

Abstract
Bacterial ParM is a homolog of eukaryotic actin and is involved in moving plasmids so that they segregate properly during cell division. Using cryo-EM and three-dimensional reconstruction, we show that ParM filaments have a different structure from F-actin, with very different subunit-subunit interfaces. These interfaces result in the helical handedness of the ParM filament being opposite to that of F-actin. Like F-actin, ParM filaments have a variable twist, and we show that this involves domain-domain rotations within the ParM subunit. The present results yield new insights into polymorphisms within F-actin, as well as the evolution of polymer families.
ParM is a bacterial actin homolog involved in plasmid segregation1,2. The structures of both ParM and MreB3, another bacterial actin homolog, as well as FtsZ4, a tubulin homolog, show that the eukaryotic cytoskeleton is similar to a prokaryotic cytoskeleton that has only recently been studied. Although the higher-order structure of FtsZ is still unknown, it has been assumed that both ParM and MreB assemble into filaments with an arrangement of subunits very similar to that found in F-actin. However, a crystal structure of monomeric ParM has unexpectedly shown that its main differences from actin are in regions expected to be involved in the filamentous subunit-subunit interface2. A previous study2 has used EM of negatively stained ParM filaments to generate a low-resolution three-dimensional reconstruction, which suggests that the ParM filament is very similar to F-actin.
We have used negative staining, cryo-EM and quick-freeze/deep-etch EM to examine filaments formed by the ParM protein from Escherichia coli. The question that we intended to answer was how similar to F-actin the ParM filaments appear at higher resolution. We show that the ParM filaments are substantially different from F-actin, with a very different subunit-subunit interface. Because of the very different interface, the helical handedness of the ParM filaments is opposite to that of F-actin.
RESULTS
Variable twist in ParM filaments
EM shows that ParM forms long filaments when incubated with the nonhydrolyzable ATP analog AMP-PNP (Fig. 1). We analyzed both negatively stained (Fig. 1a) and frozen-hydrated ParM filaments (Fig. 1b), but took advantage of the iterative helical real-space reconstruction (IHRSR) method5 to surmount the problems posed by flexible and disordered filaments. In this method, rather than individual filaments being treated as ideal helices with a uniform structure, short segments are analyzed, classified and reconstructed separately. One of the most obvious forms of disorder revealed by such a method is the presence of variable twist in the ParM filaments, even greater than what has been shown for F-actin6–8. The variability in average twist angles found in unstained frozen-hydrated ParM segments (28,886 segments, each segment ~480 Å long) is shown in Figure 2a. This variability arises from different segments within the same filaments, rather than different filaments having different twists. Strong support for the validity of this type of sorting comes from averaged power spectra generated from all of the segments in each bin. Because such power spectra do not require alignment of segments to references and do not require algorithms for averaging images or three-dimensional reconstruction, they are unbiased and model-independent. A comparison of power spectra from five different bins in Figure 2a shows that the segments behave exactly as one would expect for segments with great variability in twist, with very little variability in the axial rise per subunit (Supplementary Video 1 online).
Figure 1.

Electron micrographs of ParM. (a) Negatively stained ParM filaments. Scale bars, 1,000 Å. (b) Cryo-EM of unstained frozen-hydrated ParM filaments on a carbon substrate. (c) A power spectrum computed from 3,453 nonoverlapping cryo-EM segments of ParM filaments, where each segment was 480 Å in length. Four layer lines are labeled with their Bessel orders. The n = −2 line (arising from a left-handed two-start helix) is at ~1/(307 Å), the n = +3 line (arising from a right-handed three-start helix) is at ~1/(65 Å), the n +1 line (from a right-handed one-start helix) is at 1/(53.4 Å), and the n = −1 line (from a left-handed one-start helix) is at 1/(45.5 Å). (d) Quick-freeze/deep-etch electron micrograph of ParM filaments. Such images confirmed that the long-pitch helices repeating every ~300 Å are left-handed, not right-handed as in F-actin. As a control, F-actin filaments were imaged both separately from ParM and after preparation with ParM. (e,f) Power spectra computed from quick-freeze/deep-etch images of ParM (e) and F-actin (f) clearly show the reversal of helical handedness. Vertical lines in e,f are the meridians of the power spectra. White arrows indicate the reflections arising from the long-pitch helices, left-handed at B1/(300 Å) in ParM (e) and right-handed at ~1/(360 Å) in F-actin (f). Gray arrows indicate the reflections from the one-start helix, right-handed in ParM at ~1/(53 Å) (e) and left-handed in F-actin at B1/(59 Å) (f). (g,h) The quick-freeze/deep-etch images can be averaged, and this is shown for ParM (g) and F-actin (h). The left-handed long-pitch helix (repeating every ~300 Å) in ParM (g) and the left-handed short-pitch helix in F-actin (repeating every 59 Å) (h) are indicated with black lines.
Figure 2.

Variable twist, and overcoming it with the IHRSR approach. (a) Histogram of average twists for segments of frozen-hydrated ParM filaments. (b) The convergence of the IHRSR procedure is shown for cryo-EM segments of ParM classified as having a mean twist of ~162.5°. A solid featureless object (cylinder, left) was used as an initial reference. The IHRSR procedure was run twice, starting the search at either 162.0° (●) or 165.0° (■). After ~50 cycles, the two different sets converged to the same symmetry and the same reconstructed volume. An unsymmetrized volume from the seventieth cycle is shown as gray density at right. Each cycle, such a volume is analyzed to determine the symmetry within it, and this symmetry is imposed on the volume to generate a new symmetrized reference for the next cycle. The unsymmetrized volume shown has not been corrected for the CTF.
ParM filaments have handedness opposite to F-actin
EM reconstructions, whether from negatively stained samples or from cryo-EM of unstained, frozen-hydrated specimens, have enantiomorphic ambiguity. That is, a structure and the mirror image of a structure will both give rise to the same projection images. It has been assumed previously2 that ParM filaments have the same handedness as F-actin, with the long-pitch two-stranded helices being right-handed, but this was never directly determined. We used cryo-EM of ParM filaments (Fig. 1b) to avoid potential artifacts associated with staining and dehydration, as well as to achieve higher resolution than can be obtained by negative staining. Tilts of negatively stained ParM filaments in the electron micrographs revealed that the filaments were substantially flattened, in contrast to F-actin, which was only partially flattened (data not shown). We think that the different behavior of ParM and actin filaments during sample preparation reflects large structural differences. The relative strengths of different layer lines in the averaged power spectrum computed from ParM filaments in ice (Fig. 1c) directly shows that the structure of the ParM filament must be considerably different from that of F-actin. In any helical object, different families of helices that relate subunits to each other can be described by the pitch, the handedness (left or right) and the symmetry of the helix, which is given by the ‘start’ number— a two-start helix has two strands 180° apart, a three-start helix has three strands 120° apart, and so on. In F-actin, the strongest feature seen in such power spectra is the layer line at 1/(59 Å) arising from a left-handed one-start helix. However, in ParM power spectra, the strongest feature arises from a three-start helix at ~1/(65 Å), indicating that the arrangement of mass within the filament has substantial three-stranded character.
Single-particle helical reconstruction
The very low signal-to-noise ratio in ice images (Fig. 1b) makes traditional Fourier-Bessel reconstructions9 of weakly scattering filaments either problematic or impossible, but the IHRSR approach overcomes this problem. We can classify segments by their average twist (Fig. 2a) and then separately start reconstructions of these subsets using only a featureless solid cylinder as an initial reference model (Fig. 2b). The convergence of these reconstructions to a common symmetry and structure (Fig. 2b) shows that the sorting has been reliable, as any substantial heterogeneity results in lack of such convergence10.
As segments are sorted into smaller and smaller subsets, they become more homogeneous, but the number of segments in each class becomes smaller, which greatly limits resolution. Compromises are therefore needed to achieve homogeneous subsets. We generated more than 25 different cryo-EM reconstructions by choosing different twist states and ranges of twist to be grouped together. Owing to the variability in the ParM filaments, we have not been able to generate any reconstructions at better than 16-Å resolution. In all of the cryo-EM reconstructions, even more clearly than in the negative-stain reconstructions, the only reasonable fit of the crystal subunit into these volumes (Fig. 3a) was when the long-pitch strands were set as left-handed, the opposite to F-actin’s right-handedness. As the flattening of the ParM filaments by both negative staining and metal shadowing of air-dried specimens (data not shown) precluded determination of helical handedness, we confirmed the assignment of left-handedness (Fig. 1d–h) using quick-freeze/deep-etch EM11.
Figure 3.

Filaments of ParM have the opposite handedness to that of F-actin. (a–d) The cryo-EM reconstruction of ParM is shown as a transparent gray surface in a and d, and a model for the ParM subunit (ribbon representation) has been fit into this volume. The four subdomains of the ParM subunit are labeled in b; the strongest interface in the ParM filament is between subdomain IIa of one subunit and subdomain Ia of a subunit above it. c shows a close-up view of an alignment of ParM subdomain IIa (magenta) with actin subdomain 4 (green, with actin residues 204 and 247 in red). Actin residues 204 and 247 are expected to be part of the subunit-subunit interface in F-actin, as mutation of these residues results in loss of polymerization35. The overall structural conservation between actin and ParM does not extend to this region, consistent with different filamentous interfaces. (e) For comparison, a reconstruction of F-actin is shown. In F-actin (e), subunit 2 is rotated from subunit 0 by a right-handed rotation that is ~28° on average. In ParM (d), subunit 4 is rotated from subunit 2 by a left-handed rotation that is ~29° on average.
Although two different conformations of the ParM molecule have been crystallized previously2 (a ‘closed’ state with ADP bound, PDB 1MWM, and an ‘open’ state with no nucleotide, PDB 1MWK), neither structure fit well into the filament reconstruction as is. This was not completely unexpected, as ParM cannot be polymerized under either condition. All actin crystal structures have been obtained by either forming complexes of actin with proteins that prevent polymerization (such as DNase I, profilin or gelsolin) or modifying actin itself so that polymerization is prevented. In contrast, the unmodified wild-type ParM was crystallized2 under conditions (absence of nucleotide or presence of ADP) where ParM will not polymerize. Because the two ParM crystal structures are related almost entirely by domain-domain rotations, with only small conformational changes within each domain, we fit the two domains independently (domain I, residues 1–163 and 306–320; domain II, residues 164–305; Fig. 3). The best fit (Fig. 3a,d) arises from rotation of the domains even further apart than they are in the open state. This can be seen in Figure 4, where the two crystal structures of monomeric ParM are compared with the EM model of the protomer. The magnitude of the domain rotation between our model and that of PDB 1MWK is almost the same as the domain rotation between PDB 1MWM and PDB 1MWK.
Figure 4.
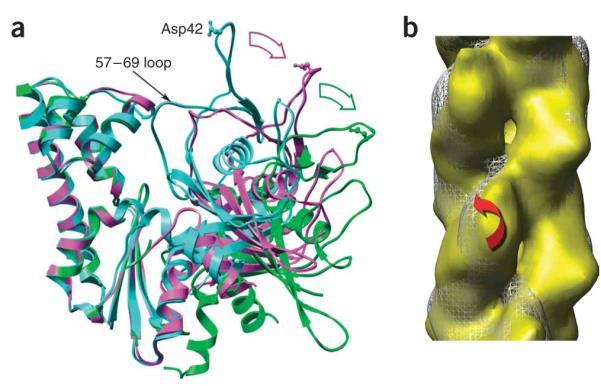
Domain rotations within ParM. (a) Crystal structures of ParM in the closed state (PDB 1MWM, cyan) and the open state (PDB 1MWK, magenta) are compared with the subunit model (green) used to fit the EM reconstruction (Fig. 3a,b). Major domain II has been aligned for all three structures. The 57–69 loop in domain I is one of the few elements that shifts between the crystal structures of the open and closed states. A single residue, Asp42, is shown in ball-and-stick representation to indicate the relative motions of domain I. The displacement of Asp42 from 1MWM to 1MWK is ~18 Å, which is about the same as the displacement from 1MWK to the EM model. (b) Reconstructions of ParM in two different twist states are compared. White mesh, twist of 165.2°; yellow surface, twist of 169.6°. Red arrow represents rotation of major domain I by ~10° between these two reconstructions while the orientation of major domain II remains relatively fixed.
ParM and F-actin have different subunit-subunit interfaces
As a control for reconstructing ParM filaments in ice, we used the same electron microscope, preparative conditions and image analysis procedures to reconstruct F-actin filaments. Starting with 30,766 image segments, and sorting by twist, we generated a reconstruction at ~12-Å resolution (using the Fourier shell correlation = 0.5 criterion) from a subset containing 4,369 segments. We were able to achieve this resolution by using conditions (phosphate buffer, 10 mM Mg2+ and no KCl) where the variability in F-actin’s twist is greatly reduced. Fitting an atomic model of the G-actin subunit into the F-actin reconstruction (Fig. 3e) shows that the nucleotide-binding cleft in the actin subunit is quite closed under these conditions, as seen previously at lower resolution12. We suggest (see below) that the variability in twist in both F-actin and ParM is related to the abilities of the two domains within these proteins to move with respect to each other.
The filament model that we have generated for ParM (Fig. 3d) shows subunit-subunit interfaces that are completely different from what is found in F-actin (Fig. 3e). In fact, the opposite helical handedness for ParM dictates that the interfaces must be different between ParM and F-actin. The strongest interface between subunits in both filaments is along the same long-pitch helical strand. In F-actin, a subunit is rotated on average approximately +28° with respect to a subunit below it on the same long-pitch helical strand. In ParM, this rotation is on average approximately −29°. As the difference between these two is a 57° rotation, there is no way that a similar interface can exist between subunits in ParM and F-actin. Whereas F-actin has substantial longitudinal contacts between subdomain 2 (corresponding to subdomain Ib in ParM) and the subunit above it along the same long-pitch helix12–14, in ParM no such contacts exist. The most substantial interface in the ParM filament involves subdomain IIb (corresponding to subdomain 4 in F-actin) with subdomain Ia of the subunit above it (Fig. 3b). The interface between the two strands is much more limited in ParM than in F-actin. The difference in subunit-subunit interfaces between actin and ParM is supported by experimental studies on actin. The hydrophobic loop in actin containing residues 262–274 has been shown to be essential for F-actin polymerization15, but this loop is completely missing from both ParM and MreB. A nonpolymerizing mutant of actin has been created by introducing two amino acid changes in subdomain 4 of actin (Fig. 3c), but the structure of the ParM subunit is completely different in this region and therefore could not form an interaction similar to that formed between two actin subunits. The very different subunit-subunit interface in ParM resolves a problem that has been raised previously2: why are the sites of the subunit-subunit interfaces in the actin protomer the regions where the ParM subunit differs the most from crystal structures of G-actin? This is no longer a contradiction, given that, contrary to what was assumed, the filament of ParM involves totally different contacts from those in F-actin. Thus, our results indicate that earlier questions16 about the difficulty of reconciling the very different polymerization properties of ParM and actin with a common filament structure were based upon wrong assumptions.
Domain-domain motions in the actin superfamily
The ParM filament structure adds evidence supporting the suggestion that all actin-like proteins undergo large domain-domain motions17. Although all actin crystal structures, with one exception18, have revealed a closed conformation, recent studies have suggested that crystallization may trap actin in a closed conformation that is independent of the bound nucleotide (ADP or ATP)19. This is similar to the conclusion reached about another actin-like protein, bacterial propionate kinase; it has been suggested that the crystal-packing interactions may have a larger effect on the conformation of this molecule than does the bound ligand20. The ATP analog AMP-PNP is bound to the ParM filaments that we examined, indicating that the nucleotide is bound to the wide-open conformation of the subunit. We determined that there is no appreciable exchange of nucleotide after ParM polymerization (Fig. 5), showing that the nucleotide must be tightly bound to subunits. ParM is no more similar to actin than it is to hexokinase, and a recent crystal structure has shown ADP bound to a wide-open conformation of an archaeal hexokinase21 that is very similar to the wide-open protomer that we observed in the ParM filament. ATP can also be bound to the wide-open archaeal hexokinase molecule (T. Wagaki, University of Tokyo, personal communication). Human hexokinase I with bound ADP has been crystallized22 in a conformation that is very similar to the open conformation of nucleotide-free ParM3, and hexokinase I also exists in a wide-open conformation23 assumed to bind ATP. It has been shown that nucleotide binds subdomains 3 and 4 of another actin structural homolog, Arp2, even though subdomains 1 and 2 are disordered in a crystal24. This suggests that interactions of the nucleotide with subdomains 3 and 4 of Arp2 provide much of the binding energy24, and the same is likely to be true for ParM.
Figure 5.
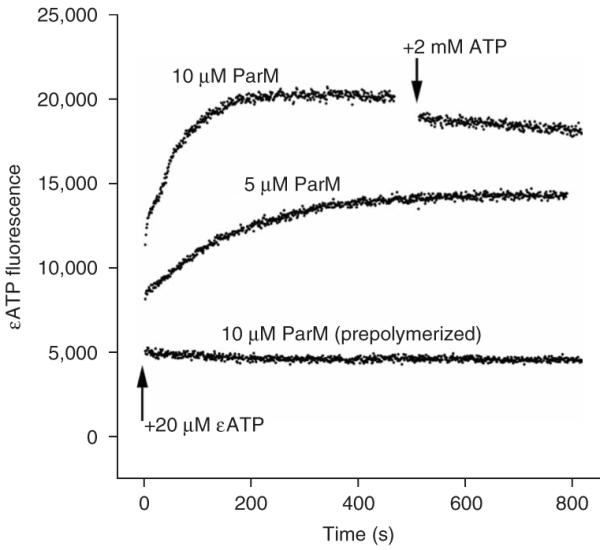
Absence of nucleotide exchange after ParM polymerization. Fluorescent etheno-ATP (εATP) was used to induce polymerization in the nonhydrolyzing ParM mutant E148A. Acrylamide was used as a collisional quencher to increase the signal from the bound nucleotide and to monitor protection of the nucleotide from interaction with solvent. Adding ParM (5 μM) to εATP caused a rapid jump in fluorescence, consistent with monomer binding, followed by a slower increase, whose time course is identical to that of polymerization. When prepolymerized ParM (10 μM) was added to εATP, there was no jump in fluorescence. When a 100-fold excess of unlabeled ATP was added to the polymerized ParM, only a very slow decrease in fluorescence (with a half-life on the order of 1 h) was seen that was not consistent with rapid nucleotide exchange on the filament. This time course is orders of magnitude slower than exchange on monomeric ParM, and the kinetics are consistent with turnover of subunits in the filament.
The opening of the two domains in ParM is comparable in extent to that seen in the tilted state of filamentous actin25, establishing that the hinge motions of the two domains are exploited similarly in these distantly related proteins. Although the cryo-EM reconstruction is from the wild-type ParM with the nonhydrolyzable ATP analog AMP-PNP, we have also done reconstructions from negatively stained images using the slowly hydrolyzable ATP analog ATP-γS and using ATP with the E148A mutant of ParM that does not hydrolyze ATP26. At a resolution of ~24 Å, there were no major differences among these negative-stain reconstructions. A large domain-domain rotation would be readily apparent at even this low resolution, suggesting that both AMP-PNP and ATP-γS are bound in the same state as we observed for ATP bound with the E148A mutant.
In contrast to ParM, in F-actin the subunit in the filament is in a closed state when ATP or ADP-Pi is bound (Fig. 3e)12. On the basis of the structures of actin, hexokinase and Arp2, we expect that the hydrolysis of ATP will occur in ParM filaments only when the nucleotide-binding cleft closes, bringing the catalytic residues in domain I of ParM into contact with the phosphates. We do not know the state of the ParM subunit within the filament after phosphate release, owing to the instability of such filaments26, but the present observations highlight another very large difference between F-actin and ParM.
The variability in twist within F-actin is now widely accepted6–8, but past observations have offered little insight into the molecular basis for this variability. Are subunits rigid bodies within F-actin, with a multiplicity of interfaces27, or is the variability in twist largely a result of internal conformational changes within the actin protomer? We can address this question in ParM by comparing two reconstructions that differ in their average twist by 4.4° (165.2° or 169.6°; Fig. 4b). Alignment of domain II of one subunit between the reconstructions reveals a rotation of domain I by ~10° (Supplementary Video 2 online). This shows that the variability in twist within ParM has a large component attributable to the domain-domain rotations within individual subunits, and it suggests that the same is likely to be true for F-actin, as conditions that lead to the closure of the nucleotide-binding cleft in actin reduce the observed variability in twist.
DISCUSSION
Actin, ParM, MreB, hexokinase, propionate kinase, acetate kinase28 and many other molecules with diverse functions share the same fold, suggesting a common evolutionary origin17. Some proteins within this family, such as ParM, MreB and actin, polymerize into filaments, whereas other members function as monomers, dimers and tetramers29. Crystal structures2,3 and phylogenetic analysis30 indicate that ParM is further removed from actin than is MreB. It is clear that the common fold does not dictate a common oligomeric organization, just as it is now clear that the helical polymers formed by actin and ParM can have quite different structures with different subunit-subunit contacts. This structural diversity raises a fundamental question. Did filaments evolve two (or perhaps three) separate times from different members of the actin superfamily? Or does the structural diversity reflect divergence from a common, polymerizing ancestor? The former case seems more likely, and our present results suggest that the different structural organization of F-actin and ParM may explain their different rates of divergence, above and beyond the greater rates expected for bacterial proteins in general. The sequence inserts that are present in actin, but absent in both MreB and ParM, seem to be key in forming subunit-subunit contacts within actin filaments and to allow for polymorphic switching of contacts25. Actin has been subject to an anomalously low rate of sequence divergence over eukaryotic evolution31, and it has been suggested that the polymorphic switching and allosteric relations within filaments may have placed a large constraint on sequence divergence25. We do not have structural information for many bacterial MreBs, but their primary sequences are not nearly as well conserved as those of eukaryotic actins. Further structural work will determine whether this sequence diversity corresponds to substantial diversity in filament structure.
METHODS
Sample preparation and electron microscopy
ParM, both the wild-type protein and the E148A mutant, were overexpressed and purified as described26. Wild-type ParM (5 μM) was polymerized by the addition of AMP-PNP (5 mM), and after 5–10 min incubation, 5 μl was applied to glow-discharged carbon-covered 300-mesh copper grids. The E148A mutant was incubated with ATP. The grids were either negatively stained with 1% (w/v) uranyl acetate or blotted and plunged into liquid ethane. Images were collected on film using a Tecnai 12 (80 kV and ×30,000 magnification) for negatively stained samples or a Tecnai 20 FEG (200 kV and ×50,000 magnification) for frozen-hydrated samples. The 31 different images used for the cryo-EM reconstruction had defocus values ranging from 2.0 to 4.0 μm. Films were scanned on a Nikon Coolscan 8000 with a sampling of 4.2 Å per pixel for negative-stain images and 2.4 Å per pixel for cryo-EM.
Image analysis
The SPIDER package32 was used for most image processing, but the EMAN package33 was used to determine the defocus values in the micrographs and to extract filament images from micrographs. The power spectrum in Figure 1e was computed from 160 nonoverlapping ParM segments (each 980 Å in length), whereas the spectrum in Figure 1f was computed from 196 F-actin segments (also 980 Å in length). The averages in Figure 1g,h were computed from 2,925 and 3,351 overlapping segments, respectively, each segment 490 Å long. The scale bars in Figure 1a,b,d are 1,000 Å.
All cryo-EM images were multiplied by the theoretical contrast transfer function (CTF) to correct for phase reversals and to optimize the signal-to-noise ratio. Final reconstructions were then divided by the weighted sum of the squared CTF functions (as the images had been multiplied by a CTF twice: once by the EM, and once by us) and corrected for the envelope function of the EM with the use of negative B-factors. The segments were classified as shown in Figure 2a using an iterative approach. First, a global reconstruction was generated using all segments. This global reconstruction was then deformed into 14 different twist states, and these models were used as references for an initial sorting. Reconstructions were independently generated from the 14 different classes of segments. These new reconstructions were then used as references for sorting. The validity of the sorting shown in Figure 2a was confirmed by two independent means. First, power spectra of segments from different bins behave exactly as predicted by the twist values shown in the histogram (Supplementary Video 1). Second, we used the IHRSR approach5 to separately reconstruct different bins. This showed that segments classified as having a mean twist of 167.5°, for example, generated a reconstructed volume that had a twist of ~167.5° (data not shown). The University of California San Francisco Chimera software34 was used to fit crystal structures into the experimental maps.
Supplementary Material
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health grants GM081303 (to E.H.E.) and GM061010 and GM675287 (to R.D.M.).
Footnotes
Accession codes. Protein Data Bank: Coordinates have been deposited with accession code 2QU4.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Jensen RB, Gerdes K. Mechanism of DNA segregation in prokaryotes: ParM partitioning protein of plasmid R1 co-localizes with its replicon during the cell cycle. EMBO J. 1999;18:4076–4084. doi: 10.1093/emboj/18.14.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Ent F, Moller-Jensen J, Amos LA, Gerdes K, Lowe J. F-actin-like filaments formed by plasmid segregation protein ParM. EMBO J. 2002;21:6935–6943. doi: 10.1093/emboj/cdf672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Ent F, Amos LA, Löwe J. Prokaryotic origin of the actin cytoskeleton. Nature. 2001;413:39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- 4.Lowe J, Amos LA. Crystal structure of the bacterial cell-division protein FtsZ. Nature. 1998;391:203–206. doi: 10.1038/34472. [DOI] [PubMed] [Google Scholar]
- 5.Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 6.Egelman EH, Francis N, DeRosier DJ. F-actin is a helix with a random variable twist. Nature. 1982;298:131–135. doi: 10.1038/298131a0. [DOI] [PubMed] [Google Scholar]
- 7.Schmid MF, Sherman MB, Matsudaira P, Chiu W. Structure of the acrosomal bundle. Nature. 2004;431:104–107. doi: 10.1038/nature02881. [DOI] [PubMed] [Google Scholar]
- 8.Galkin VE, Orlova A, Lukoyanova N, Wriggers W, Egelman EH. Actin depoly-merizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J. Cell Biol. 2001;153:75–86. doi: 10.1083/jcb.153.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore PB, Huxley HE, DeRosier DJ. Three-dimensional reconstruction of F-actin, thin filaments, and decorated thin filaments. J. Mol. Biol. 1970;50:279–295. doi: 10.1016/0022-2836(70)90192-0. [DOI] [PubMed] [Google Scholar]
- 10.Egelman EH. The iterative helical real space reconstruction method: Surmounting the problems posed by real polymers. J. Struct. Biol. 2007;157:83–94. doi: 10.1016/j.jsb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Heuser J. Preparing biological samples for stereomicroscopy by the quick-freeze, deep-etch, rotary-replication technique. Methods Cell Biol. 1981;22:97–122. doi: 10.1016/s0091-679x(08)61872-5. [DOI] [PubMed] [Google Scholar]
- 12.Belmont LD, Orlova A, Drubin DG, Egelman EH. A change in actin conformation associated with filament instability after Pi release. Proc. Natl. Acad. Sci. USA. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes KC, Popp D, Gebhard W, Kabsch W. Atomic model of the actin filament. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 14.Orlova A, et al. Probing the structure of f-actin: cross-links constrain atomic models and modify actin dynamics. J. Mol. Biol. 2001;312:95–106. doi: 10.1006/jmbi.2001.4945. [DOI] [PubMed] [Google Scholar]
- 15.Shvetsov A, Musib R, Phillips M, Rubenstein PA, Reisler E. Locking the hydrophobic loop 262–274 to G-actin surface by a disulfide bridge prevents filament formation. Biochemistry. 2002;41:10787–10793. doi: 10.1021/bi020205f. [DOI] [PubMed] [Google Scholar]
- 16.Popp D, et al. Concerning the dynamic instability of actin homolog ParM. Biochem. Biophys. Res. Commun. 2007;353:109–114. doi: 10.1016/j.bbrc.2006.11.130. [DOI] [PubMed] [Google Scholar]
- 17.Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc. Natl. Acad. Sci. USA. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chik JK, Lindberg U, Schutt CE. The structure of an open state of β-actin at 2.65 Ångstrom resolution. J. Mol. Biol. 1996;263:607–623. doi: 10.1006/jmbi.1996.0602. [DOI] [PubMed] [Google Scholar]
- 19.Klenchin VA, Khaitlina SY, Rayment I. Crystal structure of polymerization-competent actin. J. Mol. Biol. 2006;362:140–150. doi: 10.1016/j.jmb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Simanshu DK, Savithri HS, Murthy MR. Crystal structures of ADP and AMPPNP-bound propionate kinase (TdcD) from Salmonella typhimurium: comparison with members of acetate and sugar kinase/heat shock cognate 70/actin superfamily. J. Mol. Biol. 2005;352:876–892. doi: 10.1016/j.jmb.2005.07.069. [DOI] [PubMed] [Google Scholar]
- 21.Nishimasu H, Fushinobu S, Shoun H, Wakagi T. Crystal structures of an ATP-dependent hexokinase with broad substrate specificity from the hyper-thermophilic archaeon Sulfolobus tokodaii. J. Biol. Chem. 2007;282:9923–9931. doi: 10.1074/jbc.M610678200. [DOI] [PubMed] [Google Scholar]
- 22.Aleshin AE, et al. Crystal structures of mutant monomeric hexokinase I reveal multiple ADP binding sites and conformational changes relevant to allosteric regulation. J. Mol. Biol. 2000;296:1001–1015. doi: 10.1006/jmbi.1999.3494. [DOI] [PubMed] [Google Scholar]
- 23.Aleshin AE, Zeng C, Bartunik HD, Fromm HJ, Honzatko RB. Regulation of hexokinase I: crystal structure of recombinant human brain hexokinase complexed with glucose and phosphate. J. Mol. Biol. 1998;282:345–357. doi: 10.1006/jmbi.1998.2017. [DOI] [PubMed] [Google Scholar]
- 24.Nolen BJ, Pollard TD. Insights into the influence of nucleotides on actin family proteins from seven structures of Arp2/3 complex. Mol. Cell. 2007;26:449–457. doi: 10.1016/j.molcel.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galkin VE, VanLoock MS, Orlova A, Egelman EH. A new internal mode in F-actin helps explain the remarkable evolutionary conservation of actin’s sequence and structure. Curr. Biol. 2002;12:570–575. doi: 10.1016/s0960-9822(02)00742-x. [DOI] [PubMed] [Google Scholar]
- 26.Garner EC, Campbell CS, Mullins RD. Dynamic instability in a DNA-segregating prokaryotic actin homolog. Science. 2004;306:1021–1025. doi: 10.1126/science.1101313. [DOI] [PubMed] [Google Scholar]
- 27.Orlova A, Egelman EH. F-actin retains a memory of angular order. Biophys. J. 2000;78:2180–2185. doi: 10.1016/S0006-3495(00)76765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buss KA, et al. Urkinase: structure of acetate kinase, a member of the ASKHA superfamily of phosphotransferases. J. Bacteriol. 2001;183:680–686. doi: 10.1128/JB.183.2.680-686.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aleshin AE, et al. Nonaggregating mutant of recombinant human hexokinase I exhibits wild-type kinetics and rod-like conformations in solution. Biochemistry. 1999;38:8359–8366. doi: 10.1021/bi990523n. [DOI] [PubMed] [Google Scholar]
- 30.Hara F, et al. An actin homolog of the archaeon Thermoplasma acidophilum that retains the ancient characteristics of eukaryotic actin. J. Bacteriol. 2007;189:2039–2045. doi: 10.1128/JB.01454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doolittle RF. The origins and evolution of eukaryotic proteins. Phil. Trans. R. Soc. Lond. B. 1995;349:235–240. doi: 10.1098/rstb.1995.0107. [DOI] [PubMed] [Google Scholar]
- 32.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 33.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 34.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Joel PB, Fagnant PM, Trybus KM. Expression of a nonpolymerizable actin mutant in Sf9 cells. Biochemistry. 2004;43:11554–11559. doi: 10.1021/bi048899a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.