

BPGAP1 controls morphogenesis, migration, and ERK signaling by the concerted action of its multiple domains. Its BCH domain targets K-Ras and induces robust ERK activation and neuronal differentiation in a process antagonized by SmgGDS. The results highlight unique cross-talk of two regulators of GTPases in Ras/ERK signaling and differentiation.
Abstract
BPGAP1 is a Rho GTPase-activating protein (RhoGAP) that regulates cell morphogenesis, cell migration, and ERK signaling by the concerted action of its proline-rich region (PRR), RhoGAP domain, and the BNIP-2 and Cdc42GAP homology (BCH) domain. Although multiple cellular targets for the PRR and RhoGAP have been identified, and their functions delineated, the mechanism by which the BCH domain regulates functions of BPGAP1 remains unclear. Here we show that its BCH domain induced robust ERK activation leading to PC12 cell differentiation by targeting specifically to K-Ras. Such stimulatory effect was inhibited, however, by both dominant-negative mutants of Mek2 (Mek2-K101A) and K-Ras (K-Ras-S17N) and also by the small G-protein GDP dissociation stimulator (SmgGDS). Consequently SmgGDS knockdown released this inhibition and resulted in a superinduction of K-Ras activation and PC12 differentiation mediated by BCH domain. These results demonstrate the versatility of the BCH domain of BPGAP1 in regulating ERK signaling by involving K-Ras and SmgGDS and support the unique role of BPGAP1 as a dual regulator for Ras and Rho signaling in cell morphogenesis and differentiation.
INTRODUCTION
Ras and Rho small GTPases function as key molecular switches regulating cell growth, proliferation, differentiation, morphogenesis, and motility by impacting immediate cytoskeletal organization and long-term modulation of gene expression (Takai et al., 2001; Wennerberg et al., 2005; Bernards and Settleman, 2007). They cycle between an active GTP-bound form and an inactive GDP-bound form; activated by the guanine nucleotide exchange factors (GEFs), or inactivated by the GTPase-activating proteins (GAPs) or the guanine nucleotide dissociation inhibitors. Despite our good understanding of the individual signaling networks and control of Ras and Rho, little is known about how they are directly regulated by a single protein entity inside the cells and how that might impact important signaling pathways such as the Ras/ERK module that governs many aspects of cell physiology just mentioned.
BPGAP1 (BNIP-2 and Cdc42GAP homology [BCH] domain-containing, proline-rich and Cdc42GAP-like protein subtype-1) is a RhoGAP that targets the three main members of Rho GTPases—RhoA, Cdc42, and Rac1—in vitro but primarily inactivates RhoA inside the cells. Through the concerted action of its BCH and RhoGAP domains and proline-rich region (PRR), BPGAP1 induces extensive changes in cell morphologies and cell motility (Shang et al., 2003). This induction is mediated in part by the binding of cortactin to the PRR of BPGAP1 promoting their translocation to the cell periphery and to enhance cell migration (Lua and Low, 2004), whereas binding of EEN/endophilin2 to the same motif induces epidermal growth factor (EGF) receptor (EGFR) endocytosis, leading to Ras/ERK activation (Lua and Low, 2005). We further showed that this proline-rich moiety is the target of peptidylprolyl isomerase Pin1 and Mek2 that act as novel scaffold proteins in regulating Rho and ERK signaling (Pan et al., 2010). Interestingly, BPGAP1 also activates ERK1/2 independently of its interaction with EEN and Rho inactivation, perhaps implicating the involvement of its BCH domain in regulating this process (Lua and Low, 2005). Furthermore, our functional analyses of other closely related BCH domains reveal that this novel class of protein domain could act as regulatory scaffolds that engage Rho, Cdc42, RhoGEFs, or RhoGAPs in a context-dependent manner (Pan and Low, 2012; Pan et al., 2012). For example, the homologous BCH domain of BNIP-2, BNIP-Sα, and BNIP-XL can bind Cdc42, Rho, p115RhoGEF, or Lbc RhoGEF to regulate cell protrusions and cellular transformation (Zhou et al., 2005, 2006; Soh and Low, 2008), whereas the BCH domain of p50RhoGAP sequesters RhoA from being inactivated by the adjacent RhoGAP domain (Zhou et al., 2010). All these observations raise the possibility that the BCH domain of BPGAP1 might also target Ras and/or its immediate Ras regulator(s).
Here we show that BPGAP1 promotes ERK activation and neurite outgrowth in PC12 differentiation by involving K-Ras and small G-protein GDP dissociation stimulator (SmgGDS). SmgGDS, or RAP1GDS1, was first characterized as a Rap GTPase regulatory protein (Yamamoto et al., 1990), and it was believed to function like an atypical GEF, stimulating the dissociation of GDP from the small GTPases Cdc42, Rac1, RhoA, Rap1B, and K-Ras (Mizuno et al., 1991; Yaku et al., 1994). More recently, it was shown, with purified GTPases, that the GEF activity of SmgGDS is specific to RhoA and RhoC (Hamel et al., 2011). Unlike canonical GEFs, SmgGDS is not deterred from binding to the GTP-bound form of GTPases (Yamamoto et al., 1990; Kikuchi et al., 1992; Takai et al., 2001; Vikis et al., 2002). Here we show that knockdown of SmgGDS alone induced K-Ras activation and that this process was further augmented when BPGAP1 was overexpressed. However, overexpression of SmgGDS attenuated the ability of this BCH domain to interact with K-Ras, leading to inhibition of ERK1/2 activation and neuritogenesis during PC12 differentiation.
RESULTS
BCH domain of BPGAP1 promotes ERK activation and differentiation of PC12 cells
We previously showed that full-length BPGAP1 interacts with EEN, enhancing EGFR endocytosis and subsequent ERK1/2 phosphorylation in a manner dependent on its GAP activity, whereas its BCH domain (BPGAP1-BCH) alone induced chronic ERK1/2 phosphorylation in an EGFR endocytosis–independent manner (Lua and Low, 2005). To further delineate the mechanistic action of the BCH domain, an N-terminal region harboring the BCH domain (aa 1–168) (Figure 1A) was expressed in the rat pheochromocytoma cell line PC12, and its impacts on the strength and duration of ERK1/2 activation were examined under EGF stimulation. BPGAP1-BCH induced robust ERK1/2 activation that was sustained at a much higher level than the vector control for up to 15 min (Figure 1B). In PC12, increased amplitude and duration of ERK activation by EGF lead to differentiation instead of cell proliferation (Marshall, 1995). To correlate the impacts of BPGAP1-BCH on sustained ERK activation and cell physiology, mCherry-tagged BPGAP1 full-length, N-terminal BPGAP1-BCH fragment, the catalytically inactive mutant of full-length BPGAP1-R232A, or vector control was expressed individually in PC12 followed by stimulation of cells with EGF for 24 h (Figure 1C). Results show that cells expressing BPGAP1-BCH exhibited bipolar neurite structures reminiscent of nerve growth factor (NGF)-mediated differentiation of PC12, whereas the full-length protein induced multiple branched neurite protrusions. Interestingly, the catalytically inactive GAP mutant R232A exhibited morphology similar to that induced by BPGAP1-BCH (Figure 1D). These BPGAP1-mediated morphological changes were potent, with approximately 80–90% of transfected cells showing long protrusions in a background of ∼5% for vector control (Figure 1E). Consistent with their potency in inducing neurite outgrowth when stimulated with EGF, BPGAP1-BCH, BPGAP1, and BPGAP1-R232A also potentiated a similarly potent neurite outgrowth upon stimulation with a suboptimal level of NGF (10 ng/ml), which by itself did not induce PC12 differentiation (Figure 1, D and F). Data in Figure 1, E and F, are means of three independent experiments with fifty cells counted per construct per experiment.
FIGURE 1:

BPGAP1–BCH domain promotes differentiation of PC12 cells under EGF stimulation. (A) Schematic representation of BPGAP1 (containing the BCH domain [BCH], PRR [P], and GAP domain) and its constructs, BCH domain and arginine mutant, R232A; numbers are amino acids. (B) BCH causes sustained ERK1/2 activation upon EGF stimulation. PC12 cells transfected with BCH or vector controls were made quiescent before stimulation with EGF (100 ng/ml). Lysates were obtained at different time points and analyzed for levels of phosphorylated ERK (top panel) as described in Materials and Methods. Levels of total ERK (panERK) (middle panel) and tubulin (bottom panel) are also detected as loading controls. (C) PC12 cells transfected with mCherry-BCH, BPGAP1, BPGAP1-R232A, or vector constructs were made quiescent before stimulation with 100 ng/ml EGF and imaged every 30 min for 24 h. Representative images show the morphology of transfected cells over time. Scale bars, 20 μm. (D) PC12 cells were transfected with GFP vector, BCH, BPGAP1, or BPGAP1-R232A, made quiescent, and stimulated with either 100 ng/ml EGF or 10 ng/ml NGF. The observed morphological changes were consistent between the sets with BPGAP1-transfected cells showing multiple branched neurites and BCH- and BPGAP1-R232A–transfected cells showing long bipolar neurites. White arrowheads point to the tips of the neurite extensions. Scale bars, 20 μm. (E and F) Statistical analysis was carried out, counting 50 transfected cells for each construct and scoring for differentiation (neurite extensions longer than 2 cell body length), and data were plotted as mean of n = 3, p < 0.01, error bars represent SEM. (G) PC12 cells were transfected with Flag-BCH domain or Flag-vector, made quiescent, and stimulated with 100 ng/ml EGF for 48 h. Lysates were obtained at different time points and analyzed to detect phosphoERK and neuronal marker, GAP43. PanERK and tubulin were used as loading controls. Dotted line in second panel denotes missing lanes cut out from the same blot.
To further confirm that the effect of BPGAP1-BCH on PC12 extension was indeed a chronic ERK activation leading to a differentiation signal and not merely due to morphogenetic changes, we examined lysates from PC12 expressing BPGAP1-BCH for the induction profiles of ERK activation and the expression of the neuronal differentiation marker GAP43 (Figure 1G). Results show that the expression of BPGAP1-BCH alone increased the basal level of active ERK. Stimulation by EGF further enhanced and sustained ERK activation and stimulated the expression of GAP43 as early as 12 h, instead of 36 h as seen in the control cells. These results strongly indicate that the BCH domain promotes ERK activation leading to neurite outgrowth in PC12. To further confirm that BPGAP1-BCH induced PC12 differentiation via the Ras/Mek/Erk pathway, cells were treated with Mek inhibitor U0126 or cotransfected with plasmids expressing a kinase-dead mutant of Mek2 (Mek2-K101A), together with full-length BPGAP1 or BPGAP1-BCH, and their effects were examined under EGF stimulation. On inhibitor treatment, the characteristically long bipolar neurite extensions resulting from the action of BCH were greatly reduced in length (Figure 2A), with ∼85% of transfected cells showing this reduction (Figure 2B). Similarly, U0126 treatment in PC12 expressing full-length BPGAP1 also resulted in a significant reduction in the length of neurite outgrowth while retaining their branching phenotype (Figure 2C) with a similar ∼85% of transfected cells showing this reduction (Figure 2D). Furthermore, expression of Mek2-K101A with the BCH domain prevented any formation of neurite outgrowth (Figure 2E) again with ∼85% of transfected cells showing this reduction (Figure 2F). All statistical data (Figure 2, B, D, and F) are means of three independent experiments with 80–110 cells counted per construct per experiment. Taken together, these results revealed a novel role of the BCH domain in promoting the Ras/MAPK pathway, at least by activating the Mek2-ERK module, leading to PC12 differentiation.
FIGURE 2:

BCH domain–mediated differentiation of PC12 cells occurs via the Ras/MAPK pathway. PC12 cells transfected with BCH (A) and BPGAP1 (B) were made quiescent before treatment with dimethyl sulfoxide (DMSO; control) or U0126 (5 mm) either with or without EGF (100 ng/ml) for 48 h before they were processed by indirect immunofluorescence for confocal microscopy. (C) PC12 cells were cotransfected with BCH and Mek2-K101A, made quiescent, and stimulated with EGF (100 ng/ml) for 48 h before they were processed by indirect immunofluorescence for confocal microscopy. Red arrowheads point to the long bipolar neurites. The merged panel shows inhibition of BCH-mediated PC12 differentiation by Mek2-K101A with the white arrowheads pointing to lack of neurites. DIC, differential interference contrast. Scale bars, 20 μm. (D–F) Quantitative representation of the data depicted in A–C, respectively, as mean of n = 3, p < 0.01; error bars represent SEM.
BCH domain of BPGAP1 binds K-Ras and promotes its activation and neuritogenesis
As described earlier, other homologous BCH domains have been shown to regulate Rho and Cdc42 small GTPase activities inside the cells, by binding either to them or to their immediate regulators such as GEF or GAP. To explore the possible mechanisms by which BPGAP1-BCH activates the Mek-Erk signaling node, we elected to test whether this BCH domain could interfere with the activation of Ras small GTPases. This was achieved by screening for its ability to form a physiological complex with various forms of Ras GTPases (K-Ras, H-Ras, or N-Ras; Figure 3). Coimmunoprecipitation experiments in HEK293T show that the BCH domain of BPGAP1 was indeed capable of forming a weak, possibly transient interaction with H-Ras and N-Ras while it formed a much more stable complex with K-Ras (Figure 3A). Particularly, it was seen that the BCH domain preferentially bound the dominant-negative form of K-Ras, K-Ras-S17N (Figure 3B). Furthermore, coexpression of K-Ras-S17N with BCH was able to suppress the ability of BCH to promote neurite outgrowth in PC12 (Figure 3C) in approximately 86% of the transfected population of cells (Figure 3D; n = 3 with ∼100 cells counted per experiment). A K-Ras activation assay using Raf1–Ras-binding domain (RBD) as a reporter was carried out in HEK293T as described in Materials and Methods. The results revealed that BPGAP1-BCH promotes K-Ras activation by more than 80% (Figure 3, E and F). This increase of K-Ras activation was recapitulated with full-length BPGAP1 (Figure 3, G and H) with a more robust increase suggesting additional mechanisms of regulation by BPGAP1. All these results confirm our hypothesis that BCH activates K-Ras upon EGF stimulation, leading to PC12 differentiation.
FIGURE 3:

BCH domain of BPGAP1 targets K-Ras and promotes its activation. (A and B) Lysates from HEK293T cells expressing relevant FLAG- and HA-tagged constructs were immunoprecipitated (IP) with anti-FLAG beads and analyzed by SDS–PAGE and Western blotting. Appropriate antibodies were used to detect bound proteins (top panel) and loading control (bottom panel) and for verification of protein expression (middle panels). BCH domain interaction with Ras isoforms K-, H-, and N-Ras shows a strong interaction with the K-Ras isoform (A) with a preference to the dominant-negative S17N mutant (B). (C) PC12 cells overexpressing Flag-BCH and HA-K-Ras-S17N were made quiescent followed by 48 h of EGF (100 ng/ml) stimulation before they were processed by indirect immunofluorescence for confocal microscopy. K-Ras-S17N blocks bipolar neurites caused by the BCH domain. Scale bars, 20 μm. (D) Quantitative representation of the data depicted in (C) as mean of n = 3, p < 0.01, error bars represent SEM. (E and G) BCH domain (E) and full-length BPGAP1 (G) increase K-Ras activation. HEK293T cells expressing Flag-BCH (E), Flag-BPGAP1 (G), or vector control were made quiescent before stimulating with EGF (100 ng/ml) for 5 min. Lysates were incubated with Raf-1–RBD immobilized on beads for the K-Ras activation assay and subsequent SDS–PAGE and Western analysis as described in Materials and Methods. Bound active GTPases (top panel) and endogenous K-Ras protein expression were detected using K-Ras antibodies. Equal loading of GST fusion is observed by Amido Black staining. Experiments were performed in triplicate, and mean band intensities relative to control are plotted in (F) and (H) ±SEM, n = 3; p < 0.01.
SmgGDS as a Ras suppressor and novel partner of BCH domain
To establish the novel signaling network of BPGAP1, we had previously conducted proteomics pull-down assays and identified various protein partners of BPGAP1 (Lua and Low, 2004), one of which was SmgGDS isoform A. Because other BCH domain-containing proteins can also target regulators of small GTPases to modulate their activities, we further hypothesized that SmgGDS could be directly involved in modulating BPGAP1 and K-Ras signaling. The physiological interaction of SmgGDS with BPGAP1 was first established by coimmunoprecipitation studies that show that endogenous SmgGDS interacted with both Flag-tagged BCH and full-length BPGAP1 (Figure 4A). Interestingly, the interaction with the full-length protein was enhanced upon EGF stimulation, whereas the binding to BCH was already elevated at the basal state and remained constitutive, suggesting a possible intramolecular autoinhibition state of BPGAP1. SmgGDS has previously been known to possess GDP/GTP exchange activities toward Rho family small GTPases and K-Ras (Mizuno et al., 1991; Yaku et al., 1994). Its activity toward K-Ras inside the cells, however, remains unclear. To establish the physiological function of SmgGDS in K-Ras signaling, and specifically its role in BCH-mediated PC12 differentiation, short hairpin RNA (shRNA)-targeted SmgGDS depletion and its impact on Ras activation were examined upon EGF stimulation. Two shRNA oligonucleotides against SmgGDS were designed using small interfering RNA (siRNA) sequences published by Tew et al. (2008), and knockdown efficiency was determined by cotransfection with green fluorescent protein (GFP)-SmgGDS. When compared with the nontargeting negative control, both shRNA constructs (rS1 and rS2) successfully depleted SmgGDS by 60 and 50%, respectively (Figure 4B). The rS1 construct was then used for subsequent experimentation as it displayed a superior knockdown effect as compared with rS2. Using the Raf-1–RBD pulldown as the reporter for Ras activation, we saw that a knockdown of SmgGDS led to increased Ras and ERK activation upon EGF stimulation when compared with the negative control (Figure 4C). This result suggests that SmgGDS down-regulates K-Ras signaling and could have an antagonistic effect on the stimulatory role of BPGAP1 in Ras/ERK signaling.
FIGURE 4:

SmgGDS as a novel partner of BPGAP1 and suppressor of K-Ras signaling. (A) HEK293T cells were transfected with Flag-BPGAP1, vector, or BCH, made quiescent, and stimulated with 100 ng/ml of EGF. Lysates were obtained at indicated time points, subjected to immunoprecipitation (IP), and analyzed by Western blotting. Endogenous SmgGDS, bound to overexpressed BPGAP1 and BCH, was detected with anti-SmgGDS, and anti-Flag revealed equal loading of IP beads. (B) Efficiency of knockdown shRNA constructs rS1 and rS2 targeting SmgGDS were tested by transfecting HEK293T cells with shRNA constructs and HA-SmgGDS in a ratio of 1:10, and lysates were used to detect levels of SmgGDS (top panel). Tubulin was detected as a loading control (bottom panel). (C) Knockdown of SmgGDS increases K-Ras activation. HEK293T cells were transfected with pSilencer negative control or SmgGDS targeting rS1 for 48 h, made quiescent, and subjected to EGF stimulation at 100 ng/ml. Lysates obtained at indicated time points were subjected to the K-Ras activation assay.
SmgGDS antagonizes BPGAP1 activation of Ras and PC12 differentiation
Next we determined how SmgGDS could influence the action of BPGAP1 in PC12 differentiation, by coexpressing both SmgGDS and BCH. Figure 5A shows that SmgGDS inhibits BCH-mediated differentiation of PC12 as evidenced by the loss of neurites (red arrowheads) when compared with cells expressing BCH alone (white arrowheads). Statistical analysis of three independent experiments based on such overexpression studies show that the presence of SmgGDS significantly reduced the number of cells bearing protrusions, down by more than 70% (p < 0.01; Figure 5B). To further delineate how the tripartite interaction among SmgGDS, BPGAP1-BCH, and K-Ras could lead to modulation of K-Ras activity, we set out to determine how BCH domain could interfere with the interaction between SmgGDS and K-Ras. Flag-tagged SmgGDS was coexpressed with hemagglutinin (HA)-tagged K-Ras as well as with both HA-tagged K-Ras and HA-tagged BCH. The results show that SmgGDS binds to K-Ras. This binding was abrogated, however, when BPGAP1-BCH was present at the same time as K-Ras; SmgGDS switched its binding preference from K-Ras to BCH (Figure 5C). These results imply that, under specific stoichiometric conditions, BPGAP1-BCH is able to displace K-Ras effectively from being down-regulated by SmgGDS, thus eliciting its stimulatory effect. Alternatively, or simultaneously, SmgGDS could block BCH's stimulatory effect on K-Ras by sequestering BCH, thereby precluding K-Ras activation.
FIGURE 5:

SmgGDS antagonizes BCH domain–induced Ras/ERK activation and neuritogenesis in PC12 cell differentiation. (A) SmgGDS blocks BCH-induced differentiation of PC12 cells. PC12 cells transfected with BCH with or without SmgGDS were made quiescent and stimulated for 48 h with 100 ng/ml EGF before they were fixed for immunofluorescence analysis. Red arrows indicate double-transfected cells, and white arrows indicate single transfection. Scale bars, 20 μm. (B) SmgGDS reduces the percentage of transfected cells with BCH-induced protrusions by ∼70%. Cells were scored for long protrusions (> 2 cell body length) with at least 100 cells counted per sample per experiment. Experiments were performed in triplicate, and the means ± SEM (n = 3), p < 0.01, are plotted. Bars with different letters are highly significant. (C) SmgGDS binds preferentially to BCH over K-Ras, thereby removing BCH from activating K-Ras. HEK293T cells were transfected with SmgGDS and K-Ras either with or without BCH and subjected to IP. (D) SmgGDS blocks BCH activation of K-Ras. Cells transfected with K-Ras, BCH, SmgGDS, or BCH+SmgGDS were subjected to RBD analysis. The intensities of the bound active K-Ras were then quantified using ImageJ, and the means were plotted ± SEM (n = 3). Bars with different letters are significant at p < 0.01. (E) SmgGDS blocks BPGAP1 activation of endogenous K-Ras. Cells transfected with BPGAP1, SmgGDS, or BPGAP1+SmgGDS were subjected to RBD analysis. The intensities of the bound active K-Ras were then quantified using ImageJ, and the means were plotted ± SEM (n = 3). Bars with different letters are significant at p < 0.01. (F) Knockdown of SmgGDS allows a potent activation of K-Ras by BCH. HEK293T cells were transfected with pSilencer negative control (neg), pSilencer rS1 (rS1), singly or together with BCH, or pSilencer rS2 (rS2) together with BCH for 48 h before lysates were subjected to RBD assay. The lysates and bound proteins were then resolved by SDS–PAGE and subsequent Western analysis. Quantified signals were plotted as an average of three independent experiments, as fold of K-Ras activation over the (vector+neg) ± SEM (n = 3). Different letters indicate significance calculated by ANOVA; p < 0.01.
To further confirm this dynamic modulation, the RBD assay was performed in HEK293T expressing Flag-tagged K-Ras and BPGAP1-BCH either with or without overexpressing HA-SmgGDS. Results in Figure 5D show that although overexpression of BCH alone caused a ∼50% increase in K-Ras activation, this stimulatory effect was completely suppressed in the presence of SmgGDS. The presence of SmgGDS alone did not significantly affect the basal activity of K-Ras. This result implies that the binding of SmgGDS to BCH hinders the ability of BCH in activating K-Ras. SmgGDS was also able to inhibit the increase in K-Ras activation brought about by the full-length BPGAP1 (Figure 5E). Consequently it can be expected that the depletion of the endogenous level of SmgGDS would lead to a relief of SmgGDS suppression, and an augmentation of BCH-mediated K-Ras activation could ensue. Indeed, upon SmgGDS knockdown (with rS1), an overwhelming 10-fold increase in K-Ras activation was observed when the BCH domain was expressed in such a background, when compared to vector control (Figure 5F). This was verified by using another shRNA construct, rS2, targeting a secondary site on SmgGDS. The BCH-mediated K-Ras activation is also augmented in this case, albeit to a lesser extent, possibly due to the lower efficiency of knockdown of SmgGDS brought about by rS2 (Figure 4B). Consistently, knockdown of SmgGDS alone (without BCH) also produced a fourfold increase in K-Ras activation—almost double the amount of K-Ras activation brought about by BCH. This strongly indicates a possible preexisting blockage of K-Ras mediated by SmgGDS, thus preventing an otherwise robust stimulatory effect by BCH.
Knockdown of SmgGDS releases its inhibition on BCH and potentiates PC12 differentiation
To elucidate how the robust increase in K-Ras activation in the absence of endogenous SmgGDS would affect the extent and nature of PC12 neurite outgrowth (Figure 5, A and B), we monitored and quantified morphology changes in PC12 by live imaging. PC12 was transfected with either the shRNA-negative control or shRNA construct targeting SmgGDS, together with plasmids expressing the BCH domain. The processes of initiation, extension, and maturation of neurites were then monitored for 36 h after EGF stimulation. Differentiated cells from three independent experiments were scored for their neurite lengths. It can be seen that knockdown of SmgGDS resulted in a twofold increase in their final neurite lengths (Figure 6A). One representative cell from each experimental condition and time point (every 30 min for 36 h) was taken, and neurite length was measured. Representative images for the time course are shown in Figure 6B (all 36 h are represented in the movies in Supplemental Figure 1), whereas the graph in Figure 6C shows the neurite length at each time point. It can be seen that the rate of neurite outgrowth for each cell is different. The negative control (BCH+neg) shows a gradual increase at almost a constant rate throughout. The knockdown (BCH+rS1), in contrast, shows a sharp increase in the initial neurite length, as evidenced by the sharp incline of the slope representing a burst in the initial growth rate. To make valid comparisons between the two populations, cells from all three experiments were first represented as a distribution of its final neurite length in terms of cell body (Figure 6D). A normal distribution is observed for control and knockdown with an observable, distinct shift in the population mode from 3 cell-body (control) and 7 cell-body (knockdown) neurite length. Ten cells were then chosen from the pool, with the final neurite length equal to the mode. The neurite length for these cells was then measured every 5 h and plotted as shown in Figure 6E. This graph resembles the trend observed in Figure 6C for a single representative cell. It can therefore be concluded that control cells show a gradual initial increase in neurite length, whereas knockdown of SmgGDS not only advances the onset of differentiation but also shows a greater initial rate of neurite outgrowth and an increase in the final neurite length.
FIGURE 6:

Knockdown of SmgGDS expression augments BCH domain–mediated PC12 cell differentiation. PC12 cells were transfected with BCH and pSilencer negative control (BCH+neg) or BCH and pSilencer rS1 (BCH+rS1), made quiescent, subjected to 100 ng/ml EGF stimulation, and imaged every 30 min for 36 h. Three independent experiments were conducted, and differentiated cells were then analyzed for neurite length, in terms of either cell body or micrometers. (A) Average length of neurites in knockdown (BCH+rS1) is ∼50% longer when compared with control (BCH+neg). Neurite lengths were measured for each cell in terms of cell body. Experiments were performed in triplicate, and the population means ± SEM (n = 3), p = 0.004, are plotted. (B) Representative cells from BCH+neg and BCH+rS1 were selected to show the neurite outgrowth over time. Images are snapshots of a time-course movie taken over 36 h, with white arrowheads indicating the tips of the neurites. (C) Neurite induction is advanced, and the initial rate of neurite outgrowth is higher for the knockdown than for the control. The graph tracks the neurite growth for one cell each from BCH+neg and BCH+rS1 and is plotted by measuring the neurite length of one representative cell per condition at each time point imaged. (D) There is a shift in the normal distribution of the population of cells with SmgGDS knockdown, to show the increased neurite length of the transfected cells. All the cells in the three experiments were plotted together to represent the population distribution of neurite lengths. (E) The population follows the same trend observed for representative cells in (C), with advanced neurite induction and a higher rate of growth for knockdown. Neurite growth of 10 cells with neurites of three cell body length (control) and 7 cell body length (knockdown) were traced every 5 h. Curves are means of neurite length at each time point ± SEM (n = 10).
We have revealed a novel function for the BCH domain of BPGAP1 in promoting K-Ras activation, leading to sustained ERK activation and PC12 differentiation under EGF stimulation. This effect is negatively regulated by SmgGDS, thus forming a novel signaling circuitry essential for neuronal morphogenesis and differentiation.
DISCUSSION
BPGAP1 as a dual Rho and Ras regulator in neuritogenesis
Rho and Ras play important roles in the spatiotemporal control of multiple signaling pathways, affecting many downstream biological events from the acute protein activities to chronic gene expression levels. Understanding integration of upstream and downstream signaling networks mediated by various small GTPases in general, and Rho and Ras specifically, remains a key challenge, as it is usually unclear whether those signaling events are running in parallel or coordinated via a common set of regulators. For example, the GEF proteins RasGRF1 and RasGRF2 activate Rho and Ras GTPases (Fernandez-Medarde and Santos, 2011), and the GAP proteins ARAP1 and ARAP2 inactivate both Rho and Arf family GTPases (Miura et al., 2002; Yoon et al., 2006; Randazzo et al., 2007). In addition, the p200RhoGAP protein not only stimulates the GTPase activities of Rac1 and RhoA in vivo to regulate neurite outgrowth (Moon et al., 2003) but also increases the Ras-GTP levels to stimulate fibroblast cell proliferation and cell cycle progression, leading to transformation (Shang et al., 2007). These proteins may serve as a common regulator for two different GTPase pathways or act as a control switch between specific pathways leading to coordinated changes in membrane and actin cytoskeletal dynamics. For the process of axon morphogenesis, both Rho and Ras GTPases are critical regulators of axon initiation, growth, guidance, and branching (Hall and Lalli, 2010). For example, R-Ras regulates phosphoinositide 3-kinase activity, inactivating GS3K via Akt leading to microtubule polarization and axon induction (Oinuma et al., 2007). Ras also acts via Raf and Akt promoting elongation and branching (Markus et al., 2002). The Rho GTPases, Cdc42 and Rac, promote neurite outgrowth via effector Par6 or Rac GEFs Tiam1 and Tiam2 (Nishimura et al., 2005), aiding microtubule polarization and stabilization leading to axon extension (de Curtis, 2008), whereas Rho acts antagonistically via ROCK (Rho-associated protein kinase), negatively regulating neurite initiation (Da Silva et al., 2003) and outgrowth (Govek et al., 2005).
Here we provide further evidence that BPGAP1 serves as one such common regulator, not only acting as a RhoGAP to inactivate Rho for neurite branching, but also stimulating the Ras/ERK signaling required to drive neurite outgrowth. This concerted action, mediated by the interplay of the unique scaffolding function of its K-Ras–activating BCH domain and the RhoA-inactivating RhoGAP domain, is visible as long, branched neurites in BPGAP1-transfected cells (Figure 2B, top row, fourth panel). The retention of branched protrusions in U0126-treated BPGAP1-transfected cells (Figure 2B) and induction of long, mainly unbranched protrusions by BPGAP-R232A (Figure 1, C and D) support this. Interestingly, we further reveal that this signaling cross-talk involves SmgGDS as an additional regulator of Ras/ERK signaling to ensure a proper level of coordination.
SmgGDS, a novel Ras regulator that suppresses Ras-BPGAP1–induced Ras/ERK activation
Early attempts identified SmgGDS as a GTPase regulator possessing GEF activity toward Rho and Ras GTPases (Mizuno et al., 1991; Yaku et al., 1994), displaying transforming activity in NIH3T3 (Fujioka et al., 1992) and mitogenic activity in Swiss3T3 cells (Yoshida et al., 1992) in cooperation with K-Ras. More recent work, however, shows that its GEF activity is specific to RhoA and RhoC and not toward K-Ras in vitro (Hamel et al., 2011). Interestingly, we show that SmgGDS instead acts as a negative regulator of K-Ras in 293T and PC12 as the specific knockdown of SmgGDS robustly increases active K-Ras levels upon EGF stimulation. Overexpression of SmgGDS does not readily lead to suppression of K-Ras activation (Figure 5D), however, indicating that the threshold effect of SmgGDS itself is also under the influence of other cellular factors. The apparent duality in roles for SmgGDS could be context-dependent, such that in fibroblast cells, SmgGDS activates K-Ras, whereas in 293T and PC12, SmgGDS could collaborate with other partners to exert an overall negative effect on K-Ras. In this context, recombinant BPGAP1 interacts with SmgGDS, in its endogenous form and when both are coexpressed inside the cells. Furthermore, a stronger interaction was observed with the BCH domain of BPGAP1, the same domain that interacts with K-Ras. Therefore it is possible that SmgGDS could suppress Ras/Erk activation induced by BPGAP1 upon EGF stimulation, by competing away the BCH domain from K-Ras activation (Figure 7) or by recruiting other suppressors to the BPGAP1/SmgGDS complex. Alternatively, this inhibition by SmgGDS could be mediated via a yet unknown mechanism independent of their direct interaction. The detailed molecular mechanisms underlying their direct interaction or indirect effects require further investigation.
FIGURE 7:
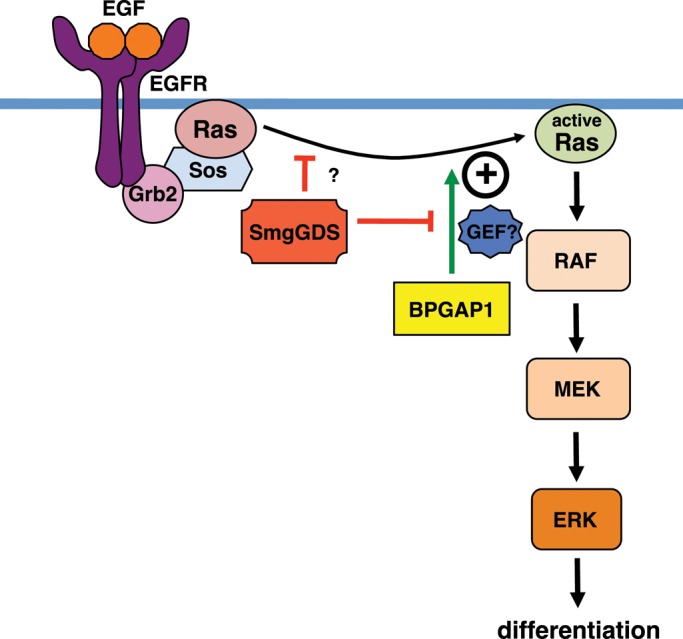
BPGAP1–SmgGDS interplay defines a novel regulatory node in Ras signaling. BPGAP1 enhances Ras/ERK activation leading to PC12 differentiation. On EGF stimulation, BPGAP1 enhances K-Ras activation—a process inhibited by SmgGDS. SmgGDS also negatively regulates K-Ras activation by an as yet unknown mechanism. The mechanism for BPGAP1–SmgGDS regulation of Ras signaling is explained. On EGF stimulation, activators of Ras stimulate Ras activation to a certain low level, which is not sufficient for differentiation. BPGAP1 recruits Ras activators (possibly GEFs) to enhance this activation. Although endogenous SmgGDS could provide a level of attenuation of Ras activation by sequestering Ras activators, this effect is more pronounced in the presence of overexpressed SmgGDS, which also blocks the effect of BPGAP1 by sequestration. When SmgGDS is knocked down, the release of Ras activators leads to enhanced Ras activation albeit insufficient for differentiation. When BPGAP1 is added to this, a pronounced increase in Ras activation occurs resulting in a higher rate of differentiation, longer neurites, and an earlier induction of neurite outgrowth.
To activate K-Ras, we hypothesize that BPGAP1 could recruit an as yet unidentified RasGEF, as the BCH domain shows a strong preference for K-Ras-S17N, a feature shared by many GEFs (Figure 3B). Although it was initially suspected to be a candidate RasGEF when identified in our exploratory proteomics screens, SmgGDS turned out to be a suppressor of BPGAP1’s function as the specific knockdown of SmgGDS further potentiates Ras/ERK activation and the induction, maturation, and maintenance of neurite outgrowth by the BCH domain of BPGAP1. Such a delicate interplay of tripartite Ras–BPGAP1–SmgGDS signalome is likely to be sensitive to their stoichiometric perturbation in addition to their spatiotemporal distribution. This mechanism should therefore offer better control of Ras and ERK signaling, as their potency and plasticity in regulating the onset and speed of differentiation could be fine-tuned by removal of the SmgGDS block and/or increase of the presence of recruiter BPGAP1. How subsets of these signaling nodes of BPGAP1 interact in the context of SmgGDS regulation remains to be further determined.
BCH as a versatile regulatory scaffold protein domain
There is compelling evidence to suggest that the BCH domain is an emerging class of regulatory domain for Rho GTPases. The BCH domain is a key player in regulating the interactions between GTPases and their regulators. For example, the BNIP-2–BCH promotes Cdc42 activation driving cell protrusion and muscle differentiation (Zhou et al., 2005; Kang et al., 2008), whereas BNIP-Sα–BCH targets RhoA, bringing about cell rounding and apoptosis by freeing RhoA from its inactivation by p50RhoGAP (Zhou et al., 2005). BNIP-XL–BCH also targets RhoA while separating RhoA from its proto-oncogenic RhoA-specific RhoGEF, Lbc, inhibiting Lbc-induced oncogenic transformation (Soh and Low, 2008), whereas p50RhoGAP–BCH sequesters RhoA from its adjacent RhoGAP domain, thus preventing its inactivation (Zhou et al., 2010). This work provides the first evidence for the involvement of the BCH domain of BPGAP1 in regulating K-Ras and the Ras regulator, SmgGDS. All these observations add to the significance and plasticity of BCH domain in regulating GTPase signaling (Pan and Low, 2012; Pan et al., 2012). To date, as many as 175 different proteins harboring BCH domains have been identified across species from slime mold to plants and humans, and the BCH domain represents a novel subclass of the CRAL-TRIO (previously named Sec14p) superfamily (Gupta et al., 2012). It therefore remains an interesting prospect to examine how the BCH domains in these and other related proteins evolved to recognize different GTPases and their immediate regulators to achieve dynamic network control. In this context, our studies further highlight the possibilities of some of these BCH domains being involved in the Ras/MAPK pathway.
In summary, we have identified a novel function for BPGAP1 in neuritogenesis. We have revealed another role for SmgGDS as a negative regulator of Ras signaling and further showed that the SmgGDS antagonizes BPGAP1 function in regulating Ras/ERK signaling in neuronal differentiation. This should provide new insights into the intricate network of Ras, Rho, and other signaling nodes in cell dynamics control.
MATERIALS AND METHODS
Plasmid construction
BPGAP1, BCH, and SmgGDS were cloned into Flag-, HA-, mCherry-, and GFP-pXJ40 mammalian expression vectors provided by E. Manser (The Institute of Medical Biology, Immunos, Singapore). The Ras constructs (K-Ras, H-Ras, N-Ras, and K-Ras-S17N) were a generous gift from Graeme Guy (Institute of Molecular and Cell Biology). HA-mMek2-K101A was provided by L. L. Chew (National University of Singapore, Singapore). Raf-1–RBD construct was generated by PCR of human cDNA and cloned into pGEX-4T-1 vector (Amersham Biosciences, Uppsala, Sweden). Constructs were sequenced to confirm sequence fidelity.
Cell culture and transfection
HEK293T cells were grown in RPMI 1640 medium (HyClone, Logan, UT) supplemented with 10% (vol/vol) fetal bovine serum (PAA Cell Culture, Pasching, Austria), 10 mM HEPES (HyClone), and 2 mM l-glutamine (HyClone). Cells at 60–80% confluence in six-well plates were transfected with 1–2 μg of plasmid using FuGENE 6 (Roche Applied Science, Indianapolis, IN) or TransIT LT1 Transfection Reagent (Mirus, Madison, WI), according to the manufacturer's instructions. PC12 cells were cultured in DMEM supplemented with 4500 mg of glucose (HyClone), 10 mM HEPES (HyClone), 5% fetal bovine serum (PAA Cell Culture), and 10% horse serum (Life Technologies, Carlsbad, CA). PC12 cells were transfected at 70–80% confluence with Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions for 8 h before they were reseeded onto six-well plates or poly-d-lysine (Sigma, St. Louis, MO)-coated coverslips. All cells were maintained at 37°C in 5% CO2. EGF (Sigma) and NGF (Sigma) stimulation was carried out after 12-h starvation (0% serum for HEK293T and 0.5% serum for PC12) at 100 and 10 ng/ml, respectively, in starvation medium. Where indicated, PC12 cells were treated with 5 μM Mek inhibitor, U0126 (Promega, Fitchburg, WI; Kasai et al., 2005) concomitantly with EGF.
Coimmunoprecipitation
HEK293T and PC12 cells were lysed in 250 μl of RIPA lysis buffer (50 mM Tris-HCl, pH 7.3; 150 mM NaCl, 0.75 mM EDTA, 1% [wt/vol] sodium deoxycholate, 1% [vol/vol] Triton X-100, 0.2% [wt/vol] sodium fluoride with freshly added protease inhibitors (Roche Applied Science, Penzberg, Germany), and 5 mM sodium orthovanadate) per six-well plate. Aliquots were either directly analyzed by Western blotting or were used for protein binding studies. For coimmunoprecipitation, lysates were incubated with anti-FLAG antibody conjugated to agarose beads (Sigma, St. Louis, MO) at 4°C for 3 h. The beads were washed with lysis buffer and analyzed by Western blotting. Primary antibodies; polyclonal anti-FLAG (Sigma), anti-HA (Zymed, South San Francisco, CA), anti-Growth Associated Protein-43 (GAP43; Millipore, Billerica, MA), monoclonal anti-phospho-ERK (Sigma), monoclonal anti-pan-Erk1/2 (BD Transduction Laboratories, Franklin Lakes, NJ), monoclonal anti-K-Ras (Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-α/β tubulin (Cell Signaling Technology, Danvers, MA), polyclonal SmgGDS, and polyclonal BPGAP1 were used.
Immunofluorescence and confocal microscopy
PC12 cells grown on poly-d-lysine–coated glass coverslips were washed twice with PBS and fixed with 3.7% formaldehyde for 15 min at 37°C. Fixed cells were washed twice with PBS, twice with 4 mM NH4Cl in PBS, twice with PBS, and permeabilized with 0.2% Triton X-100 (Bio-Rad Laboratories, Hercules, CA) in PBS for 15 min at room temperature. Blocking was carried out with 2% bovine serum albumin and 7% fetal bovine serum in PBS for 30–60 min at room temperature. Cells were incubated with anti-FLAG (Sigma, St. Louis, MO) and anti-HA (Zymed) antibodies in blocking solution. Samples were washed three times with 0.1% Triton X-100–containing PBS before incubation with Alexa Fluor 488–conjugated donkey anti–mouse immunoglobulin G (IgG) and Alexa Fluor 555–conjugated donkey anti–rabbit IgG (all Molecular Probes, Eugene, OR). Confocal fluorescence images were captured on a Zeiss (Oberkochen, Germany) LSM 510 META microscope. At least 100 transfected cells per construct were evaluated for long protrusions (> 2 cell body length). Data are means ± SEM from three independent experiments. Statistical significance was analyzed using analysis of variance (ANOVA) and Student's t test (paired, two-tailed).
shRNA knockdown
Two shRNA oligonucleotides against rat SmgGDS were designed using siRNA sequences published by Tew et al. (2008); rS1: 5′-AAG CAA AGA TGT TAT CAG CCG-3′ (nt 911–931) and rS2: 5′-ATG TTA ATA GAT GCA CAA GCA-3′ (nt 1009–1029), synthesized (Eurofins MWG Operon, Ebersberg, Germany) and cloned into the pSilencer 2.1-U6 hygro vector (Ambion, Carlsbad, CA). Constructs were sequenced to confirm sequence fidelity, and knockdown efficiency was determined by cotransfection with GFP–SmgGDS plasmid at a 10:1 ratio. Lysates were analyzed 48 h posttransfection. When compared with the nontargeting negative control, both shRNA constructs (rS1 and rS2) successfully depleted SmgGDS by 60 and 50%, respectively. The rS1 construct was then used for subsequent experimentation as it displayed a superior knockdown effect as compared with rS2.
K-Ras activation assay
HEK293T cells were lysed in lysis buffer (50 mM HEPES, pH 7.4; 150 mM NaCl, 1.5 mM MgCl2, 5 mM EDTA, 10% [vol/vol] glycerol, 1% [vol/vol] Triton X-100) with freshly added protease inhibitors (Roche Applied Science, Penzberg, Germany) and 5 mM sodium orthovanadate, and 60 μg of lysates was incubated with 10 μg of GST–Raf-1–RBD for 50 min at 4°C (Young et al., 2004). The beads were washed three times with lysis buffer, and bound proteins were separated by SDS–PAGE for Western blot analyses. The membranes were then stained with Amido Black to reveal equal loading of GST and GST–Raf-1–RBD.
Live imaging for differentiation
Eight hours posttransfection, PC12 cells were reseeded onto poly-d-lysine-coated, six-well plates at a confluence of 0.5 × 105 cells/ml and allowed to settle for 4 h, followed by 12-h starvation with medium containing 0.5% serum before 50% of the medium was replaced with EGF (final concentration 100 ng/ml) containing 0.5% serum medium. Cells were then imaged for 36 h with a wide field Olympus IX81 inverted microscope. Images were analyzed with the help of ImageJ.
Supplementary Material
Acknowledgments
We thank Ed Manser (The Institute of Medical Biology, Immunos, Singapore), Graeme Guy (formerly of the Institute of Molecular and Cell Biology, Singapore), and Li Li Chew, Fu Ling Soh, and Bee Leng Lua (formerly of the National University of Singapore, Singapore) for their valuable constructs. This work was supported by a grant from the Ministry of Education, Singapore (Academic Research Fund R-154-000-236-112), and in part by the Mechanobiology Institute, cofunded by the National Research Foundation and the Ministry of Education, Singapore, to B.C.L.
Abbreviations used:
- ANOVA
analysis of variance
- BCH
BNIP-2 and Cdc42GAP homology
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- HA
hemagglutinin
- NGF
nerve growth factor
- PRR
proline-rich region
- RBD
Ras-binding domain
- RhoGAP
Rho GTPase-activating protein
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SmgGDS
small G-protein GDP dissociation stimulator
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-04-0300) on November 14, 2012.
A.R. and B.C.L. conceived the ideas, analyzed data, and wrote the manuscript.
A.R. performed the experiments.
REFERENCES
- Bernards A, Settleman J. GEFs in growth factor signaling. Growth Factors. 2007;25:355–361. doi: 10.1080/08977190701830375. [DOI] [PubMed] [Google Scholar]
- Da Silva JS, Medina M, Zuliani C, Di Nardo A, Witke W, Dotti CG. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J Cell Biol. 2003;162:1267–1279. doi: 10.1083/jcb.200304021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis I. Functions of Rac GTPases during neuronal development. Dev Neurosci. 2008;30:47–58. doi: 10.1159/000109851. [DOI] [PubMed] [Google Scholar]
- Fernandez-Medarde A, Santos E. The RasGrf family of mammalian guanine nucleotide exchange factors. Biochim Biophys Acta. 2011;1815:170–188. doi: 10.1016/j.bbcan.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Fujioka H, Kaibuchi K, Kishi K, Yamamoto T, Kawamura M, Sakoda T, Mizuno T, Takai Y. Transforming and c-fos promoter/enhancer-stimulating activities of a stimulatory GDP/GTP exchange protein for small GTP-binding proteins. J Biol Chem. 1992;267:926–930. [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Gupta AB, Wee LE, Zhou YT, Hortsch M, Low BC. Cross-species analyses identify the BNIP-2 and Cdc42GAP homology (BCH) domain as a distinct functional subclass of the CRAL_TRIO/Sec14 superfamily. PLoS One. 2012;7:e33863. doi: 10.1371/journal.pone.0033863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, Lalli G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb Perspect Biol. 2010;2:a001818. doi: 10.1101/cshperspect.a001818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel B, Monaghan-Benson E, Rojas RJ, Temple BR, Marston DJ, Burridge K, Sondek J. SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J Biol Chem. 2011;286:12141–12148. doi: 10.1074/jbc.M110.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Bae GU, Yi MJ, Yang YJ, Oh JE, Takaesu G, Zhou YT, Low BC, Krauss RS. A Cdo-Bnip-2-Cdc42 signaling pathway regulates p38alpha/beta MAPK activity and myogenic differentiation. J Cell Biol. 2008;182:497–507. doi: 10.1083/jcb.200801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai A, Shima T, Okada M. Role of Src family tyrosine kinases in the down-regulation of epidermal growth factor signaling in PC12 cells. Genes Cells. 2005;10:1175–1187. doi: 10.1111/j.1365-2443.2005.00909.x. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Kaibuchi K, Hori Y, Nonaka H, Sakoda T, Kawamura M, Mizuno T, Takai Y. Molecular cloning of the human cDNA for a stimulatory GDP/GTP exchange protein for c-Ki-ras p21 and smg p21. Oncogene. 1992;7:289–293. [PubMed] [Google Scholar]
- Lua BL, Low BC. BPGAP1 interacts with cortactin and facilitates its translocation to cell periphery for enhanced cell migration. Mol Biol Cell. 2004;15:2873–2883. doi: 10.1091/mbc.E04-02-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lua BL, Low BC. Activation of EGF receptor endocytosis and ERK1/2 signaling by BPGAP1 requires direct interaction with EEN/endophilin II and a functional RhoGAP domain. J Cell Sci. 2005;118:2707–2721. doi: 10.1242/jcs.02383. [DOI] [PubMed] [Google Scholar]
- Markus A, Zhong J, Snider WD. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 2002;35:65–76. doi: 10.1016/s0896-6273(02)00752-3. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Miura K, Jacques KM, Stauffer S, Kubosaki A, Zhu K, Hirsch DS, Resau J, Zheng Y, Randazzo PA. ARAP1: a point of convergence for Arf and Rho signaling. Mol Cell. 2002;9:109–119. doi: 10.1016/s1097-2765(02)00428-8. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kaibuchi K, Yamamoto T, Kawamura M, Sakoda T, Fujioka H, Matsuura Y, Takai Y. A stimulatory GDP/GTP exchange protein for smg p21 is active on the post-translationally processed form of c-Ki-ras p21 and rhoA p21. Proc Natl Acad Sci USA. 1991;88:6442–6446. doi: 10.1073/pnas.88.15.6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SY, Zang H, Zheng Y. Characterization of a brain-specific Rho GTPase-activating protein, p200RhoGAP. J Biol Chem. 2003;278:4151–4159. doi: 10.1074/jbc.M207789200. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- Oinuma I, Katoh H, Negishi M. R-Ras controls axon specification upstream of glycogen synthase kinase-3beta through integrin-linked kinase. J Biol Chem. 2007;282:303–318. doi: 10.1074/jbc.M607979200. [DOI] [PubMed] [Google Scholar]
- Pan CQ, Liou YC, Low BC. Active Mek2 as a regulatory scaffold that promotes Pin1 binding to BPGAP1 to suppress BPGAP1-induced acute Erk activation and cell migration. J Cell Sci. 2010;123:903–916. doi: 10.1242/jcs.064162. [DOI] [PubMed] [Google Scholar]
- Pan CQ, Low BC. Functional plasticity of the BNIP-2 and Cdc42GAP Homology (BCH) domain in cell signaling and cell dynamics. FEBS Lett. 2012;586:2674–2691. doi: 10.1016/j.febslet.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Pan CQ, Sudol M, Sheetz M, Low BC. Modularity and functional plasticity of scaffold proteins as p(l)acemakers in cell signaling. Cell Signal. 2012;24:2143–2165. doi: 10.1016/j.cellsig.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Randazzo PA, Inoue H, Bharti S. Arf GAPs as regulators of the actin cytoskeleton. Biol Cell. 2007;99:583–600. doi: 10.1042/bc20070034. [DOI] [PubMed] [Google Scholar]
- Shang X, Moon SY, Zheng Y. p200 RhoGAP promotes cell proliferation by mediating cross-talk between Ras and Rho signaling pathways. J Biol Chem. 2007;282:8801–8811. doi: 10.1074/jbc.M609375200. [DOI] [PubMed] [Google Scholar]
- Shang X, Zhou YT, Low BC. Concerted regulation of cell dynamics by BNIP-2 and Cdc42GAP homology/Sec14p-like, proline-rich, and GTPase-activating protein domains of a novel Rho GTPase-activating protein, BPGAP1. J Biol Chem. 2003;278:45903–45914. doi: 10.1074/jbc.M304514200. [DOI] [PubMed] [Google Scholar]
- Soh UJ, Low BC. BNIP2 extra long inhibits RhoA and cellular transformation by Lbc RhoGEF via its BCH domain. J Cell Sci. 2008;121:1739–1749. doi: 10.1242/jcs.021774. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- Tew GW, Lorimer EL, Berg TJ, Zhi H, Li R, Williams CL. SmgGDS regulates cell proliferation, migration, and NF-kappaB transcriptional activity in non-small cell lung carcinoma. J Biol Chem. 2008;283:963–976. doi: 10.1074/jbc.M707526200. [DOI] [PubMed] [Google Scholar]
- Vikis HG, Stewart S, Guan KL. SmgGDS displays differential binding and exchange activity towards different Ras isoforms. Oncogene. 2002;21:2425–2432. doi: 10.1038/sj.onc.1205306. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- Yaku H, Sasaki T, Takai Y. The Dbl oncogene product as a GDP/GTP exchange protein for the Rho family: its properties in comparison with those of Smg GDS. Biochem Biophys Res Commun. 1994;198:811–817. doi: 10.1006/bbrc.1994.1116. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kaibuchi K, Mizuno T, Hiroyoshi M, Shirataki H, Takai Y. Purification and characterization from bovine brain cytosol of proteins that regulate the GDP/GTP exchange reaction of smg p21s, ras p21-like GTP-binding proteins. J Biol Chem. 1990;265:16626–16634. [PubMed] [Google Scholar]
- Yoon HY, Miura K, Cuthbert EJ, Davis KK, Ahvazi B, Casanova JE, Randazzo PA. ARAP2 effects on the actin cytoskeleton are dependent on Arf6-specific GTPase-activating-protein activity and binding to RhoA-GTP. J Cell Sci. 2006;119:4650–4666. doi: 10.1242/jcs.03237. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Kawata M, Miura Y, Musha T, Sasaki T, Kikuchi A, Takai Y. Microinjection of smg/rap1/Krev-1 p21 into Swiss 3T3 cells induces DNA synthesis and morphological changes. Mol Cell Biol. 1992;12:3407–3414. doi: 10.1128/mcb.12.8.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TW, Mei FC, Yang G, Thompson-Lanza JA, Liu J, Cheng X. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004;64:4577–4584. doi: 10.1158/0008-5472.CAN-04-0222. [DOI] [PubMed] [Google Scholar]
- Zhou YT, Chew LL, Lin SC, Low BC. The BNIP-2 and Cdc42GAP homology (BCH) domain of p50RhoGAP/Cdc42GAP sequesters RhoA from inactivation by the adjacent GTPase-activating protein domain. Mol Biol Cell. 2010;21:3232–3246. doi: 10.1091/mbc.E09-05-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YT, Guy GR, Low BC. BNIP-2 induces cell elongation and membrane protrusions by interacting with Cdc42 via a unique Cdc42-binding motif within its BNIP-2 and Cdc42GAP homology domain. Exp Cell Res. 2005;303:263–274. doi: 10.1016/j.yexcr.2004.08.044. [DOI] [PubMed] [Google Scholar]
- Zhou YT, Guy GR, Low BC. BNIP-Salpha induces cell rounding and apoptosis by displacing p50RhoGAP and facilitating RhoA activation via its unique motifs in the BNIP-2 and Cdc42GAP homology domain. Oncogene. 2006;25:2393–2408. doi: 10.1038/sj.onc.1209274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.