

Desiccation is thought to impose many stresses. Which of these stresses is responsible for desiccation-induced death and how the stress response is regulated are unknown, however. Here we use Saccharomyces cerevisiae to show that reduction of a 60S biogenesis intermediate via RAS or TOR down-regulation increases desiccation tolerance.
Abstract
Tolerance to desiccation in cultures of Saccharomyces cerevisiae is inducible; only one in a million cells from an exponential culture survive desiccation compared with one in five cells in stationary phase. Here we exploit the desiccation sensitivity of exponentially dividing cells to understand the stresses imposed by desiccation and their stress response pathways. We found that induction of desiccation tolerance is cell autonomous and that there is an inverse correlation between desiccation tolerance and growth rate in glucose-, ammonia-, or phosphate-limited continuous cultures. A transient heat shock induces a 5000–fold increase in desiccation tolerance, whereas hyper-ionic, -reductive, -oxidative, or -osmotic stress induced much less. Furthermore, we provide evidence that the Sch9p-regulated branch of the TOR and Ras-cAMP pathway inhibits desiccation tolerance by inhibiting the stress response transcription factors Gis1p, Msn2p, and Msn4p and by activating Sfp1p, a ribosome biogenesis transcription factor. Among 41 mutants defective in ribosome biogenesis, a subset defective in 60S showed a dramatic increase in desiccation tolerance independent of growth rate. We suggest that reduction of a specific intermediate in 60S biogenesis, resulting from conditions such as heat shock and nutrient deprivation, increases desiccation tolerance.
INTRODUCTION
Desiccation tolerance is the ability of an organism to withstand removal of as much as 95% of its water and resume normal metabolism after rehydration (Crowe et al., 1992). Desiccation tolerance is commonly found in seeds, which contain desiccated plant embryos, and also rarely in adult extremophiles such as bacteria (Deinococcus radiodurans), fungi (Saccharomyces cerevisiae), plants (resurrection plants and bryophytes), and animals (nematodes, tardigrades, brine shrimp, and rotifers) (Potts, 1994). Desiccation is thought to impose a number of stresses, including hyperosmolarity, hyperoxidation, hyperionicity, and protein misfolding/aggregation (Franca et al., 2005; Chakrabortee et al., 2007). All these stresses have the potential to inflict lethal damage upon essential cellular components such as DNA, proteins, and membranes (Herrero et al., 2008; Hohmann, 2009; Verghese et al., 2012). However, which of these stresses (alone or in combination) is actually responsible for desiccation-induced death and how extremophiles counter the relevant lethal stresses are unknown. A molecular understanding of desiccation tolerance is likely to provide key insights into water homeostasis in all cells and to generate powerful tools to address important agricultural problems such as crop loss due to drought and biomedical problems such as long-term blood storage.
Desiccated organisms have been studied for many years and have been found to harbor a number of molecules thought to mitigate different stresses. These putative protective molecules include osmolytes, heat shock proteins (HSPs), redox balance enzymes, trehalose, short unstructured hydrophilic proteins, and others (Crowe et al., 1992; Billi and Potts, 2002; Tunnacliffe and Wise, 2007). However, in many cases, their presence remains only correlated to desiccation tolerance without a demonstration of causality. An ideal tool to assess causality in vivo is the budding yeast S. cerevisiae, a conditionally desiccation-tolerant organism especially amenable to genetic analyses.
Our laboratory and others have shown that S. cerevisiae is more desiccation tolerant during stationary phase than exponential phase (Ratnakumar and Tunnacliffe, 2006; Calahan et al., 2011). One method to predict the stress response pathways responsible for stress tolerance in stationary-phase cells is to examine changes in gene expression levels. However, previous studies in yeast showed that any one of a panel of environmental stresses triggers a stereotypical change in expression of some 900 genes, including those encoding products necessary for combating other stresses (Gasch et al., 2000; Causton et al., 2001). This broad gene expression response, named the environmental stress response (ESR), is also strongly induced during entry into stationary phase, obfuscating the importance of individual genes in surviving a particular stress, including desiccation tolerance. Furthermore, studies investigating the importance of stress-activated genes and stress-tolerance genes have found little overlap (Giaever et al., 2002; Berry and Gasch, 2008). Thus analyses of gene expression responses to stresses have shown limited predictive value for identifying the genes essential for survival.
In light of these discoveries, researchers have begun to exploit genetic tools in yeast to assess the functional importance of specific genes and pathways in desiccation tolerance. A survival analysis of a mixture of 5000 mutant strains subjected to desiccation, each deleted for a different nonessential gene, suggested that 653 genes may be important for desiccation tolerance of cells growing after diauxic shift (Ratnakumar et al., 2011). Strains lacking MSN2, MSN4, or GIS1, genes encoding transcriptional regulators of HSPs and trehalose biosynthetic enzymes, were underrepresented, as were some of the regulators of the Ras-cAMP signaling pathway. Although this screen provided potential clues, we decided to make further inquiries to clarify the signaling mechanisms regulating desiccation tolerance.
Using a more sensitive assay for desiccation tolerance capable of assaying up to 108 cells for viability (see Materials and Methods), we surveyed mutants defective in many stress response factors and regulators (DNA damage repair, oxidative and salt homeostasis, as well as global stress regulators of osmolarity) for desiccation tolerance in saturated cultures (Calahan et al., 2011). Surprisingly, desiccation tolerance in these mutants was minimally affected, if at all. The failure of the candidate gene approach prompted a screen of the yeast deletion collection of nonessential genes to identify deletions that reduced desiccation tolerance of stationary-phase cultures by several orders of magnitude. Intriguingly, only deletions that abrogated respiration were identified, exhibiting a reduction of 10,000-fold in desiccation tolerance. Further analysis demonstrated that processes associated with respiration before desiccation allow desiccation tolerance.
This respiration requirement for desiccation tolerance can be bypassed by activation of Med2p, a subunit of the activator submodule of the mediator complex involved in regulation of transcription, or inhibition of Ras2p, a small GTPase that is responsive to glucose and stress signals. Taken together, these results suggest that the induction of stress effectors by Ras2p or Med2p before desiccation is necessary for subsequent tolerance. Unfortunately, exploiting these insights to find the stress effectors has proven difficult. Ras2p and Med2p regulate the expression of hundreds of genes. Furthermore, several Ras2p-/Med2p-regulated genes are likely involved because the deletion of any single nonessential gene (other than those required for respiration) does not reduce tolerance beyond a few fold.
Here we narrow down potential candidates for effectors by defining the specific stress responses that contribute to desiccation tolerance. We show that desiccation-sensitive, exponential-phase cells can acquire desiccation tolerance by pretreatment with heat stress, or in a graded manner by decreasing growth rate due to nutrient limitation in continuous culture, but very little by osmotic, reductive, oxidative, or salt stress. We further connect desiccation and heat tolerance by showing that they are both inhibited by TORC1. We provide evidence that TOR and Ras-cAMP signaling inhibit desiccation tolerance in exponential phase via Sch9p and the cAMP-dependent protein kinase A (PKA), respectively. We show that Sch9p and PKA modulate desiccation tolerance by modulating the activity of Msn2p, Mns4p, and Gis1p (transcription factors critical for heat stress response) and Sfp1p (involved in ribosome biogenesis). Finally, examination of 41 ribosome biogenesis mutants reveals mutants defective in 60S ribosome biogenesis that exhibit increased desiccation tolerance independent of growth rate. We suggest a model in which reduced 60S biogenesis, resulting from conditions such as heat shock and nutrient deprivation, is necessary for increased desiccation tolerance.
RESULTS
Acquisition of desiccation tolerance is cell autonomous
Only one in a million cells from an exponential culture survives desiccation, whereas one in five survives desiccation from a stationary-phase culture (Calahan et al., 2011). We wondered whether the difference in desiccation tolerance of these two cultures might be the result of cell-to-cell communication. For example, quorum-sensing molecules secreted by bacteria accumulate in proportion to cell density and cause their constituents to alter gene expression in response to signal concentration (Miller and Bassler, 2001). Additionally, fungi including S. cerevisiae exhibit quorum-sensing activity in response to nutrient limitation (Chen and Fink, 2006).
To test for the presence of a secreted desiccation tolerance–inducing factor, we pelleted exponential-phase cells and resuspended them in either dilute phosphate-buffered saline (PBS) or conditioned medium from a stationary-phase culture. Cells were assayed for desiccation tolerance immediately after resuspension and 6 h after resuspsension (Figure 1, lanes 3, 4, 9, and 10). A 6-h incubation was likely to be more than sufficient time to induce desiccation tolerance, because a change in carbon source can induce tolerance in 2 h (Calahan et al., 2011). Brief incubation of exponential-phase cells in conditioned medium did not change desiccation tolerance significantly (Figure 1, lanes 1 and 3). A 6-h incubation caused desiccation tolerance to increase 36-fold (Figure 1, lanes 3 and 4); however, cells resuspended in dilute PBS for 6 h showed a similar fold increase (Figure 1, lanes 4 and 10). Thus the small increase observed due to conditioned medium appeared to be independent of the medium itself and likely due to time spent in a nondividing, starving state. We conclude that conditioned medium is unlikely to contain a soluble factor that induces desiccation tolerance.
FIGURE 1:
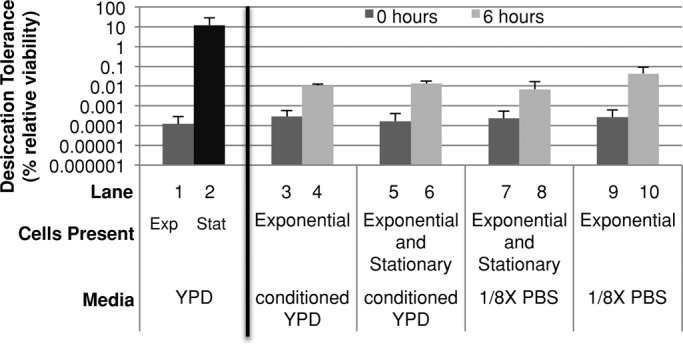
Desiccation tolerance is acquired in a cell-autonomous manner. Exponential-phase G418R cells (gal3Δ) were resuspended in four conditions: medium from saturated cells, medium and cells from stationary-phase culture, cells from stationary-phase culture in dilute PBS, or in dilute PBS alone. These were then assayed for desiccation tolerance at the indicated times. Controls (left) are G418R cells (gal3Δ) in either exponential or stationary phase.
Secreted factors are not the only means by which cells communicate. Examples of cell signaling due to direct contact (juxtacrine) are common in mammalian cells and also cause gene expression changes (Singh and Harris, 2005). We considered that an extracellular factor important for desiccation tolerance induction may be cell bound, and thus signal an increase in desiccation tolerance when cells received sufficient direct-contact stimulation. To test this, we mixed stationary-phase G418S cells (1.6 × 108 cells) with exponential-phase G418R cells (4 × 107 cells) in a low volume of dilute PBS (optical density [OD]600 = 44), and assessed desiccation tolerance of the exponential-phase cells immediately after mixing and 6 h later (Figure 1, lanes 7 and 8). We were able to distinguish the desiccation tolerance of exponential-phase cells from stationary-phase cells by their ability to grow in medium containing the antibiotic G418. The increase in desiccation tolerance of the exponential-phase cells was not significantly different from that of exponential-phase cells resuspended in dilute PBS (Figure 1, lanes 8 and 10). Thus direct contact with stationary-phase cells does not confer desiccation tolerance on exponential-phase cells, indicating that a cell-bound factor capable of inducing desiccation tolerance does not exist. In this experiment the exponential-phase culture was incubated at a cell density similar to those of stationary-phase cultures for 6 h before desiccation. The failure to induce tolerance under these conditions also suggests that high cell density is not sufficient to induce desiccation tolerance.
To test whether a cell-bound factor may require its native medium to be active, we replaced a small fraction of the cells in a stationary-phase culture with the same number of G418R exponential-phase cells and allowed this mixed culture to incubate for 6 h. Again this condition failed to elevate the desiccation tolerance of exponential-phase cells, either immediately or after 6 h, above that observed for exponential-phase cells in dilute PBS (Figure 1, lanes 5, 6, 9, and 10). This experiment also eliminated the possibility that stationary cells could protect exponential cells by forming a structural cage. Taken together, these experiments suggest that the increase in desiccation tolerance of a culture as it grows to saturation is a cell-autonomous process.
Induction of desiccation tolerance via stress treatment
As cells transition from exponential phase to stationary phase, they exhaust energy-containing carbon sources. Thus a potential cell-autonomous inducer of desiccation tolerance in stationary-phase cultures is nutrient starvation. We began investigating this by first asking whether glucose starvation might induce desiccation tolerance. We transferred exponentially growing cells in rich medium with glucose to rich medium lacking glucose and measured desiccation tolerance over time (Figure 2A). Desiccation tolerance increased a millionfold within 16 h, whereas cell density increased by 1.5-fold. This result indicates that glucose starvation can induce desiccation tolerance and that extensive cell doubling is not required for the acquisition of desiccation tolerance.
FIGURE 2:

Starvation can induce desiccation tolerance. (A) Exponential-phase BY4742 cells were transferred from rich medium containing glucose to the same medium without glucose, and samples were taken at the indicated times and assayed for desiccation tolerance. (B) Strain DBY12000 was grown under defined media conditions in a chemostat at constant doubling time at the times and nutrient-limiting conditions indicated until steady-state conditions were reached, and then were tested for desiccation tolerance using the described protocol (see Materials and Methods). The experiment was performed in triplicate, and representative results are shown.
To assess whether desiccation tolerance could be induced by decreasing growth rate by nutrient limitation, we used a continuous culture system. Cultures of protorophic yeast were grown in chemostat vessels under three different medium regimes at three different exponential growth rates for each medium: glucose-limited, nitrogen-limited, and phosphate-limited. In the continuous culture system, the culture growth rate is set by the rate of fresh medium (containing a defined limiting nutrient) influx: an increased flow rate results in an increased growth rate, whereas a decreased flow rate results in a decreased growth rate (Monod, 1950; Novick and Szilard, 1950). When at steady state, desiccation tolerance was assessed.
Regardless of the limiting nutrient used to set the growth rate, decreased growth rate resulted in increased desiccation tolerance (Figure 2B). Desiccation tolerance for cultures with the slowest growth rate (13.8-h doubling time) reached levels similar to those seen in stationary-phase cultures. The tight correlation between nutrient-imposed slow growth and desiccation tolerance suggests that slow growth in general or a component(s) associated with nutrient-imposed slow growth results in desiccation tolerance.
A potential explanation for these observations is that increased respiration, due to reduced growth rate, is responsible for the induction of desiccation tolerance in continuous culture. We think not, for several reasons. First, under sufficiently nutrient-limiting conditions, yeast cells will respire regardless of glucose abundance (van Hoek et al., 1998, 2000), yet we see a graded increase in dessication tolerance as the growth rate is lowered, and desiccation tolerance is not fully induced until much longer doubling times (Figure 2B). Second, in the case of heat shock resistance, Lu et al. (2009) were able to show a graded increase of resistance with lower growth rate in respiratory-deficient (ρ−) strains of yeast.
We next assessed whether stresses other than nutrient limitation could induce desiccation tolerance. We asked whether tolerance could be induced after exponential-phase cells were treated with heat, ionic, osmotic, or oxidative stress. A previous study suggested that heat stress gave a modest twofold increase, and the other stresses had even smaller increases (Ratnakumar and Tunnacliffe, 2006). However, the assay used to measure desiccation tolerance in this study was compromised by the high basal rate of desiccation tolerance of the untreated control (∼5%), likely caused by the slow desiccation method used. During slow desiccation, samples undergo nutrient starvation, thereby inducing desiccation tolerance. We decided to reinvestigate the ability of other stresses to induce desiccation tolerance using our more sensitive assay in which the basal rate of desiccation tolerance was one in a million cells.
Exponential-phase cells growing at 30°C were subjected to sublethal stresses by resuspending them in rich medium that either was preheated to 39°C or contained sublethal amounts of sodium chloride, sorbitol, dithiothreitol, or hydrogen peroxide (Figure 3, A– C). At several time points, aliquots were resuspended in dilute PBS and then immediately desiccated (Figure 3, A and C). Additionally, at these same time points, thermally, ionically, or osmotically stressed cultures were treated with lethal heat shock (52°C) and then assayed for viability (Figure 3B). We chose concentrations of these stressors and treatment time frames that had previously been shown to induce significant changes in the transcription profile of genes implicated in alleviating stress (Gasch et al., 2000). We found that untreated control cultures remained both thermosensitive and desiccation sensitive. Pretreatment of cells with sodium chloride or sorbitol increased desiccation tolerance 70- to 100-fold, a significant but small increase compared with the 5000-fold increase caused by heat shock (Figure 3A). This trend is observed at a later time point as well (Supplemental Figure S1). No increase in desiccation tolerance was observed when cells were treated with dithiothreitol or hydrogen peroxide (Figure 3C). Thus pretreatment of cells with different sublethal stressors appears to have dramatically different effects on the level of desiccation tolerance induced.
FIGURE 3:
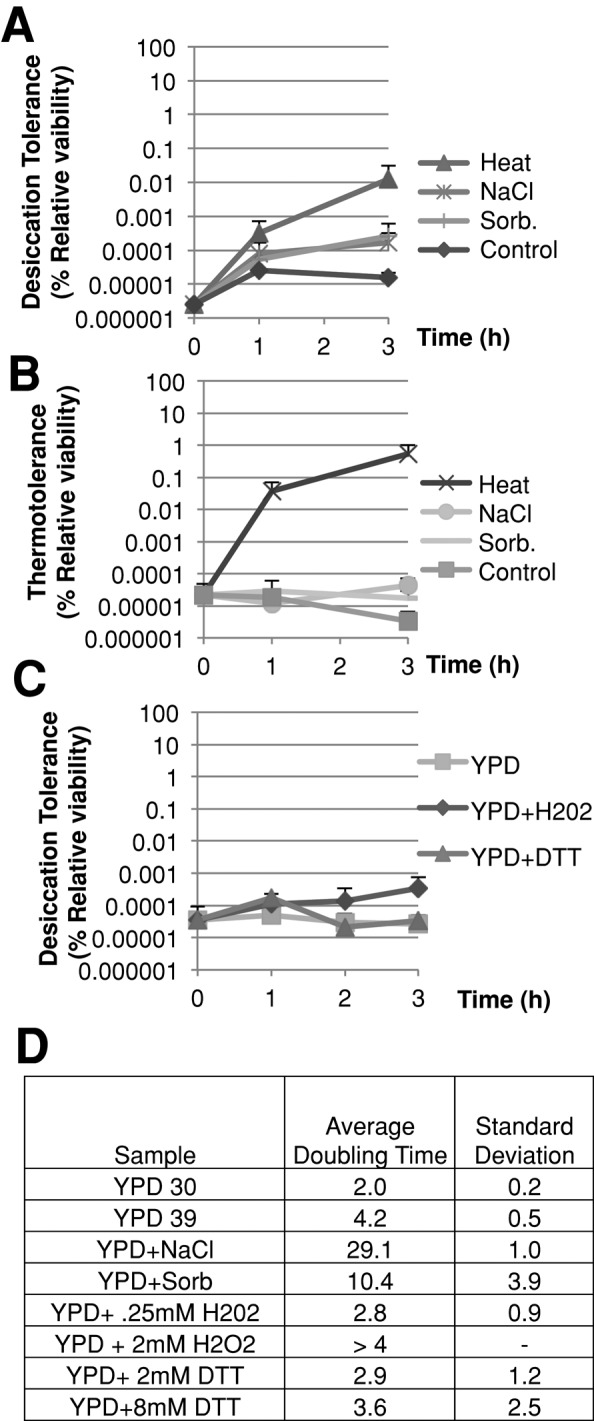
Pretreatment with heat, but not other stresses, highly induces desiccation tolerance. (A) Exponential-phase BY4742 cells growing at 30°C in rich medium were pelleted and resuspended in the same medium with or without 1 M sodium chloride or 1.5 M sorbitol added or preheated to 39°C in rich medium at an OD600 < 0.6 and desiccated at the indicated time points and then assessed for viability. (B) Cells from the same experiment indicated in (A) were also treated for thermotolerance at the same time points by incubating at 52°C for 15 min and then assessed for viability. (C) Exponential-phase BY4742 cells from rich medium were collected and resuspended in rich medium with or without dithiothreitol or H2O2 added, and samples were assayed for desiccation tolerance at the indicated time points. (D) Growth rates of indicated cultures. Growth rate was calculated using the following formula: ΔTime/(Log2[Cell densityfinal/Cell densityinital]). The time period used to assess growth rate for each culture was between 1 and 3 h.
This conclusion depends on the assumption that the level of stressors used in our study indeed induced significant amounts of the relevant stress and stress response. This assumption was supported by our knowledge that levels of stressors used in our experiments were known to cause similar induction of the ESR (Gasch et al., 2000). Beyond ESR induction, we validated this assumption in several ways. Thermotolerance increased in the 39°C culture 30,000-fold within 3 h (Figure 3B). These results corroborate previous studies in which heat shock increased thermotolerance and induced sweeping gene expression changes within 15–30 min (Sanchez and Lindquist, 1990; Gasch et al., 2000). Hyperosmotic treatment induced gene expression changes indicative of osmotic stress, and showed a significant decrease in growth rate (Figure 3D and Supplemental Figure S2). Treatment with hydrogen peroxide and dithiothreitol at the concentrations used here and in previous transcription studies failed to produce any change in growth rate. To investigate whether higher levels of stress imposed by these two stressors increased desiccation tolerance, we treated cells with sufficiently higher concentrations of these stressors to slow growth (Figure 3D and Supplemental Figure S4, A and B). We still failed to observe an increase in desiccation tolerance. Thus, the difference in the type of stress used to confer cross-tolerance to desiccation is unlikely to be due to differences in the degree of stress experienced.
Finally, to make sure that the stress did not dissipate during desiccation, we tested whether the presence of these stressors with the desiccating samples would affect desiccation tolerance. A culture of exponential-phase cells growing at 30°C was transferred to fresh medium with or without sodium chloride, dithothreitol, or hydrogen peroxide and desiccated in dilute PBS with or without the same stressor (Supplemental Figure S3, A–C). We found no significant change in desiccation tolerance due to the presence of the stressor during desiccation. Sorbitol was not tested in this manner because it forms a glass at the bottom of the tube and blocks desiccation. These results strongly suggest that our conditions were sufficient to induce the relevant stresses and their stress responses.
Taken together, these experiments suggest a number of important conclusions. The minimal cross-protection conferred by salt, osomotic, or redox stresses suggests that effectors of these stress responses, unlike heat shock, contribute minimally to desiccation tolerance. Consistent with a unique relationship between desiccation tolerance and heat shock, the other stressors also failed to induce heat tolerance as well as desiccation tolerance (Figure 3B). However, cross-protection due to heat shock is only partial, as the increase in desiccation tolerance is still several orders of magnitude lower than the level of desiccation tolerance in a stationary-phase culture. This finding suggests that acquisition of desiccation tolerance requires additional stress effectors beyond those sufficient for heat tolerance and/or higher levels of the common stress effectors. Finally, because these other stresses imposed slow growth but failed to induce desiccation tolerance, the stress/slow growth imposed by nutrient limitation (and possibly heat shock) is distinct. This distinction suggests that desiccation tolerance arises from a component or components that are associated with nutrient-imposed slow growth rather than slow growth per se.
Regulation of desiccation tolerance by the TOR pathway
The TOR pathway, conserved from yeast to humans, is one of the major integrators of information regarding the nutrient status within a cell (Zaman et al., 2008). The TOR complex 1 (TORC1) is active in exponential-phase cells, phosphorylating downstream targets to repress stress responses and to promote ribosome biogenesis and cell-cycle progression. Interestingly, a recent study showed similar regulation of certain proteins by heat shock and TOR signaling (Bandhakavi et al., 2008). Thus TORC1 regulates responses to both nutrient abundance and heat, two factors that we showed correlated with desiccation tolerance.
On the basis of these reports, we hypothesized that TORC1 might mediate the increase in desiccation tolerance exhibited by nutrient-deprived or heat-stressed cells. Manipulation of TORC1 activity is possible using the macrolide drug rapamycin. Rapamycin binds to Fpr1p (FKBP12 in humans), forming a complex that binds Tor1p, thereby inhibiting TORC1. Using rapamycin, we investigated whether TORC1 may regulate heat shock and desiccation tolerance. A culture of exponential-phase cells was treated with rapamycin and then assayed for both desiccation and heat tolerance (Figure 4A). Rapamycin was washed out of the culture before assessment of desiccation or heat tolerance. Rapamycin treatment elevated both desiccation and heat tolerance three to four orders of magnitude; these absolute values approach the maximum values of desiccation and heat tolerance seen in stationary-phase cultures or those pretreated with a heat stress. This dual induction by rapamycin of desiccation and heat tolerance is consistent with their putative use of common stress mitigators.
FIGURE 4:
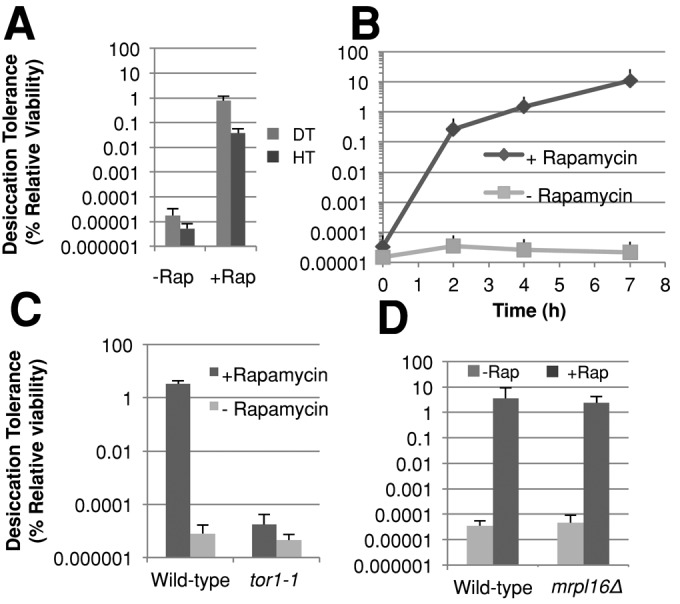
The TOR pathway inhibits desiccation tolerance in exponential-phase cells. (A) Exponential-phase cells growing in rich medium were transferred to the same media containing either rapamycin or the vehicle (ethanol) and then assayed for desiccation tolerance or heat tolerance after 6–10 h incubation while diluting to maintain OD600 < 0.6. (B) Exponential-phase cells growing in rich medium were transferred to the same media with either rapamycin or the vehicle (ethanol) added and then assayed for desiccation tolerance at the indicated time points, while maintaining OD600 < 0.6. (C) Cells containing a rapamycin-insensitive allele of TOR1 (tor1-1) and wild-type cells were assayed for desiccation tolerance as described in (A). (D) Exponential-phase cells of the indicated genotype were assayed for desiccation tolerance as described in (A).
We further characterized the induction of desiccation tolerance by rapamycin by assessing the kinetics of desiccation tolerance induction. Cell aliquots were removed at various times after the addition of rapamycin and assayed for desiccation tolerance (Figure 4B). This treatment induced a dramatic increase in desiccation tolerance of 7700-fold in 2 h, with desiccation tolerance reaching an absolute level of 10% by 7 h. Throughout this time course, control cell viability of rapamycin-treated cultures did not drop below 50% as compared with the untreated control culture's viability. Both the rapid response and significant induction mirror the desiccation tolerance response of cells switched from glucose to glycerol medium (Calahan et al., 2011). To verify that the desiccation tolerance induction by rapamycin was in fact due to inhibition of Tor1p and not an off-target effect of this drug, we used the tor1-1 allele that is insensitive to rapamycin. A strain bearing the tor1-1 allele as the only copy of TOR1 was assayed for desiccation tolerance in the presence or absence of rapamycin (Figure 4C). Rapamycin failed to induce any desiccation tolerance in this mutant, confirming that rapamycin induces desiccation tolerance by Tor1p inhibition.
It has been reported that tor1Δ cells induce a mild stress response (Wei et al., 2008, 2009). However, we did not observe any change in desiccation tolerance in tor1Δ cells during growth in exponential phase (Supplemental Figure S5A). This lack of change may be due to the ability of Tor2p to substitute for Tor1p in TORC1.
Finally, we asked whether the induction of desiccation tolerance by Tor1p inhibition is dependent on respiration. To do this, we examined desiccation tolerance in respiration-incompetent mrpl16∆ cells treated with or without rapamycin (Figure 4D). MRPL16 encodes a mitochondrially specific ribosomal protein of the large subunit, and absence of this protein causes cells to lose respiration capability. Indeed, this strain exhibited robust desiccation tolerance after treatment with rapamycin. Thus the induction of desiccation tolerance by Tor1p inhibition is independent of the respiratory state of the cell.
The TORC1 pathway regulates a multitude of downstream targets. For example, TORC1 inhibits genes required for autophagy, retrograde signaling to the mitochondria, and nitrogen catabolite repression (NCR; Zaman et al., 2008). Indeed, a recent study suggested that autophagy is important for desiccation tolerance (Ratnakumar et al., 2011). TORC1 inhibits these processes by phosphorylating Tap42p (Zaman et al., 2008; Figure 5A). Tap42p is an essential protein that forms several distinct complexes with protein phosphatase 2A or Sit4p. To test whether Tor1p inhibits effectors of desiccation tolerance through Tap42p, we grew a strain bearing a temperature-sensitive allele of TAP42 to exponential phase at permissive (23°C) and semipermissive (30°C) temperatures and tested desiccation tolerance (Figure 5B). Desiccation tolerance increased 120-fold in this mutant. However, the total level of desiccation tolerance was still four orders of magnitude below those of stationary-phase cultures. Thus genes inhibited by Tap42p may play a role in desiccation tolerance.
FIGURE 5:
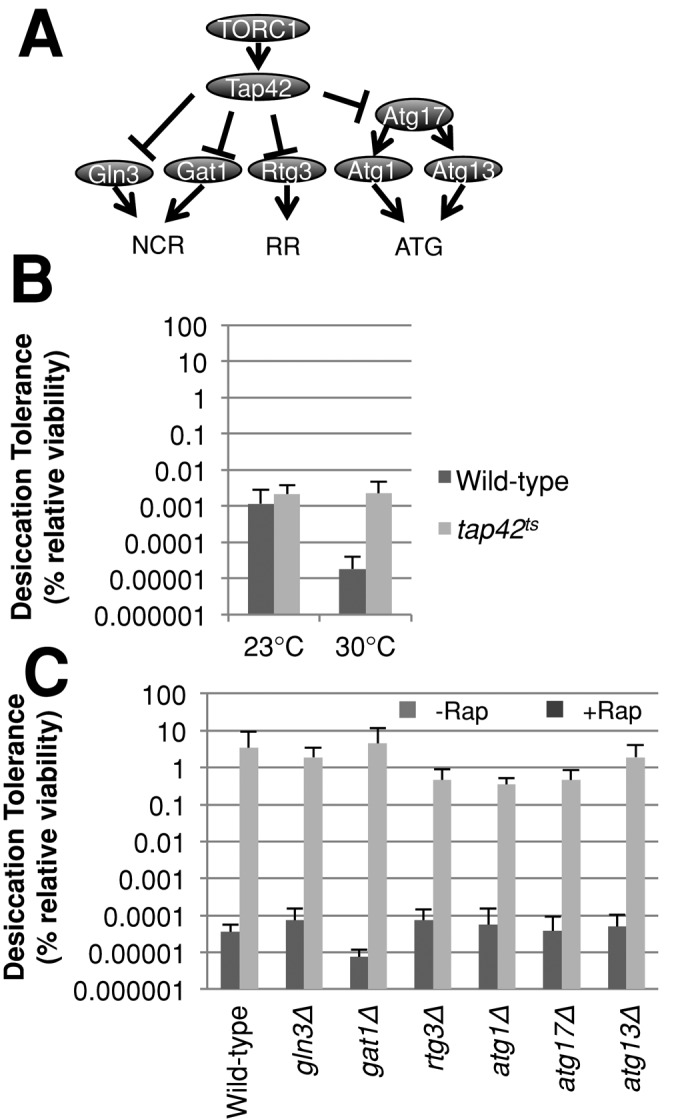
Effectors of Tap42 branch do not significantly affect desiccation tolerance. (A) Genetic pathways model depicting TORC1’s downstream effectors. (B) Cells of the indicated genotype were grown to exponential phase in rich medium at 23°C and either assayed for desiccation tolerance or transferred to the same medium at 30°C for 6 h, then assayed for desiccation tolerance. (C) Exponential-phase cells of the indicated genotype were assayed for desiccation tolerance as described in Figure 4A.
To test whether any of the known targets of Tap42p inhibition were effectors of desiccation tolerance, we asked whether strains deleted for individual genes encoding these targets compromised rapamycin-induced desiccation tolerance (Figure 5C). Mutants lacking RTG3, ATG1, or ATG17 exhibited minor suppression of rapamycin-induced desiccation tolerance (∼10-fold), whereas the remaining deletions had no effect. These results indicate that autophagy, retrograde signaling, and NCR signaling have no role in rapamycin-induced desiccation tolerance. Additional experiments will be required to assess the targets of Tap42 necessary for desiccation tolerance.
In addition to Tap42p, TORC1 activates Sch9p, which inhibits stress-responsive transcription factors and activates ribosome biogenesis. Because TORC1 activates Sch9p, the expectation was that deletion of SCH9 should mimic TORC1 inhibition. To test this expectation, exponential-phase sch9∆ cells were grown in rich medium in the presence or absence of rapamycin alongside wild-type cells (Figure 6, B and C). Indeed, we found that exponential sch9∆ cells had an 800-fold increase in desiccation tolerance above wild type, up to 0.03%, with this strain having a minor increase in doubling time (3 ± 0.8 h). Rapamycin treatment of sch9∆ cells further increased desiccation tolerance to a level similar to that of wild-type cells treated with rapamycin. These results indicate that TORC1 activation of Sch9p can account for a significant portion of desiccation sensitivity in exponential-phase cells.
FIGURE 6:
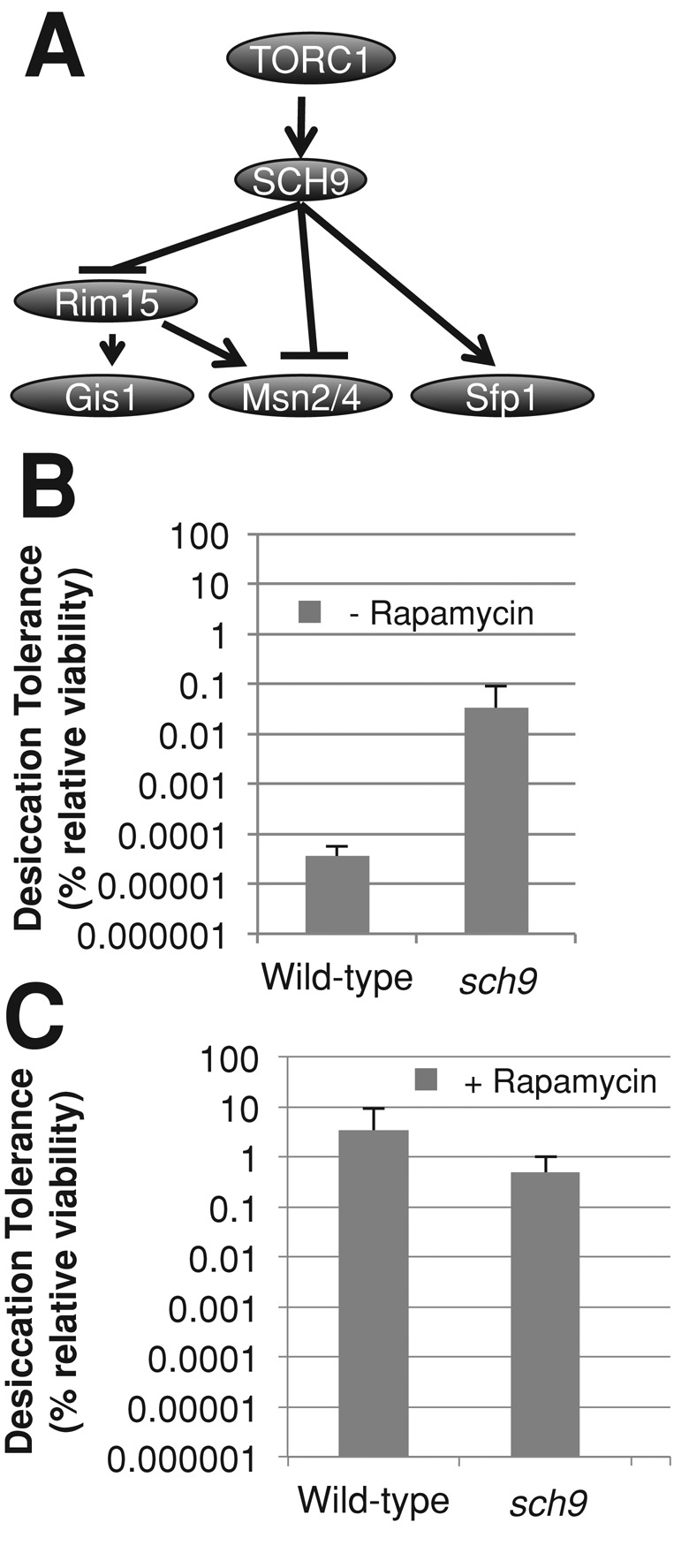
The Sch9 branch of the TOR pathway inhibits desiccation tolerance. (A) Genetic pathways model depicting TORC1’s downstream effectors. (B) Exponential-phase cultures of the indicated genotypes grown in rich medium were assayed for desiccation tolerance. (C) Exponential-phase cells of the indicated genotype were assayed for desiccation tolerance as described in Figure 4A.
Regulation of desiccation tolerance by the Ras/PKA pathway
Many of the proteins regulated by TORC1 through Sch9p are also regulated by the Ras-cAMP pathway via PKA. Ras2p is a small GTP-binding protein and homologous to the mammalian RAS proto-oncogenes. We showed in a previous study that deletion of RAS2 restores desiccation tolerance of a stationary-phase culture of respiration-defective cells to nearly wild-type levels (Calahan et al., 2011). However, ras2∆ cells do not exhibit increased desiccation tolerance in exponential phase (Figure 7B; Calahan et al., 2011). We reasoned that exponential-phase cells might fail to acquire desiccation tolerance because of functionally redundant mechanisms that compensate for loss of Ras2p. Ras2p functions in the Ras-cAMP pathway by activating adenylate cyclase (Cyr1p); however, the glucose-sensing transmembrane receptors Gpr1p and Gpa2p and the Ras2p homolog Ras1p also activate Cyr1p and are capable of compensating for loss of Ras2p.
FIGURE 7:

The Ras-cAMP pathway modulates desiccation tolerance. (A) Model depicting Ras-cAMP regulation of desiccation tolerance. (B) Exponential-phase cultures of the indicated genotype grown in rich medium were assayed for desiccation tolerance. (C) Exponential-phase ras2Δ cells carrying either a vector expressing a dominant-negative allele of RAS2 or an empty vector were grown in SC medium lacking leucine and methionine for 24 h then assayed for desiccation tolerance. (D) Exponential-phase cells carrying a vector expressing PDE2, MSI1, or an empty-vector control were grown in yeast extract peptone (YEP) with 2% galactose for 24 h then assayed for desiccation tolerance. Exponential-phase cells carrying a vector expressing BCY1 or an empty-vector control were grown in SC medium lacking histidine and assayed for desiccation tolerance. (E) Cells with either cyr1ts allele or the wild-type allele of CYR1 were grown to exponential phase at 23°C and then assayed for desiccation tolerance or transferred to the same medium at 30°C for 6 h and then assayed for desiccation tolerance.
To overcome these potential compensatory effects, we used a dominant-negative allele (ras2ala22) that locks Ras2p into a complex with Cdc25p, inactivating both Ras1p and Ras2p (Powers et al., 1989; Budovskaya et al., 2004). We constructed strains containing deletions for endogenous RAS2 and carrying either a vector expressing the dominant-negative allele of RAS2 (ras2a22) or an empty vector. These strains were grown to exponential phase in medium selecting for the vector and subsequently tested for desiccation tolerance (Figure 7C). The strain bearing the empty vector was desiccation sensitive as expected, but not as sensitive as previously observed for cultures grown in rich medium. This finding is consistent with the idea that nutrient deprivation (due to selective medium) is positively correlated with desiccation tolerance (Figure 2B). Nonetheless, the strain bearing the ras2a22 allele exhibited desiccation tolerance 3000-fold greater than that of cells bearing the empty vector alone. The absolute level of desiccation tolerance approached that of stationary-phase cultures. These results suggest that Ras1p and Ras2p activities inhibit desiccation tolerance in exponential-phase yeast cells.
Ras2p is known to function in several pathways, including the Ras-cAMP pathway (Sobering et al., 2004). To test whether the Ras-cAMP pathway inhibits desiccation tolerance in exponential-phase cultures, we examined the desiccation tolerance of exponential-phase cells overexpressing negative regulators of the Ras-cAMP pathway (MSI1, PDE2, BCY1). Msi1p, first characterized as a subunit of chromatin assembly factor 1, has a distinct function as a suppressor of hyperactive Ras mutants, acting downstream of Pde2p but upstream of Bcy1p (Zhu et al., 2000; Johnston et al., 2001). Bcy1p binds to the subunits of the PKA complex and renders them inactive in the absence of cAMP (Toda et al., 1987). Wild-type cells carrying plasmids that overexpress MSI1 or BCY1 were grown to exponential phase in inducing medium, then assayed for desiccation tolerance (Figure 7D). The elevated levels of desiccation tolerance in the vector-alone controls are likely due to nutrient deprivation (Figure 2B). Despite the high basal tolerance of the controls, overexpression of MSI1 and BCY1 induced a 10- to 40-fold additional increase in desiccation tolerance, consistent with the hypothesis that modulation of Ras-cAMP signaling affects desiccation tolerance.
The activity of the Ras-cAMP pathway is ultimately determined by the kinase activity of the PKA complex, which is regulated by the level of intracellular cAMP. To test whether cellular cAMP concentration affects desiccation tolerance, we overexpressed PDE2, which encodes a cAMP-phosphodiesterase that degrades cAMP. Wild-type cells carrying a plasmid that overexpresses PDE2 were grown to exponential phase in rich medium containing galactose and then assayed for desiccation tolerance (Figure 7D). Cells overexpressing PDE2 exhibited an increase in desiccation tolerance of 400-fold over the empty-vector controls, to levels approaching stationary-phase cultures.
To further test the effect of modulation of cAMP levels, we inhibited cAMP production by compromising Cyr1p, the essential adenylate cyclase protein responsible for converting AMP into cAMP. To do this, the CYR1 gene was replaced with a temperature-sensitive allele. The cyr1ts strain was grown to exponential phase at the permissive temperature (23°C), transferred to a semipermissive temperature (30°C) for 6 h, and assayed for desiccation tolerance (Figure 7E). While at 23°C, the temperature-sensitive strain exhibited desiccation tolerance equivalent to wild type; at 30°C it showed an increase of 30,000-fold over wild type. Interestingly, the wild type grown at 23°C exhibited 100-fold increased desiccation tolerance as compared with the culture grown at 30°C, indicating that cold stress may induce higher desiccation tolerance. These results show that the Ras-cAMP pathway is an important inhibitor of desiccation tolerance in exponential cells.
To test whether the Ras-cAMP pathway and the TOR pathway may have a synergistic effect on desiccation tolerance, we tested the same cyr1ts strain at 30°C with or without rapamycin added (Supplemental Figure S5B). Cells inhibited by both the Ras-cAMP and TOR pathways did not show a further increase in desiccation tolerance. This result suggests that these pathways target the same effectors to modulate desiccation tolerance.
Common targets of Tor and Ras/PKA pathways in desiccation tolerance
The targets of the Ras-cAMP and TOR pathways, PKA and Sch9p, respectively, regulate common targets, including Msn2p, Msn4p, Gis1p, Rim15p, and Sfp1p (Zaman et al., 2008). These common targets represent good candidates for regulators of desiccation tolerance. On the basis of our earlier results, we expected that factors inhibited by these two pathways might be positive effectors and those activated might be negative effectors of desiccation tolerance.
To investigate the role of these candidate targets in desiccation tolerance, we first assayed whether individual deletions of these genes impacted the desiccation tolerance of rapamycin-treated, exponential-phase cells (Figure 8B). Deletion of targets activated by TORC1, Sch9p, and Sfp1p had no effect on desiccation tolerance as expected. However, rim15Δ cells exhibited 100-fold reduced desiccation tolerance as compared with wild type. Rim15p regulates Msn2p, Msn4p, and Gis1p, thus we predicted that these proteins may be playing a role in desiccation tolerance. To test whether Msn2p, Msn4p, and Gis1p were needed for desiccation tolerance, we examined desiccation tolerance in the msn2∆ msn4∆ double mutant and gis1∆ single mutant (Figure 8B). Neither of these mutant strains showed a significant decrease in desiccation tolerance, which was not surprising as these transcription factors are thought to be partially redundant. To assess possible redundancy, we constructed a gis1∆ msn2∆ msn4∆ strain and tested it for rapamycin-induced desiccation tolerance (Figure 8B). This strain exhibited reduced rapamycin-induced desiccation tolerance by 100-fold similar to that seen in the rim15∆ strain, suggesting that Rim15p was acting through MSN2, MSN4, and GIS1. These results taken together suggest that Rim15p activates Msn2p, Msn4p, and Gis1p, which in turn induce expression of common stress effectors of desiccation tolerance. By inhibiting Rim15p, TORC1 and the Ras-cAMP pathway inhibit the activation of Msn2p, Msn4p, and Gis1p and their common targets of stress effectors.
FIGURE 8:

Common effectors of PKA and Sch9p affect desiccation tolerance. (A) Genetic pathways model depicting PKA and Sch9p's downstream effectors. (B) Exponential-phase cells grown in rich medium with rapamycin of the indicated genotype were assayed for desiccation tolerance as described in Figure 4A. (C) Exponential-phase cells carrying a vector expressing MSN2, MSN4, GIS1, or an empty-vector control were grown in YEP with 2% galactose for 20 h and then assayed for desiccation tolerance. (D) Exponential-phase cultures of the indicated genotype grown in rich medium were assayed for desiccation tolerance.
This model predicts that overexpression of MSN2, MSN4, or GIS1 might bypass the inhibitory effects of TORC1 and the Ras-cAMP pathway's activities on Rim15p in exponential-phase cells and allow desiccation tolerance. To test this prediction, we introduced plasmids into wild-type cells to overexpress MSN2, MSN4, or GIS1 under control of the galactose promoter. Strains bearing these plasmids were grown to exponential phase in rich medium containing galactose and were compared with the strain carrying the vector alone. Strains expressing these transcription factors exhibited increased desiccation tolerance 100- to 1000-fold, as compared with strains carrying the empty vector, to an absolute level 10-fold less than a stationary-phase culture (Figure 8C). These results taken together suggest that Ras-cAMP and TORC1 signaling through PKA and Sch9p, respectively, inhibit Rim15p and the activity of Msn2p, Msn4p, and Gis1p to block desiccation tolerance.
Sch9p and PKA not only inhibit these positive effectors, they also enhance transcription of SFP1. Sfp1p is a positive regulator of ribosome biogenesis. Thus we hypothesized that Sfp1p activation by Sch9p and PKA in exponential-phase cells inhibits desiccation tolerance. If so, deletion of SFP1 should partially induce desiccation tolerance in exponential cells. Indeed, in the absence of rapamycin, sfp1∆ exponential-phase cells are 1000-fold more desiccation tolerant than are wild-type cells and grow more slowly (3.9 ± 0.5 h; Figure 8D). Although this induction is significant, it does not reach the same level of desiccation tolerance seen in wild-type or sfp1∆ cells treated with rapamycin (Figure 8B), consistent with our observations that TORC1 inhibits desiccation tolerance by other means as well–in particular, by inhibiting expression of Msn2p, Msn4p, and Gis1p.
To explain the role of Sfp1p in desiccation tolerance, we first examined gene expression profiles. Unlike cells treated with rapamycin, cells containing a deletion of SFP1 do not exhibit a robust increase in stress tolerance genes except for two outliers–the small HSPs Hsp12 and Hsp26 (Marion et al., 2004). On the basis of these data, we hypothesized that the increase in desiccation tolerance in sfp1∆ cells may be dependent on Hsp12p and Hsp26p. To test this hypothesis, we constructed strains containing deletions of these genes in combination with sfp1∆ and tested these strains for desiccation tolerance in exponential phase (Supplemental Figure S6). Strains lacking SFP1, HSP12, and HSP26 showed no decrease in desiccation tolerance as compared with the sfp1∆ strain. This finding indicates that the increase in desiccation tolerance of sfp1∆ strains is not dependent on the small HSPs Hsp12 and Hsp26.
We next asked whether the sfp1∆ strain exhibited increased desiccation tolerance by reducing translation. To test this possibility, we grew wild-type cells in low amounts of cycloheximide and monitored desiccation tolerance over the course of 6 h (Figure 9A). At the highest concentration tested (1 μg/ml), translation was inhibited sufficiently to reduce the cell division rate to 12.5 ± 2 h. Nonetheless, slow growth and inhibition of translation did not increase desiccation tolerance over the course of 6 h. This result suggests that the inhibition of ribosome biogenesis, and not translation per se, is an important contributor to desiccation tolerance.
FIGURE 9:

Decreases in ribosome biogenesis can increase desiccation tolerance. (A) Wild-type cells grown in rich medium were transferred to the same medium containing the indicated concentrations of cycloheximide (CHX) and were assayed for desiccation tolerance at the indicated time points. (B) Strains containing a temperature-sensitive allele of a ribosome biogenesis gene or the wild-type strain were grown in rich medium to exponential phase at 23°C, shifted to 33°C for 6 h, then assayed for desiccation tolerance and growth rate. Presented here is only the desiccation tolerance. (C) Here the same data for desiccation tolerance are shown as in (B), but they are now plotted against the doubling time. The wild-type strain has a striped box, whereas all mutant strains have a solid black box. A logarithmic-regression line was fitted to these data. The equation for this line is y = 0.0093e–0.337x, and the R2 value of this line is displayed.
Next we wanted to test more directly whether inhibition of ribosome biogenesis could increase desiccation tolerance. To do this, we obtained 41 strains containing temperature-sensitive alleles of ribosome biogenesis genes. These strains were grown to exponential phase at a permissive temperature (23°C), transferred to a semipermissive temperature (33°C), and then assayed for desiccation tolerance (Figure 9B). These strains exhibited a range of desiccation tolerance, from wild-type levels up to 40,000-fold greater. The robust tolerance in these mutants compared with that in the sfp1∆ strain indicates that ribosome biogenesis may account for all of the tolerance induced by the inactivation of Sfp1.
The fact that only a subset of mutants exhibited high desiccation tolerance had two possible explanations. One is that, at the semipermissive temperature, mutants that compromised ribosome function the most became the most tolerant. Alternatively, the tolerant subset of mutants may have compromised specific components or steps in ribosome biogenesis that modulated desiccation tolerance. To test the former explanation, we examined the correlation between desiccation tolerance and growth rate of our mutants at the semipermissive temperature, assuming that growth rate was a proxy for ribosome function. Therefore we would expect the most desiccation-tolerant strains to grow the slowest. However, comparison of growth rate with desiccation tolerance of these strains does not support this hypothesis (Figure 9C). Desiccation tolerance did not increase with slower growth rate at the semipermissive temperature. In fact, many mutants exhibiting >1000-fold increases in desiccation tolerance were in the quartile with the fastest growth rate, whereas no mutants with >80-fold increases in desiccation tolerance were found in the slowest growing quartile. Indeed, based on the logarithmic regression line fitted to these data, there is no correlation between growth rate and desiccation tolerance (R2 = 0.037). These data provide the strongest evidence yet that specific components or steps in ribosome biogenesis modulate desiccation tolerance. Furthermore these mutants corroborate our previous observations with osmotic, redox, and ionic stress in which desiccation tolerance does not correlate with slow growth.
To begin to understand the specific components or steps in ribosome biogenesis that modulate desiccation tolerance, we categorized the mutants by their function in general ribosome biogenesis or specifically in large 60S or small 40S biogenesis (Supplemental Figure S7). The most desiccation-tolerant mutants were highly enriched for defects in 60S biogenesis. The fact that other mutants defective in 60S biogenesis grew slower but had much lower tolerance indicated that any defect in 60S biogenesis was not sufficient to induce tolerance. Thus, to investigate the roles of these genes more specifically, we used AmiGo (version 1.8) to search for enrichment of Gene Ontology (GO) categories in the most desiccation-tolerant mutants. This examination revealed genes involved in 5.8S rRNA maturation (Figure 9C, black boxes). This process is a critical step in the biogenesis of the 60S subunit. We were unable to find enrichment for any other GO categories specific to any other quartiles.
DISCUSSION
Here we have shown that the desiccation sensitivity of exponential-phase cells is not altered by the addition of cells and/or medium from a desiccation-tolerant, stationary-phase culture. These results suggest that, at least in a liquid culture, the acquisition of desiccation tolerance occurs through a cell-autonomous mechanism. The cell autonomy of desiccation tolerance is consistent with the observed heterogeneity of desiccation tolerance among cells in a given culture. This heterogeneity may allow a culture to contain at least some cells poised and ready for emergencies, such as rapid water loss, while the remainder of the culture continues to grow and divide as rapidly as possible. Whether or how a cell senses water availability to make the autonomous decision to become desiccation tolerant or sensitive is not clear. One clue may be that desiccation tolerance is coupled to nutrient limitation (Calahan et al., 2011). We hypothesize that nutrients normally serve as a proxy for water abundance in nature. Yeast are immobile; they acquire nutrients largely by diffusion through their aqueous environment. Thus nutrient limitation may often occur as a consequence of evaporation, leaving a yeast colony in isolated water droplets separated from the nutrient source, and serve as a harbinger of impending drought and desiccation.
Our study reveals that the exposure of exponentially growing yeast cells to different environmental stresses gives a graded induction of desiccation tolerance. Nutrient limitation or exposure to heat stress increased desiccation tolerance 100,000- and 5000-fold, respectively. These observations corroborate a previous study, although our study produced effects several hundredfold greater, likely due to the improved sensitivity of the assay we developed (Ratnakumar and Tunnacliffe, 2006; Calahan et al., 2011). Our results indicate that nutrient limitation, heat, and desiccation produce common (or shared) stresses that are mitigated by common (or shared) stress factors. This connection is strengthened greatly by the observation that tolerance to nutrient limitation, heat, and desiccation are all induced by inhibition of TORC1 (Bandhakavi et al., 2008). As the damage from heat stress is thought largely to result from protein misfolding and aggregation, this connection suggests that protein misfolding and aggregation are stresses limiting desiccation tolerance. We previously showed that HSP104 was not needed for desiccation tolerance in stationary-phase cultures (Calahan et al., 2011). Our current study suggests that we should revisit HSP function in desiccation tolerance in the context of exponential-phase cells.
In contrast to heat stress, pretreatment of cells with other stresses like osmotic, reductive, oxidative, and salt minimally (if at all) increased desiccation tolerance. This finding is surprising on two accounts. First, many studies have assumed that these stresses must be responsible for desiccation sensitivity (Franca et al., 2005). Our observations suggest that the level of osmotic and salt stresses that occur during desiccation may be unimportant to cell viability. Indeed, we previously showed that removing oxidative stress by desiccating in the absence of oxygen did not increase desiccation tolerance in exponential-phase cultures (Calahan et al., 2011). Alternatively, the levels or activities of the effectors responding to these stresses may be already sufficiently high in unstressed exponential cells to mitigate these stresses during desiccation.
Second, the inability of stresses such as osmotic, reductive, and salt to provide cross-protection to desiccation has important implications for the ESR. The concept of an ESR is derived from observations that many different stresses cause a common change in the pattern of gene expression (Lu et al., 2009). Despite this common change in gene expression, the failure to observe universal cross-protection here and in a previous study (Berry and Gasch, 2008) suggests that cells suffering different stresses must be in different physiological states. Therefore, although the gene products of the ESR may be necessary for protection against all stresses, they are clearly not sufficient. Identifying the factors important for desiccation tolerance and heat shock, be they within or outside of the ESR, remains an important goal.
We also show that exponential cells can become desiccation tolerant by the inhibition of Ras-cAMP as well as TORC1 activity. The regulation of desiccation tolerance by these factors is concordant with their key roles as regulators of cell metabolism in response to environmental changes. We identify common targets of Ras/cAMP/PKA and TOR/Sch9 as potential effectors of desiccation tolerance. Msn2p and Msn4p were likely candidates for stress response factors; however, demonstrating a causal role for them in desiccation tolerance was problematic because knockouts were only mildly reduced in desiccation tolerance (Calahan et al., 2011). Here we provide more compelling phenotypic evidence for causality. First, deletion of RIM15, a regulator of Msn2p, Msn4p, and Gis1p, gives a 100-fold reduction in desiccation tolerance. Second, we show that overexpression of any one of the three effectors of Rim15p induces significant desiccation tolerance in the culture. Third, we show that deletion of all three of these genes results in a significant reduction in rapamycin-induced desiccation tolerance. By establishing a significant redundant role for these three transcription factors in desiccation tolerance, assessing their target genes for desiccation tolerance effectors becomes a higher priority.
An even more interesting observation from our studies of Ras-cAMP and TORC1 pathways is that they inhibit desiccation tolerance by activating Sfp1p. Sfp1p seems to function almost completely in ribosome biogenesis rather than repressing known stress response factors (except for Hsp12 and Hsp26) based on gene expression profiles of strains deleted for SFP1 (Marion et al., 2004). Despite the exciting link between the increase in desiccation tolerance due to heat shock and the fact that HSPs are induced in an sfp1∆ strain, we were unable to uncover any role for HSPs in desiccation tolerance thus far.
The hypothesis that the down-regulation of ribosome biogenesis in the sfp1∆ strain enhances desiccation tolerance is strongly supported by our ability to phenocopy this tolerance with mutations in a subset of ribosome biogenesis components. Moreover, this subset is highly enriched for mutants compromised in 60S biogenesis and particularly the 5.8S rRNA maturation step. Interestingly, the abundance of 60S subunits in Caenorhabditis elegans and S. cerevisiae has been implicated as an important factor in the stress-associated process of aging (Hansen et al., 2007; Steffen et al., 2008). We speculate that assembly intermediates of the 60S subunit induce stress upon desiccation perhaps directly through their aggregation or indirectly through their competition for chaperones that are needed to protect other proteins. This propensity to aggregate could be counteracted by increases in chaperone abundance, thereby explaining one mechanism of how transcriptional activators of chaperones such as Msn2, Msn4, Gis1, and heat shock may also facilitate desiccation tolerance. Thus reduction of specific ribosome intermediates and increased chaperones would cooperate to increase desiccation tolerance.
The connection between desiccation tolerance and ribosome biogenesis informs on the inconsistent relationship between desiccation tolerance and growth rate. We show here that tolerance correlates with slow growth imposed by nutrient deprivation but not with slow growth imposed by osmotic, salt, or redox stresses. This conundrum can be resolved by postulating that stresses or mutants can grow slowly by different mechanisms and result in different physiological states. Only growth conditions that lead to a reduction in the abundance of specific ribosome intermediates will increase desiccation tolerance. This model predicts that the slow growth associated with hyperosmotic or oxidative stress will behave like the 40S mutants and not decrease the abundance of a specific ribosome intermediate, whereas stresses such as heat shock or nutrient deprivation will behave like 60S mutants and cause a decrease in the abundance of this important ribosome intermediate.
The regulation of desiccation tolerance by TOR and RAS pathways in yeast is intriguing given their evolutionary conservation in all eukaryotes. The role of TOR and RAS pathways in desiccation tolerance has not yet been investigated in other organisms. However, studies in the moss Physcomitrella patens show that abscisic acid (ABA) induces desiccation tolerance and may inhibit TOR in Arabidopsis thaliana (Deprost et al., 2007; Khandelwal et al., 2010). A mutant in DAF-2 causing C. elegans to arrest in the dauer larva stage is desiccation tolerant (Erkut et al., 2011). DAF-2 regulates expression of a component of CeTORC1 (DAF-15), and deletion of DAF-15 or the C. elegans homolog of TOR1 (let-363) results in developmental arrest in a dauer-like stage (Jia et al., 2004). On the basis of these examples, we suggest that TOR pathways in plants and animals may inhibit desiccation tolerance and that down-regulation of TOR activity may increase desiccation tolerance possibly via the mechanism just described. Collectively these studies make the TOR and RAS pathways appealing targets for agricultural or medical research (e.g., for the purpose of developing drought-resistant crops or improving blood storage).
MATERIALS AND METHODS
Strain construction
Standard methods were used for strain construction (Burke et al., 2000). The Ras2a22-containing vector (pPHY2701) was a gift from Paul Herman (Department of Molecular Genetics, Program in Molecular, Cellular and Developmental Biology, The Ohio State University, Columbus, OH; Budovskaya et al., 2004). The vectors containing Bcy1 (HTP20, HTP21) were a gift from Kevin Morano (Department of Microbiology and Molecular Genetics, University of Texas Medical School at Houston, Houston, TX). The tor1-1 allele was amplified from a plasmid given to us by Ted Powers (Department of Molecular and Cellular Biology, College of Biological Sciences, University of California, Davis, Davis, CA), transformed into BY4742, and selected on plates containing rapamycin to obtain tor1-1–stable integrant. A galactose-inducible plasmid library was a gift from Rebecca Butcher from which the MSI1-containing strain was obtained (Butcher et al., 2006). All other galactose-inducible promoter containing plasmids came from the FLEXGene collection from Harvard Institute of Proteomics (Hu et al., 2007).
Yeast strains and plasmids used in this study are shown in Tables 1 and 2, respectively.
TABLE 1:
Yeast strains used in this study.
| Name | Genotype | Source |
|---|---|---|
| AW6400 | MATα wild-type | This study |
| DBY12000 | MATa HAP1+ wild-type | Hickman et al., 2011 |
| BY4742 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 | American Type Culture Collection (ATCC) |
| AW6410 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 URA3 | This study |
| AW6413 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 sch9::LEU2 | This study |
| AW6421 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 URA3 PGAL1-MSI1 | This study |
| AW6423 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 HIS3 | This study |
| AW6430 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 URA3 PGAL1-MSN2 | This study |
| AW6431 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 URA3 PGAL1-MSN4 | This study |
| AW6433 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 ras2::G418R PMET3-Empty LEU2 | This study |
| AW6434 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 ras2::G418R PMET3-RAS2a22 LEU2 | This study |
| AW6473 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 msn2:: NATR msn4:: HYGR | This study |
| AW6481 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 PGAL1-PDE2 URA3 | This study |
| AW6500 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 rtg3:: NATR | This study |
| AW6507 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 sfp1:: NATR | This study |
| AW6510 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 gln3::NATR | This study |
| AW6514 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 gln3::NATR ure2::G418R | This study |
| AW6517 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 sch9::HYGR | This study |
| AW6518 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 atg17::HYGR | This study |
| AW6531 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 PGPD-BCY1 URA3 | This study |
| AW6532 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 PGPD-BCY1 HIS3 | This study |
| AW6540 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 msn2:: NATR msn4:: HYGR gis1:: G418R | This study |
| atg1Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 atg1::G418R | ATCC |
| atg13Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 atg13::G418R | ATCC |
| gal3Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 gal3::G418R | ATCC |
| gat1Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 gat1::G418R | ATCC |
| gis1Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 gis1::G418R | ATCC |
| rim15Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 rim15::G418R | ATCC |
| rtg3Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 rtg3::G418R | ATCC |
| sfp1Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 sfp1::G418R | ATCC |
| tor1Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 tor1::URA3 | This study |
| tor1-1 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 tor1-1 | This study |
| ure2Δ | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 ure2::G418R | ATCC |
| cyr1ts | MATα his3∆1 leu2∆0 met15∆0 ura3∆0 G418R cyr1-ts | Li et al., 2011 |
TABLE 2:
Plasmids used in this study.
| Name | Marker | Description | Promoter | Copy number | Source |
|---|---|---|---|---|---|
| pPHY2701 | LEU2 | PMET3-Ras2a22 | Met3 | Centromeric | Budovskaya et al., 2004 |
| pAW14 | LEU2 | PMET3-Empty | Met3 | Centromeric | This study |
| pAW15 | URA3 | PGAL1-MSN2 | Gal1-10 | Centromeric | This study |
| pAW16 | URA3 | PGAL1-MSN4 | Gal1-10 | Centromeric | This study |
| pGal-GIS1 | URA3 | PGAL1-GIS1 | Gal1-10 | Centromeric | Harvard Institute of Proteomics FLEXGene Collection |
| pBY011 | URA3 | PGAL1-Empty | Gal1-10 | Centromeric | Harvard Institute of Proteomics FLEXGene Collection |
| pGal-MSI1 | URA3 | PGAL1-MSI1 | Gal1-10 | Centromeric | Harvard Institute of Proteomics FLEXGene Collection |
| pGal-PDE2 | URA3 | PGAL1-PDE2 | Gal1-10 | Centromeric | Harvard Institute of Proteomics FLEXGene Collection |
| pRS303 | HIS3 | HIS3 | None | Integrating | LabLife™ |
| HTP21 | HIS3 | PGPD-BCY1 | GAPDH | Centromeric | Trott et al., 2005 |
| HTP22 | HIS3 | PGPD-BCY1 | GAPDH | Centromeric | Trott et al., 2005 |
Media and growth conditions
Unless otherwise stated, rich medium signifies yeast extract, peptone, and dextrose (YPD), which was made with Bacto Yeast Extract at 10 g/l (Becton Dickinson, Franklin Lakes, NJ) and Bacto Peptone at 20 g/l (Becton Dickinson) and dextrose at 20 g/l (Thermo Fisher Scientific, Waltham, MA). SC medium was prepared as recommended in Burke et al. (2000). Dithiothreitol (DTT; Sigma-Aldrich, St. Louis, MO) was used at a concentration of 2 mM. Hydrogen peroxide (H2O2; Sigma) was used at a concentration of 250 μM. Rapamycin (Sigma) was used at a concentration of 250 ng/ml. G418 sulfate (Invitrogen Life Technologies, Carlsbad, CA) was used in plates or liquid media at 200 μg/ml. Log-phase cultures were inoculated as a single colony, or from a liquid culture kept at 4°C, into 5 ml of liquid medium in a 20-ml glass tube at a cell density such that overnight incubation yielded a cell density less than OD600 = 0.6. This culture was then used as the zero time point for subsequent experiments that day. Stationary-phase cultures were made the same way, except that they were left to incubate at 30°C for 2–7 d. Cultures used for time courses were maintained at an OD600 < 0.6 by dilution with fresh medium throughout the time course. Control-cell viabilities were >10% unless otherwise mentioned. The strain BY4742 is referred to as wild type. All experiments were repeated at least two times on separate days with separate isolates when appropriate.
Gene expression changes
RNA was extracted from cells using an RNeasy Kit (Qiagen, Chatsworth, CA) and digested with DNase, and then quantitative reverse-transcription PCR (qRT-PCR) was performed using SYBR Green and Opticon Monitor (Geneworks, Hindmarsh, Australia).
Chemostat
All cultures were monitored for changes in cell density and dissolved oxygen and were grown until these values remained steady for at least 24 h. To achieve different rates of growth, the frequency of medium addition containing a defined limiting nutrient was modified (Monod, 1950; Novick and Szilard, 1950). For example, to achieve slower growth rates, less medium was added per unit of time, resulting in greater nutrient deprivation. Chemostat medium, which is essentially standard minimal medium with minor changes, was prepared as described in Brauer et al. (2008) with some modifications. Tables 3 and 4 list the components of each chemostat medium formulation.
TABLE 3:
Composition of synthetic medium used in continuous cultures.
| Glucose-limited medium | Phosphate-limited medium | Nitrogen-limited medium |
|---|---|---|
| 1X vitamins* | 1X vitamins* | 1X vitamins* |
| 1X metals** | 1X metals** | 1X metals** |
| 0.8 g/l glucose (4.4 mM) | 20.0 g/l glucose (111 mM) | 20.0 g/l glucose (111 mM) |
| 1.0 g/l CaCl2 (6.8 mM) | 1.0 g/l CaCl2 (6.8 mM) | 1.0 g/l CaCl2 (6.8 mM) |
| 1.0 g/l NaCl (17.1 mM) | 1.0 g/l NaCl (17.1 mM) | 1.0 g/l NaCl (17.1 mM) |
| 5.0 g/l MgSO4 (20.3 mM) | 5.0 g/l MgSO4 (20.3 mM) | 5.0 g/l MgSO4 (20.3 mM) |
| 50.0 g/l (NH4)2SO4 (378.4 mM) | 50.0 g/l (NH4)2SO4 (378.4 mM) | 10.0 g/l KH2PO4 (73.5 mM) |
| 10.0 g/l KH2PO4 (73.5 mM) | 1.0 g/l KCl (13.4 mM) | 40.0 mg/l (NH4)2SO4 (303 μM) |
| 20.0 mg/l KH2PO4 (147 μM) |
The limiting nutrient is shown in bold. See Table 4 for asterisk descriptions.
TABLE 4:
Composition of vitamins or metals used in continuous cultures.
| *1X vitamins (prepared as 1000X stock) | **1X metals (prepared as 1000X stock) |
|---|---|
| 8.19 nM biotin | 8.09 μM boric acid |
| 0.86 μM calcium panthothenate | 0.16 μM copper sulfate |
| 4.53 nM folic acid | 0.60 μM potassium iodide |
| 11.1 μM inositol (myo-inositol) | 0.74 μM ferric chloride |
| 3.25 μM niacin (nicotinic acid) | 2.37 μM manganese chloride |
| 1.46 μM p-aminobenzoic acid | 0.83 μM sodium molybdate |
| 1.95 μM pyridoxine HCl | 1.39 μM zinc sulfate |
| 0.53 μM riboflavin | |
| 1.19 μM thiamine HCl |
Determination of cell viability
Controls and samples containing 4 × 107 cells were resuspended in 200 μl of dilute PBS or YPD, respectively, in a 96-well tissue culture plate (Becton Dickinson Labware) and serially diluted 1:10 vol/vol seven times in successive wells containing 180 μl of YPD. Tissue culture plates were placed at 30°C for 2–3 d until individual colonies were visible in the most dilute well. Because we were diluting 1:10, the typical number of colonies counted in the final well was <10. These colonies were counted and multiplied by the appropriate factor for total viable cells.
Data were entered into a spreadsheet (Microsoft Excel 2012 for Mac, Redmond, WA), and the “% Desiccation Tolerance” or “% Viability” of a sample was determined by dividing the viable colonies for the treated (desiccated, heat shocked) sample by the viable colonies for the untreated (control) sample. Averages and standard deviations were computed using the AVERAGE and STDEV worksheet functions, respectively.
Cultures grown in YEP+galactose to overexpress a gene on a vector were not under selective pressure to maintain plasmids, and thus some of them were lost. We measured desiccation tolerance in these cultures based on the number of cells retaining the plasmid, which we could determine by their ability to grow in selective medium.
Desiccation tolerance assay
Approximately 108 cells were withdrawn from a liquid culture and washed twice in 1 ml of dilute PBS (a stock solution of 50X PBS was diluted to 1/8X with deionized reverse-osmosis water) then brought to a final volume of 500 μl. Then a 200-μl aliquot was transferred to a 1.5-ml microcentrifuge tube and pelleted at 14,000 rpm in a microcentrifuge. Supernatant was aspirated or decanted, and the tubes were placed into a vacuum desiccator (Centrivap Concentrator; Labconco, Kansas City, MO) connected to a rotary vane vacuum pump (Labconco) producing 67 kPa of vacuum at 35°C. The vacuum centrifuge was operated, without added heat, for 12–36 h. Samples were removed and resuspended in YPD, and diluted as described earlier in the text. For the undesiccated controls, 200-μl aliquots were diluted in 96-well plates as described earlier in the text.
Supplementary Material
Acknowledgments
Support for A. W. and D. K. is from the Howard Hughes Medical Institute. P. G. is supported by a National Institutes of Health (NIH) National Research Service Award (GM097852). D. B. is supported by NIH R01 (GM046406) and by the National Institute of General Medical Sciences Center for Quantitative Biology (GM071508). We thank Donna Storton and Jessica Buckles in the Princeton Microarray Facility for allowing us to use their equipment. We also thank Nathaniel Krefman, Lamia Wahba, Thomas Eng, Jeremy D. Amon, Hugo Tapia, Gamze Çamdere, Anjali D. Zimmer, Jeremy Thorner, and Vincent Guacci for critical reading and advice on the manuscript.
Abbreviations used:
- ABA
abscisic acid
- GO
gene ontology
- HSP
heat shock protein
- NCR
nitrogen catabolite repression
- PKA
protein kinase A
- TORC1
TOR complex 1
- YPD
yeast extract, peptone, and dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-07-0524) on November 21, 2012.
REFERENCES
- Bandhakavi S, Xie H, O'Callaghan B, Sakurai H, Kim DH, Griffin TJ. Hsf1 activation inhibits rapamycin resistance and TOR signaling in yeast revealed by combined proteomic and genetic analysis. PloS One. 2008;3:e1598. doi: 10.1371/journal.pone.0001598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DB, Gasch AP. Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol Biol Cell. 2008;19:4580–4587. doi: 10.1091/mbc.E07-07-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billi D, Potts M. Life and death of dried prokaryotes. Res Microbiol. 2002;153:7–12. doi: 10.1016/s0923-2508(01)01279-7. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budovskaya YV, Stephan JS, Reggiori F, Klionsky DJ, Herman PK. The Ras/cAMP-dependent protein kinase signaling pathway regulates an early step of the autophagy process in Saccharomyces cerevisiae. J Biol Chem. 2004;279:20663–20671. doi: 10.1074/jbc.M400272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Dawson D, Stearns T. Plainview, NY: Cold Spring Harbor Laboratory Press; 2000. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. [Google Scholar]
- Butcher RA, Bhullar BS, Perlstein EO, Marsischky G, LaBaer J, Schreiber SL. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat Chem Biol. 2006;2:103–109. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]
- Calahan D, Dunham M, Desevo C, Koshland DE. Genetic analysis of desiccation tolerance in Saccharomyces cerevisiae. Genetics 189, 507–519. 2011 doi: 10.1534/genetics.111.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabortee S, Boschetti C, Walton LJ, Sarkar S, Rubinsztein DC, Tunnacliffe A. Hydrophilic protein associated with desiccation tolerance exhibits broad protein stabilization function. Proc Natl Acad Sci USA. 2007;104:18073–18078. doi: 10.1073/pnas.0706964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fink GR. Feedback control of morphogenesis in fungi by aromatic alcohols. Genes Dev. 2006;20:1150–1161. doi: 10.1101/gad.1411806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe JH, Hoekstra FA, Crowe LM. Anhydrobiosis. Annu Rev Physiol. 1992;54:579–599. doi: 10.1146/annurev.ph.54.030192.003051. [DOI] [PubMed] [Google Scholar]
- Deprost D, Yao L, Sormani R, Moreau M, Leterreux G, Nicolai M, Bedu M, Robaglia C, Meyer C. The Arabidopsis TOR kinase links plant growth, yield, stress resistance and mRNA translation. EMBO Rep. 2007;8:864–870. doi: 10.1038/sj.embor.7401043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkut C, Penkov S, Khesbak H, Vorkel D, Verbavatz JM, Fahmy K, Kurzchalia TV. Trehalose renders the dauer larva of Caenorhabditis elegans resistant to extreme desiccation. Curr Biol. 2011;21:1331–1336. doi: 10.1016/j.cub.2011.06.064. [DOI] [PubMed] [Google Scholar]
- Franca MB, Panek AD, Eleutherio EC. The role of cytoplasmic catalase in dehydration tolerance of Saccharomyces cerevisiae. Cell Stress Chaperon. 2005;10:167–170. doi: 10.1379/CSC-103R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Herrero E, Ros J, Belli G, Cabiscol E. Redox control and oxidative stress in yeast cells. Biochim Biophys Acta. 2008;1780:1217–1235. doi: 10.1016/j.bbagen.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Hickman MJ, Petti AA, Ho-Shing O, Silverman SJ, McIsaac RS, Lee TA, Botstein D. Coordinated regulation of sulfur and phospholipid metabolism reflects the importance of methylation in the growth of yeast. Mol Biol Cell. 2011;22:4192–4204. doi: 10.1091/mbc.E11-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S. Control of high osmolarity signalling in the yeast Saccharomyces cerevisiae. FEBS Lett. 2009;583:4025–4029. doi: 10.1016/j.febslet.2009.10.069. [DOI] [PubMed] [Google Scholar]
- Hu Y, et al. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 2007;17:536–543. doi: 10.1101/gr.6037607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Johnston SD, Enomoto S, Schneper L, McClellan MC, Twu F, Montgomery ND, Haney SA, Broach JR, Berman J. CAC3(MSI1) suppression of RAS2(G19V) is independent of chromatin assembly factor I and mediated by NPR1. Mol Cell Biol. 2001;21:1784–1794. doi: 10.1128/MCB.21.5.1784-1794.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal A, Cho SH, Marella H, Sakata Y, Perroud PF, Pan A, Quatrano RS. Role of ABA and ABI3 in desiccation tolerance. Science. 2010;327:546. doi: 10.1126/science.1183672. [DOI] [PubMed] [Google Scholar]
- Li Z, et al. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol. 2011;29:361–367. doi: 10.1038/nbt.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Brauer MJ, Botstein D. Slow growth induces heat-shock resistance in normal and respiratory-deficient yeast. Mol Biol Cell. 2009;20:891–903. doi: 10.1091/mbc.E08-08-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O'Shea EK. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc Nat Acad Sci USA. 2004;101:14315–14322. doi: 10.1073/pnas.0405353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- Monod J. La technique de culture continue, theorie et applications. Annales Institut Pasteur. 1950;79:390–410. [Google Scholar]
- Novick A, Szilard L. Description of the chemostat. Science. 1950;112:715–716. doi: 10.1126/science.112.2920.715. [DOI] [PubMed] [Google Scholar]
- Potts M. Desiccation tolerance of prokaryotes. Microbiol Rev. 1994;58:755–805. doi: 10.1128/mr.58.4.755-805.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers S, O'Neill K, Wigler M. Dominant yeast and mammalian RAS mutants that interfere with the CDC25-dependent activation of wild-type RAS in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:390–395. doi: 10.1128/mcb.9.2.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumar S, Hesketh A, Gkargkas K, Wilson M, Rash BM, Hayes A, Tunnacliffe A, Oliver SG. Phenomic and transcriptomic analyses reveal that autophagy plays a major role in desiccation tolerance in Saccharomyces cerevisiae. Mol Biosyst. 2011;7:139–149. doi: 10.1039/c0mb00114g. [DOI] [PubMed] [Google Scholar]
- Ratnakumar S, Tunnacliffe A. Intracellular trehalose is neither necessary nor sufficient for desiccation tolerance in yeast. FEMS Yeast Res. 2006;6:902–913. doi: 10.1111/j.1567-1364.2006.00066.x. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- Singh AB, Harris RC. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal. 2005;17:1183–1193. doi: 10.1016/j.cellsig.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Sobering AK, Watanabe R, Romeo MJ, Yan BC, Specht CA, Orlean P, Riezman H, Levin DE. Yeast Ras regulates the complex that catalyzes the first step in GPI-anchor biosynthesis at the ER. Cell. 2004;117:637–648. doi: 10.1016/j.cell.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Steffen KK, et al. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell. 2008;133:292–302. doi: 10.1016/j.cell.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Cameron S, Sass P, Zoller M, Scott JD, McMullen B, Hurwitz M, Krebs EG, Wigler M. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:1371–1377. doi: 10.1128/mcb.7.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott A, Shaner L, Morano KA. The molecular chaperone Sse1 and the growth control protein kinase Sch9 collaborate to regulate protein kinase A activity in Saccharomyces cerevisiae. Genetics. 2005;170:1009–1021. doi: 10.1534/genetics.105.043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunnacliffe A, Wise MJ. The continuing conundrum of the LEA proteins. Die Naturwissenschaften. 2007;94:791–812. doi: 10.1007/s00114-007-0254-y. [DOI] [PubMed] [Google Scholar]
- van Hoek P, van Dijken JP, Pronk JT. Effect of specific growth rate on fermentative capacity of baker's yeast. Appl Environ Microbiol. 1998;64:4226–4233. doi: 10.1128/aem.64.11.4226-4233.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoek P, van Dijken JP, Pronk JT. Regulation of fermentative capacity and levels of glycolytic enzymes in chemostat cultures of Saccharomyces cerevisiae. Enzyme Microb Technol. 2000;26:724–736. doi: 10.1016/s0141-0229(00)00164-2. [DOI] [PubMed] [Google Scholar]
- Verghese J, Abrams J, Wang Y, Morano KA. Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol Mol Biol Rev. 2012;76:115–158. doi: 10.1128/MMBR.05018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008;4:e13. doi: 10.1371/journal.pgen.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Madia F, Hu J, Ge H, Li LM, Longo VD. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet. 2009;5:e1000467. doi: 10.1371/journal.pgen.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces responds to nutrients. Annu Rev Genet. 2008;42:27–81. doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
- Zhu X, Demolis N, Jacquet M, Michaeli T. MSI1 suppresses hyperactive RAS via the cAMP-dependent protein kinase and independently of chromatin assembly factor-1. Curr Genet. 2000;38:60–70. doi: 10.1007/s002940000133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.