE-C or BSE-H (Torres et al., 2011). BSE-H and BASE were originally described in France (Biacabe et al., 2004) and Italy (Casalone et al., 2004), respectively; however, they have since been documented in other European countries (Jacobs et al., 2007), Japan (Hagiwara et al., 2007) and North America (Dudas et al., 2010; Richt et al., 2007). While BSE-C is thought to be the result of feeding cattle prion-contaminated meat and bone meal, the origin of BASE and BSE-H remains unknown.
Several previous studies examining the transmission characteristics of these atypical BSE isolates compared with BSE-C revealed an increased transmissibility of BASE to transgenic mice overexpressing bovine PrP compared with BSE-C. However, these studies are difficult to compare as they have been performed in different laboratories using different transgenic mouse lines that express varying levels of bovine PrP on mixed murine genetic backgrounds. It has previously been shown that genetic background of inbred mouse strains can significantly alter BSE incubation time in wild-type mice (Manolakou et al., 2001). Additionally, only two studies have been published in which all three isolates were compared in the same transgenic line (Buschmann et al., 2006; Nicot & Baron, 2011), which had high expression levels of PrPc and lacked a genetically compatible wild-type control. In order to control for expression level and genetic background, here we carried out a fully comprehensive comparison of the neuropathological and molecular properties of BASE, BSE-C and BSE-H when transmitted into both 129/Ola wild-type mice and Bov6 mice, which contain a targeted substitution of murine PrP in the mouse genome with bovine PrP, and are maintained on an inbred 129/Ola background. This is the first description of atypical BSE transmission to transgenic mice expressing bovine PrP under the same spatial and temporal control as wild-type murine PrP. We found that Bov6 mice were susceptible to all three BSE agents; however, unlike previous studies using overexpressing mice (Béringue et al., 2006, 2007; Buschmann et al., 2006; Capobianco et al., 2007; Nicot & Baron, 2011), Bov6 mice challenged with BASE did not produce shorter survival times than mice inoculated with BSE-C and presented a pathological profile, which was less severe than BSE-C. Our results show that expression levels of bovine PrP in these animals can influence transmission characteristics and neuropathology of classical and atypical BSE infection. Thus, while mouse lines overexpressing bovine PrP are valuable, sensitive models for the analysis of these diseases, a combination of overexpressing- and gene-targeted mouse models may give a more detailed understanding of transmissibility and neuropathogenesis of these diseases.
Results
Transmissibility of cattle TSE isolates to gene-targeted bovine PrP transgenic mice
Following intracerebral inoculation of Bov6 mice with BASE-, BSE-C- or BSE-H-infected cattle brain, a TSE was induced in 24/24, 22/22 and 17/23 of the mice, respectively, defined by the presence of either PrP deposition (using immunohistochemistry) in the brain or vacuolar pathology (Table 1). Bov6 mice inoculated with BASE, BSE-C or BSE-H first showed histopathological characteristics of disease (PrP deposition and vacuolar pathology) at 387, 328 and 476 days post-inoculation (p.i.), respectively (Fig. 1a). The average survival time, which was calculated from those mice displaying both PrP deposition in the brain and vacuolar pathology, was similar between BSE agents (BASE, 547±18; BSE-C, 518±18; BSE-H, 561±15 days) (Table 1, Fig. 1a). Interestingly, despite the observation of neuropathological changes in the majority of mice challenged with BASE and BSE-H, signs of clinical disease were virtually absent in these mice, which was not the case for BSE-C, where 15/22 mice showed clinical TSE (Table 1). Additionally, 8/8 129/Ola control mice inoculated with BSE-C showed clinical signs of disease and were both positive for vacuolation and PrP deposition in the brain (Table 1). However, very few neuropathological changes were detected in 129/Ola mice inoculated with BASE or BSE-H.
Table 1. Transmission of BASE, BSE-C and BSE-H to Bov6 mice and wild-type 129/Ola controls.
| TSE isolate | Bov6 | 129/Ola | |||||||
| Survival time* | Incubation time† | Clinical signs | Vacuolar pathology | PrP deposition | Incubation time | Clinical signs | Vacuolar pathology | PrP deposition | |
| BASE | 547±18 | 534, 541 | 2/24 | 21/24 | 24/24 | na‡ | 0/24 | 1/24 | 0/24 |
| BSE-C§ | 518±18 | 551±12 | 15/22 | 20/22 | 22/22 | 498±43 | 8/8 | 8/8 | 8/8 |
| BSE-H | 561±15 | 506 | 1/23 | 17/23 | 15/23 | na‡ | 0/24 | 0/24 | 5/24 |
Measured as days±sem and calculated from mice showing both vacuolar pathology and PrP deposition.
Measured as days±sem and calculated from mice showing clinical signs of disease.
No mice showed clinical signs of disease or both vacuolar pathology and PrP deposition in the brain.
Reanalysis of data from Bishop et al. (2006) and Plinston et al. (2011). Here, we have calculated survival time (see definition above) in addition to incubation time.
Fig. 1.
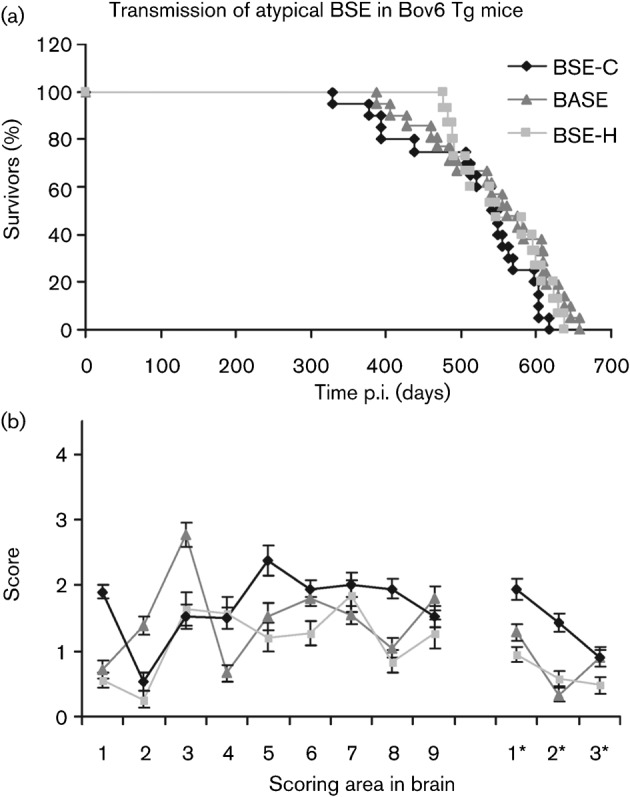
Kaplan–Meier survival curve showing percentage survival of Bov6 mice following challenge with BSE-C, BASE and BSE-H. All cases used were both positive for PrP deposition in the brain and showed vacuolar pathology (a). Pattern of vacuolation observed in brains of Bov6 mice inoculated with BASE, BSE-C and BSE-H. A profile was produced from nine grey matter areas (1, medulla; 2, cerebellum; 3, superior colliculus; 4, hypothalamus; 5, thalamus; 6, hippocampus; 7, septum; 8, retrospinal cortex; and 9, cingulate and motor cortex) and three white matter areas (1*, cerebellar white matter; 2*, midbrain white matter; and 3*, cerebral peduncle). Mean scores were taken from a minimum of 15 mice per group and plotted against brain area±sem (b).
The lesion profiles, which define areas of vacuolation and their degree of severity in the brain, were determined for Bov6 mice inoculated with all three of BSE agents (Fig. 1b). Although, the production of lesion profiles in preclinical mice is not standard practice, all mice challenged with BASE and BSE-H that scored positively for vacuolation pathology were included to give an indication of vacuolation profile in comparison to BSE-C. While Bov6 mice inoculated with BSE-C and BSE-H had a similar lesion score in the cerebellum, superior colliculus and hypothalamus, overall lesion profiles of BSE-C, BASE and BSE-H were clearly distinct, indicating three different strains. In general, the degree of vacuolation present was not obviously greater in any individual BSE agent.
Neuropathological characterization of BSE-C, BASE and BSE-H in Bov6 mice
Pattern of PrP deposition.
Widespread PrP deposition, detected by using immunohistochemistry, was observed in Bov6 mice challenged with all three BSE agents. However, Bov6 mice challenged with BSE-C clearly showed heavier PrP deposition at earlier time points, which was also more widespread throughout the brain as compared with BASE or BSE-H (Table 2, Fig. 2a–c). Over time, PrP deposition continued to be more pronounced in BSE-C than BASE or BSE-H (Table 2, Fig. 2d–f). Fine punctate and coarse staining was observed in Bov6 mice challenged with BSE-C and was most abundant in the corpus callosum, thalamus and hippocampal region (Table 2). In contrast, Bov6 mice challenged with BASE showed plaque-like PrP deposits and milder staining. Interestingly, punctate PrP deposits were observed in the ventral tegmental area of the brainstem in a number of these mice, which was not the case for BSE-C or BSE-H. Overall Bov6 mice inoculated with BSE-H showed small amounts of PrP immunopositivity and similarly to BASE, PrP deposition was plaque-like and was represented mostly in the hippocampus and thalamus (Fig. S1, available in JGV Online). Thioflavin-S fluorescence confirmed the presence of PrP amyloid plaques in BASE- and BSE-H-challenged Bov6 mice (Fig. 2g–i). In BASE-challenged Bov6 mice, PrP amyloid plaques were observed in several regions of the brain. No thioflavin-S fluorescent PrP amyloid plaques were observed in Bov6 mice challenged with BSE-C (Fig. 2g).
Table 2. Onset and severity of PrP deposition in Bov6 mice challenged with BASE, BSE-C and BSE-H.
| TSE isolate | PrP score* | ||||||
| Caudate | Septum | Corpus callosum | Hippocampus | Thalamus | Midbrain | Brainstem | |
| <400 days p.i. | |||||||
| BASE | − | − | − | + | + | +/++ | + |
| BSE-C | ++ | + | +++ | ++ | ++ | +/++ | +/++ |
| BSE-H† | − | − | − | + | −/+ | −/+ | − |
| >400 days p.i. | |||||||
| BASE | + | + | ++ | ++ | ++ | +/++ | +/++ |
| BSE-C | +++ | ++/+++ | +++ | ++ | +++ | ++/+++ | ++/+++ |
| BSE-H | − | + | + | +/++ | +/++ | + | + |
PrP deposition scored as the following: −, no deposition; +, low deposition; ++, moderate deposition; +++, heavy deposition. Results were based on mean of four mice per group.
Bov6 mice from 476 days p.i.
Fig. 2.

Comparative analysis of variation in intensity of PrPTSE and presence of amyloid plaques in the hippocampus and thalamus regions of brains from Bov6 mice challenged with BSE-C, BASE and BSE-H. BSE-C: 378 days p.i. (a), 604 days p.i. (d); BASE: 406 days p.i. (b), 645 days p.i. (e); BSE-H: 476 days p.i. (c), 637 days p.i. (f). Images obtained after staining with anti-PrP antibody 6H4 and counterstained with haematoxylin. Magnification, ×4. Thioflavin-S fluorescent amyloid plaques (arrows) are clearly visible in the hippocampal region of Bov6 mice challenged with BASE (h) and BSE-H (i), but were not observed with BSE-C (g). Magnification, ×4.
Glial neuropathology.
Mild gliosis was present throughout the brains of aged Bov6 control mice (Fig. 3k and l). In contrast to gliosis due to ageing, an obvious increase in the appearance of astrogliosis and microgliosis was clearly evident throughout the brains of Bov6 mice inoculated with all three BSE agents and was most apparent in the hippocampus (Fig. 3). Furthermore, areas of PrP deposition in the hippocampus clearly correlated with microgliosis in Bov6 mice challenged with all three BSE agents (Fig. 3).
Fig. 3.

Comparative analysis of the CA1 region of the hippocampus of Bov6 mice challenged with BSE-C (604 days p.i.), BASE (645 days p.i.) and BSE-H (637 days p.i.). PrP deposition is visible by anti-6H4 antibody (a–c). Astro- and microgliosis is present in all mice, detected by anti-GFAP (e–g) and anti-Iba1 (i–k), respectively. Uninfected aged matched Bov6 mice showing mild gliosis were used as controls (d), (h) and (l). Magnification, ×10.
Biochemical characterization of PrPTSE in Bov6 mice inoculated with BSE-C, BASE and BSE-H
Molecular profile of BSE agents in Bov6 mice.
Brains from Bov6 mice challenged with BSE-C, BASE or BSE-H (two brains per isolate) were examined for the presence of PrPTSE by Western blotting. The brains selected for analysis were from old mice (581–610 days p.i.) and immunohistochemical analysis showed moderate to heavy PrP deposition in all selected animals. Following Western blot analysis, PK-resistant PrPTSE was present in the brains of Bov6 mice challenged with BSE-C, BASE and BSE-H. Furthermore, these agents produced distinct PrPTSE profiles (Fig. 4 and Fig. S2). We found that the BSE-H PrPTSE unglycosylated isoform had a higher molecular mass than BSE-C or BASE, and a lower molecular unglycosylated isoform was observed for BASE PrPTSE. These results are consistent with studies using transgenic overexpressing bovine PrP mice (Béringue et al., 2006, 2007; Buschmann et al., 2006; Capobianco et al., 2007; Okada et al., 2011a). Ratios of all three glycoforms (un-, mono- and diglycosylated), calculated by a Kodak 440CF digital imager, were similar between BSE-C PrPTSE and BSE-H PrPTSE; however, BASE PrPTSE had a reduced percentage of diglycosylated PrPSc as compared with BSE-C and BSE-H (data not shown). Similar differences in glycoform pattern between BSE-C and BASE have also been documented previously (Béringue et al., 2007; Capobianco et al., 2007).
Fig. 4.
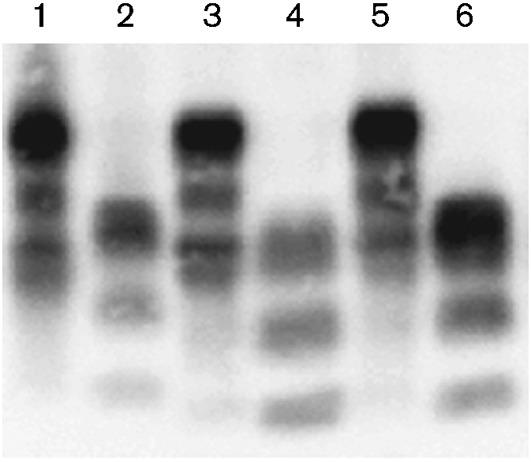
PK-resistant fragment (PrPTSE) of the prion protein in Bov6 mice challenged with BSE-H, 581 days p.i. (lanes 1 and 2), BASE, 610 days p.i. (lanes 3 and 4) and BSE-C, 604 days p.i. (lanes 5 and 6). Lanes 1, 3 and 5 represent untreated brain homogenate and lanes 2, 4 and 6 show PK-treated brain homogenate. Anti-PrP mAb 6H4 was used to detect bands.
Peripheral accumulation of PrPTSE in spleen.
Similarly to classical BSE, others have found an absence of PrPTSE in the lymphoid tissues of cattle infected with BASE and H-type BSE (Balkema-Buschmann et al., 2011b; Okada et al., 2011b; Iwamaru et al., 2010). However, studies have shown differences in lymphoid involvement in various mouse models challenged with classical and atypical BSE (Baron et al., 2011; Kong et al., 2008; Torres et al., 2011). Due to the different lymphoid involvement of classical and atypical BSE found in these mouse models, analysis of PrP accumulation in the spleen was performed on mice inoculated with each agent. To identify the presence of aggregated PrP in Bov6 mice challenged with BSE-C, BASE or BSE-H, spleen tissue was analysed by using the HerdChek* Bovine BSE Antigen Test kit (IDEXX), which is an antigen capture enzyme immunoassay (EIA) and utilizes a unique Seprion ligand capture technology (Microsens Biotechnologies). This assay permits the identification of PrPTSE without the requirement for digestion with PK. Two to three spleens derived from mice over 500 days p.i. with vacuolar pathology and moderate to heavy PrP deposition in the brain were analysed from each group (BASE, BSE-C and BSE-H). Positive assay readouts were obtained in spleens from all BSE agents (data not shown). Our findings indicate that these agents are lymphotrophic when passed into Bov6 mice.
Discussion
In this study, we examined the transmission characteristics, neuropathology and biochemical characteristics of BASE, BSE-C and BSE-H within gene-targeted Tg mice expressing bovine PrP. While previous studies have conducted similar experiments using transgenic mice overexpressing bovine PrP (Béringue et al., 2006, 2007; Buschmann et al., 2006; Capobianco et al., 2007; Nicot & Baron, 2011), this is the first study in which gene-targeted bovine Tg mice have been used to model BASE, BSE-C and BSE-H transmission and neuropathology. As these transgenic mice are produced by gene replacement, they do not suffer from any adverse phenotypes that are associated with overexpression or ectopic expression of the transgene in standard transgenic lines, and may more closely represent what happens in nature. Furthermore, previous studies have shown efficient transmission of TSE agents into gene-targeted transgenic PrP mouse lines, demonstrating that these mice do live long enough to show signs of infection, supporting the use of targeted mouse models to analyse TSE disease transmission (Bishop et al., 2006, 2010; Plinston et al., 2011). In the study described here, we found that all three BSE agents transmitted to Bov6 mice, confirming results found previously by using overexpressing bovine PrP transgenic mouse lines. However, an important difference in our study to those previously published was that, despite obvious neurodegenerative pathology, less than 10 % of Bov6 mice challenged with either BASE or BSE-H displayed any clinical signs of TSE disease, compared with 65 % of Bov6 mice inoculated with BSE-C. This was surprising, as all previous BASE inoculations using mice overexpressing bovine PrP showed clinical signs of disease, with shorter incubation times and increased levels of vacuolar pathology compared with BSE-C inoculated mice (Béringue et al., 2007; Buschmann et al., 2006; Capobianco et al., 2007; Nicot & Baron, 2011). Indeed, studies have shown that cattle inoculated intracranially with BASE have longer incubation times than those challenged with classical BSE (Balkema-Buschmann et al., 2011a; Dawson et al., 1990). In keeping with previous studies, we inoculated 20 µl of a 10 % brain homogenate for both BSE-C and BASE that were prepared from a brainstem pool and frontal/parietal cortex pool, respectively, which are known to contain high levels of infectivity for each strain. Indeed, BASE-infected tissue from the same case as was used in these studies has previously been used (Béringue et al., 2007, 2008; Biacabe et al., 2004; Casalone et al., 2004; Kong et al., 2008), and shown to produce shorter incubations times than BSE-C in overexpressing bovine Tg mice (Béringue et al., 2006, 2007; Buschmann et al., 2006; Capobianco et al., 2007; Okada et al., 2011a). Our previous studies (C. Plinston, personal communication) whereby groups of Bov6 mice were challenged with several isolates of natural scrapie, which did not induce disease, show an average lifespan of Bov6 mice is consistent with the average survival time in the mice inoculated with BASE and BSE-H. Hence, despite the presence of TSE pathology in the majority of these mice, they were still able to reach normal lifespan. Our findings were thus in accordance with studies in cattle, showing that cattle infected with atypical BSE were found to be healthy at slaughter (Biacabe et al., 2004; Casalone et al., 2004).
In addition to survival time and vacuolar degeneration, we also observed other findings which were not consistent with similar experiments in overexpressing bovine PrP mice. Using the IDEXX kit, PrPTSE was detected in spleen tissues in Bov6 mice challenged with all three BSE agents. Interestingly, other studies have shown the absence of PrPTSE in spleens derived from overexpressing bovine PrP transgenic mice challenged with BSE-H (Torres et al., 2011). Thioflavin-S fluorescent PrP deposits were observed in BASE- and BSE-H-challenged Bov6 mice; however, they were not detected with BSE-C, which is consistent with studies showing amyloid plaques in both BASE- and BSE-H-infected cattle brain (Casalone et al., 2004; Okada et al., 2011b) as opposed to classical cattle BSE, which is not associated with amyloid plaque formation. However, the opposite was found in other studies using overexpressing bovine PrP transgenic mice in which amyloid plaques were present in brains of BSE-C challenged mice, but not for BASE (Capobianco et al., 2007). Interestingly, transgenic mice overexpressing bovine PrP challenged with BASE that lack amyloid plaques display clinical signs of disease (Capobianco et al., 2007; Okada et al., 2011a). It is possible that BASE challenged Bov6 mice do not display clinical signs of disease due to the amyloid plaque pathology playing a protective role in these diseases. It has been proposed that amyloid plaques can sequester the pathogenic agent and prevent the mechanisms that lead to the development of clinical disease. Furthermore, other studies have shown that the seeding of amyloid plaques can occur in the absence of disease transmission (Piccardo et al., 2007). Interestingly, PrP deposition is heavier and has an earlier onset in BSE-C-challenged mice compared with BASE. Strikingly, significantly less PrP deposition was detected in BSE-H-challenged mice.
As part of this study we also investigated the susceptibility of wild-type mice (129/Ola) of the same genetic background as Bov6 mice to BASE, BSE-C and BSE-H. All of the mice challenged with BSE-C developed signs of disease, unlike mice challenged with BASE or BSE-H. Indeed, others have shown that while BSE-C transmits very efficiently to a variety of inbred mouse strains (SJL, C57B1/6), BASE does not (Baron et al., 2011; Capobianco et al., 2007). Our results may indicate a substantial transmission barrier between both BSE-H and BASE to wild-type mice, and this also supports the theory that BASE and BSE-H agents are distinct from BSE-C. Interestingly, other studies have shown that BSE-H readily transmits to wild-type mice (C57B1/6) and following serial passages the emergence of BSE-C strain properties is observed (Baron et al., 2011). In these transmissions, PrPTSE only accumulates as amyloid plaques in C57Bl/6 mice (Baron et al., 2006, 2011). BASE has shown no disease pathology on primary transmission in wild-type mice, but it too can produce BSE-C-like properties on subpassage (Capobianco et al., 2007). The lack of transmission of BASE and BSE-H in our study may be due to differences in inbred strains and subpassages are ongoing to determine any subclinical disease in the 129/Ola mice.
Our data confirm previous reports on transmissibility of different bovine prion isolates to transgenic mice expressing bovine PrP and support the conclusions that these isolates are distinct strains. Ongoing subpassage experiments will further analyse the relationship between BASE and BSE-H with BSE-C, and determine whether the isolates breed true or BSE-C-like phenotypes are observed. However, important contrasting differences between disease transmission of BASE in Bov6 mice compared to other transgenic lines overexpressing bovine PrP were identified. The reasons why BASE does not produce a shorter survival period in our mice and presents less neuropathology than BSE-C, which is contrary to results obtained in overexpression models, are unknown. However, our results would fit more closely with the disease profile of both classical and atypical BSE found in cattle. Interestingly, other studies investigating transmission of atypical scrapie have shown that the risk of transmission is lower for atypical scrapie than classical scrapie and this finding is closely linked to the expression level of the prion protein (Arsac et al., 2009). Transgenic mice overexpressing bovine PrP have been widely used to model bovine prion disease susceptibility in mice, and indeed the high levels of PrP expression produce sensitive assays in mice with short survival times. However, vast overexpression of PrP does not occur in nature and it is unknown if high levels of PrP alter the normal course of disease in these mice. Thus, we show here that mice with wild-type levels of bovine PrP expression live long enough to succumb to disease and may provide a more accurate representation of what happens in nature. Thus, combining data obtained from both overexpressing and gene-targeted transgenic mice may better aid our understanding on the transmissibility and neuropathology of these BSE isolates, and provide more accurate information allowing better management of these diseases.
Methods
Preparation of inocula.
Brain tissue from the frontal and parietal cortices of a cow infected with BASE was supplied by Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle D’Aosta, Torino, Italy. The brainstem of a BSE-H-infected cow was supplied by the French TSE Reference Laboratory [Agence Française de Sécurité Sanitaire des Ailments (AFSSA), Lyon, France]. The cattle BSE brainstem pool used in comparative experiments was supplied by The Animal Health Veterinary Laboratories Agency (AHVLA), Weybridge, UK [infectivity titre, 103.3 50 % infective doses (ID50)U g−1 tissue, measured in RIII mice; M. Simmons and R. Lockey, personal communication]. All inocula were prepared from brain tissue in sterile saline at a concentration of 10 % (w/v). Full pathological characterization of source tissues (BASE, C. Casalone; BSE-H, T. Baron; BSE-C, VLA) was previously performed to confirm disease status.
Inoculation of transgenic mice with classical and atypical BSE.
Gene-targeted Bov6 transgenic mice have been described previously (Bishop et al., 2006). Wild-type 129/Ola mice were used as controls (Bishop et al., 2006). Transgenic mice were grouped (24 mice each) prior to intracerebral inoculation and then injected with 0.02 ml 10−1 brain homogenate (BASE or BSE-H) into the right cerebral hemisphere under halothane anaesthesia. All inocula were prepared by maceration of the brain tissue in sterile saline to a dilution of 10−1. From 100 days mice were scored each week for signs of disease. Mice were killed by cervical dislocation at a pre-defined clinical end-point or due to welfare reasons (Dickinson et al., 1968). Due to the low number of culls for clinical TSE disease, survival times were calculated for mice showing both PrP deposition and vacuolar pathology. Brains and spleens were recovered at post-mortem. The spleen and half the brain were snap-frozen in liquid nitrogen for biochemical analysis and the remaining half brain was fixed for histological processing. Data shown (for comparison) from the inoculation of Bov6 and 129/Ola wild-type mice with a cattle BSE brainstem pool (provided by AHVLA) was generated and published previously (Bishop et al., 2006; Plinston et al., 2011). Data produced in these studies were recalculated to produce survival times as detailed above for comparison with BASE and BSE-H data. All mouse experiments were reviewed and approved by the Local Ethical Review Committee and performed under licence from the UK Home Office in accordance with the UK Animals (Scientific Procedures) Act 1986.
Vacuolation scoring.
Sections were cut (6 µm) from each mouse brain and stained using haematoxylin and eosin. TSE-related vacuolation was assessed at nine grey-matter regions (medulla, cerebellum, superior colliculus, hypothalamus, thalamus, hippocampus, septum, retrospinal cortex, cingulated and motor cortex) and three regions of white matter (cerebellar white matter, midbrain white matter and cerebral peduncle). Sections were scored on a scale of 0 (no vacuolation) to 5 (severe vacuolation) for the presence and severity of vacuolation and mean vacuolation scores for each mouse group in each experiment were calculated and plotted with sem against scoring areas to produce a lesion profile, as described previously (Bruce et al., 1997; Fraser & Dickinson, 1967). While, the production of lesion profiles in preclinical mice is not standard practice, all mice which scored positively for vacuolation pathology were included whether or not clinical signs were present, due to very few mice challenged with BASE and BSE-H displaying clinical signs of disease.
Immunohistochemical analysis of PrP deposition and glial activation in the brain.
PrPTSE localization in the brain was assessed by using immunohistochemistry. Following fixation in 10 % formal saline, brains were treated for 1.5 h in 98 % formic acid, dissected to expose several brain regions and embedded in paraffin. Sections (6 µm) were then autoclaved for 15 min at 121 °C and immersed in 95 % formic acid for 10 min prior to incubation with 0.44 µg anti-PrP mAb 6H4 (Prionics) ml−1 at room temperature overnight. Secondary anti-mouse biotinylated antibody (Jackson Immuno Research Laboratories) was added at 2.5 µg ml−1 and incubated for 1 h at room temperature. Immunolabelling was performed using the ABC Elite kit (Vector Laboratories) and the signal was visualized by a reaction with hydrogen peroxidize-activated diaminobenzidine (DAB). The presence of astrogliosis, a hallmark of prion disease, was assessed by incubating brain sections (6 µm) with 1.45 µg anti-glial fibrillary acidic protein (GFAP; Dako) ml−1 antibody at room temperature for 1 h. To detect microglial activation, brain sections were pretreated by using hydrated microwaving for 10 min prior to incubation with 0.05 µg anti-Iba1 antibody (Wako Chemicals) ml−1 at room temperature for 1 h. For both GFAP and anti-Iba-1 antibodies, 2.6 µg biotinylated secondary anti-rabbit antibody (Jackson Immno Research Laboratories) ml−1 was added for 1 h at room temperature. Both astrocytes and microglia were visualized by a reaction with hydrogen peroxidize-activated DAB.
Detection of amyloid plaques by thioflavin fluorescence.
Amyloid deposits in tissue sections were observed using Thioflavin-S. Briefly, following haematoxylin staining, sections (6 µm) were immersed in 1 % Thioflavin-S (Sigma) solution as described previously (Piccardo et al., 1998). Sections were mounted and viewed under a fluorescence microscope.
Immunoassay for detection of spleen PrPTSE.
The HerdChek* BSE Antigen Test kit (IDEXX) is an antigen capture EIA for the detection of PrPTSE in post-mortem tissues. Spleens from mice inoculated with all three BSE agents were homogenized in sterile saline in a Rybolyser (Hybaid) to achieve a 30 % homogenate. Protocol was performed following manufacturer’s instructions.
Identification of PrPTSE by immunoblotting.
Frozen brain samples from Bov6 mice challenged with BSE-C, BASE and BSE-H were homogenized at 10 % in an NP-40 buffer [0.5 % (v/v) NP-40, 0.5 % (w/v) sodium deoxycholate, 0.9 % (w/v) sodium chloride, 50 mM Tris/HCl pH 7.5] and clarified at 11 000 g for 15 min. Brain homogenate supernatant from the transgenic mice and controls was incubated with or without 20 µg proteinase K ml−1 for 1 h at 37 °C. The products were denatured and separated on a 12 % Novex Tris/glycine gel (Invitrogen) before transfer to PVDF membrane by Western blotting. The amount of brain tissue loaded onto the gels varied between 0.6 and 3 mg. PrP was identified with mAb 6H4 (0.1 µg ml−1) and bands visualized by using HRP-labelled anti-mouse secondary antibody (Jackson Immuno Research Laboratories) and a chemiluminescence substrate (Roche). Images were captured on radiographic film and with a Kodak 440CF digital imager.
PCR genotyping of mouse tail DNA.
All mice were analysed by PCR post-mortem to confirm PrP genotype. Mouse tail DNA was extracted and genotyped as described previously (Bishop et al., 2006).
Acknowledgements
The authors would like to acknowledge Professor J. Manson for the gene-targeted bovine Tg line; I. McConnell, V. Thomson, S. Cumming, S. Carpenter, R. Greenan and K. Hogan for experimental setup, care and scoring of the animals; A. Coghill, A. Boyle, S. Mack and G. McGregor for histology processing and scoring; and the Veterinary Laboratories Agency, Weybridge, UK, for providing the cattle BSE brain pool. These studies were partly funded by the Food Standards Agency (FSA) UK (contract M03054) and NIH-NIAID agreement no.Y1-AI-4893-02 and Food and Drug Administration (FDA) agreement no. 224-05-1307. The findings and conclusions in this article have not been formally disseminated by the FDA and should not be construed to represent any Agency determination or policy.
Footnotes
Supplementary figures are available with the online version of this paper.
References
- Arsac J. N., Bétemps D., Morignat E., Féraudet C., Bencsik A., Aubert D., Grassi J., Baron T. (2009). Transmissibility of atypical scrapie in ovine transgenic mice: major effects of host prion protein expression and donor prion genotype. PLoS ONE 4, e7300 10.1371/journal.pone.0007300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkema-Buschmann A., Ziegler U., McIntyre L., Keller M., Hoffmann C., Rogers R., Hills B., Groschup M. H. (2011a). Experimental challenge of cattle with German atypical bovine spongiform encephalopathy (BSE) isolates. J Toxicol Environ Health A 74, 103–109 10.1080/15287394.2011.529060 [DOI] [PubMed] [Google Scholar]
- Balkema-Buschmann A., Fast C., Kaatz M., Eiden M., Ziegler U., McIntyre L., Keller M., Hills B., Groschup M. H. (2011b). Pathogenesis of classical and atypical BSE in cattle. Prev Vet Med 102, 112–117 10.1016/j.prevetmed.2011.04.006 [DOI] [PubMed] [Google Scholar]
- Baron T. G., Biacabe A. G., Bencsik A., Langeveld J. P. (2006). Transmission of new bovine prion to mice. Emerg Infect Dis 12, 1125–1128 10.3201/eid1207.060107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron T., Vulin J., Biacabe A.-G., Lakhdar L., Verchere J., Torres J.-M., Bencsik A. (2011). Emergence of classical BSE strain properties during serial passages of H-BSE in wild-type mice. PLoS ONE 6, e15839 10.1371/journal.pone.0015839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béringue V., Bencsik A., Le Dur A., Reine F., Laï T. L., Chenais N., Tilly G., Biacabé A.-G., Baron T. & other authors (2006). Isolation from cattle of a prion strain distinct from that causing bovine spongiform encephalopathy. PLoS Pathog 2, e112 10.1371/journal.ppat.0020112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béringue V., Andréoletti O., Le Dur A., Essalmani R., Vilotte J. L., Lacroux C., Reine F., Herzog L., Biacabé A. G. & other authors (2007). A bovine prion acquires an epidemic bovine spongiform encephalopathy strain-like phenotype on interspecies transmission. J Neurosci 27, 6965–6971 10.1523/JNEUROSCI.0693-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béringue V., Herzog L., Reine F., Le Dur A., Casalone C., Vilotte J. L., Laude H. (2008). Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis 14, 1898–1901 10.3201/eid1412.080941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacabe A. G., Laplanche J. L., Ryder S., Baron T. (2004). Distinct molecular phenotypes in bovine prion diseases. EMBO Rep 5, 110–115 10.1038/sj.embor.7400054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop M. T., Hart P., Aitchison L., Baybutt H. N., Plinston C., Thomson V., Tuzi N. L., Head M. W., Ironside J. W., Will R. G. (2006). Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol 5, 393–398 10.1016/S1474-4422(06)70413-6 [DOI] [PubMed] [Google Scholar]
- Bishop M. T., Will R. G., Manson J. C. (2010). Defining sporadic Creutzfeldt–Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107, 12005–12010 10.1073/pnas.1004688107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce M. E. (1996). Strain Typing Studies of Scrapie and BSE, vol. 3, pp. 223–236 Springer [Google Scholar]
- Bruce M. E., Will R. G., Ironside J. W., McConnell I., Drummond D., Suttie A., McCardle L., Chree A., Hope J. & other authors (1997). Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389, 498–501 10.1038/39057 [DOI] [PubMed] [Google Scholar]
- Buschmann A., Gretzschel A., Biacabe A. G., Schiebel K., Corona C., Hoffmann C., Eiden M., Baron T., Casalone C., Groschup M. H. (2006). Atypical BSE in Germany – proof of transmissibility and biochemical characterization. Vet Microbiol 117, 103–116 10.1016/j.vetmic.2006.06.016 [DOI] [PubMed] [Google Scholar]
- Capobianco R., Casalone C., Suardi S., Mangieri M., Miccolo C., Limido L., Catania M., Rossi G., Di Fede G. & other authors (2007). Conversion of the BASE prion strain into the BSE strain: the origin of BSE? PLoS Pathog 3, e31 10.1371/journal.ppat.0030031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalone C., Zanusso G., Acutis P., Ferrari S., Capucci L., Tagliavini F., Monaco S., Caramelli M. (2004). Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt–Jakob disease. Proc Natl Acad Sci U S A 101, 3065–3070 10.1073/pnas.0305777101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J., Sidle K. C., Meads J., Ironside J., Hill A. F. (1996). Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 383, 685–690 10.1038/383685a0 [DOI] [PubMed] [Google Scholar]
- Dawson M., Wells G. A., Parker B. N. (1990). Preliminary evidence of the experimental transmissibility of bovine spongiform encephalopathy to cattle. Vet Rec 126, 112–113 [PubMed] [Google Scholar]
- Dickinson A. G., Meikle V. M., Fraser H. (1968). Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol 78, 293–299 10.1016/0021-9975(68)90005-4 [DOI] [PubMed] [Google Scholar]
- Dudas S., Yang J., Graham C., Czub M., McAllister T. A., Coulthart M. B., Czub S. (2010). Molecular, biochemical and genetic characteristics of BSE in Canada. PLoS ONE 5, e10638 10.1371/journal.pone.0010638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser H., Dickinson A. G. (1967). Distribution of experimentally induced scrapie lesions in the brain. Nature 216, 1310–1311 10.1038/2161310a0 [DOI] [PubMed] [Google Scholar]
- Hagiwara K., Yamakawa Y., Sato Y., Nakamura Y., Tobiume M., Shinagawa M., Sata T. (2007). Accumulation of mono-glycosylated form-rich, plaque-forming PrPSc in the second atypical bovine spongiform encephalopathy case in Japan. Jpn J Infect Dis 60, 305–308 [PubMed] [Google Scholar]
- Hill A. F., Desbruslais M., Joiner S., Sidle K. C. L., Gowland I., Collinge J., Doey L. J., Lantos P. (1997). The same prion strain causes vCJD and BSE. Nature 389, 448–450, 526 10.1038/38925 [DOI] [PubMed] [Google Scholar]
- Iwamaru Y., Imamura M., Matsuura Y., Masujin K., Shimizu Y., Shu Y., Kurachi M., Kasai K., Murayama Y. & other authors (2010). Accumulation of L-type bovine prions in peripheral nerve tissues. Emerg Infect Dis 16, 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J. G., Langeveld J. P., Biacabe A. G., Acutis P. L., Polak M. P., Gavier-Widen D., Buschmann A., Caramelli M., Casalone C. & other authors (2007). Molecular discrimination of atypical bovine spongiform encephalopathy strains from a geographical region spanning a wide area in Europe. J Clin Microbiol 45, 1821–1829 10.1128/JCM.00160-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q., Zheng M., Casalone C., Qing L., Huang S., Chakraborty B., Wang P., Chen F., Cali I. & other authors (2008). Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 82, 3697–3701 10.1128/JVI.02561-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczius T., Groschup M. H. (1999). Differences in proteinase K resistance and neuronal deposition of abnormal prion proteins characterize bovine spongiform encephalopathy (BSE) and scrapie strains. Mol Med 5, 406–418 [PMC free article] [PubMed] [Google Scholar]
- Kuczius T., Haist I., Groschup M. H. (1998). Molecular analysis of bovine spongiform encephalopathy and scrapie strain variation. J Infect Dis 178, 693–699 10.1086/515337 [DOI] [PubMed] [Google Scholar]
- Manolakou K., Beaton J., McConnell I., Farquar C., Manson J., Hastie N. D., Bruce M., Jackson I. J. (2001). Genetic and environmental factors modify bovine spongiform encephalopathy incubation period in mice. Proc Natl Acad Sci U S A 98, 7402–7407 10.1073/pnas.121172098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot S., Baron T. (2011). Strain-specific barriers against bovine prions in hamsters. J Virol 85, 1906–1908 10.1128/JVI.01872-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H., Masujin K., Imamaru Y., Imamura M., Matsuura Y., Mohri S., Czub S., Yokoyama T. (2011a). Experimental transmission of H-type bovine spongiform encephalopathy to bovinized transgenic mice. Vet Pathol 48, 942–947 [DOI] [PubMed] [Google Scholar]
- Okada H., Iwamaru Y., Imamura M., Masujin K., Matsuura Y., Shimizu Y., Kasai K., Mohri S., Yokoyama T., Czub S. (2011b). Experimental H-type bovine spongiform encephalopathy characterized by plaques and glial- and stellate-type prion protein deposits. Vet Res 42, 79 10.1186/1297-9716-42-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccardo P., Dlouhy S. R., Lievens P. M., Young K., Bird T. D., Nochlin D., Dickson D. W., Vinters H. V., Zimmerman T. R. & other authors (1998). Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 57, 979–988 10.1097/00005072-199810000-00010 [DOI] [PubMed] [Google Scholar]
- Piccardo P., Manson J. C., King D., Ghetti B., Barron R. M. (2007). Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A 104, 4712–4717 10.1073/pnas.0609241104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plinston C., Hart P., Chong A., Hunter N., Foster J., Piccardo P., Manson J. C., Barron R. M. (2011). Increased susceptibility of human-PrP transgenic mice to bovine spongiform encephalopathy infection following passage in sheep. J Virol 85, 1174–1181 10.1128/JVI.01578-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richt J. A., Kunkle R. A., Alt D., Nicholson E. M., Hamir A. N., Czub S., Kluge J., Davis A. J., Hall S. M. (2007). Identification and characterization of two bovine spongiform encephalopathy cases diagnosed in the United States. J Vet Diagn Invest 19, 142–154 10.1177/104063870701900202 [DOI] [PubMed] [Google Scholar]
- Stack M. J., Focosi-Snyman R., Cawthraw S., Davis L., Chaplin M. J., Burke P. J. (2009). Third atypical BSE case in Great Britain with an H-type molecular profile. Vet Rec 165, 605–606 10.1136/vr.165.20.605-c [DOI] [PubMed] [Google Scholar]
- Stimson E., Hope J., Chong A., Burlingame A. L. (1999). Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 38, 4885–4895 10.1021/bi982330q [DOI] [PubMed] [Google Scholar]
- Torres J. M., Andréoletti O., Lacroux C., Prieto I., Lorenzo P., Larska M., Baron T., Espinosa J. C. (2011). Classical bovine spongiform encephalopathy by transmission of h-type prion in homologous prion protein context. Emerg Infect Dis 17, 1636–1644 10.3201/eid1709.101403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells G. A., Scott A. C., Johnson C. T., Gunning R. F., Hancock R. D., Jeffrey M., Dawson M., Bradley R. (1987). A novel progressive spongiform encephalopathy in cattle. Vet Rec 121, 419–420 10.1136/vr.121.18.419 [DOI] [PubMed] [Google Scholar]