

During protein targeting by the signal recognition particle (SRP), signals from cargo binding in the SRP's M domain must be communicated to its GTPase domain to initiate the membrane delivery of cargo. In this study, a conserved fingerloop lining the signal sequence–binding groove of SRP is shown to provide a key link in this molecular communication.
Abstract
During cotranslational protein targeting by the signal recognition particle (SRP), information about signal sequence binding in the SRP's M domain must be effectively communicated to its GTPase domain to turn on its interaction with the SRP receptor (SR) and thus deliver the cargo proteins to the membrane. A universally conserved “fingerloop” lines the signal sequence–binding groove of SRP; the precise role of this fingerloop in protein targeting has remained elusive. In this study, we show that the fingerloop plays important roles in SRP function by helping to induce the SRP into a more active conformation that facilitates multiple steps in the pathway, including efficient recruitment of SR, GTPase activation in the SRP•SR complex, and most significantly, the unloading of cargo onto the target membrane. On the basis of these results and recent structural work, we propose that the fingerloop is the first structural element to detect signal sequence binding; this information is relayed to the linker connecting the SRP's M and G domains and thus activates the SRP and SR for carrying out downstream steps in the pathway.
INTRODUCTION
Membrane and secretory proteins, whose syntheses are initiated in the cytosol, must be efficiently localized to their correct cellular destinations to assume their function. The signal recognition particle (SRP) is part of the essential cellular machinery responsible for the cotranslational recognition and delivery of proteins destined to the eukaryotic endoplasmic reticulum (ER) or the bacterial plasma membrane (Walter and Johnson, 1994). As a nascent polypeptide emerges from a translating ribosome, SRP recognizes the ribosome nascent chain complex (termed RNC or the cargo), through interaction with both the ribosome exit site and with N-terminal signal sequences on its substrate protein (Pool et al., 2002; Halic et al., 2004, 2006; Schaffitzel et al., 2006). The cargo is delivered to the membrane via the interaction of SRP with the SRP receptor (SR; called FtsY in bacteria). Subsequently, the RNC is transferred to the protein translocation machinery (Sec61p in eukaryotes or SecYEG in bacteria), where the nascent protein is either translocated across or integrated into the membrane (Gilmore et al., 1982a, b; Keenan et al., 2001).
The composition of the SRP varies across different species, but its functional core is highly conserved and composed of two essential components: the SRP54 protein subunit and the SRP RNA (called Ffh and 4.5S, respectively, in bacteria; Walter and Johnson, 1994). SRP54 (Ffh) contains two structurally and functionally distinct domains connected by an ∼30-amino-acid-long linker: 1) a methionine-rich M domain containing a hydrophobic groove that serves as the signal sequence–binding site and a helix-turn-helix motif that binds the 4.5S RNA (Freymann et al., 1997; Keenan et al., 1998; Batey et al., 2000; Janda et al., 2010); and 2) a special GTPase, NG domain responsible for interacting with the SR (Egea et al., 2004; Focia et al., 2004) and for contacting the ribosome exit site (Pool et al., 2002; Schaffitzel et al., 2006). The SR, FtsY, also contains an NG domain highly homologous to that in Ffh (Montoya et al., 1997). During protein targeting, the GTP-dependent assembly of a stable complex between the NG domains of Ffh and FtsY mediates the delivery of cargo proteins to the target membrane (Egea et al., 2004; Focia et al., 2004). Subsequent rearrangements in the Ffh•FtsY complex further induce the reciprocal activation of their GTPase activity; this late rearrangement is essential for driving the unloading of cargo to the translocation machinery (Zhang et al., 2008, 2009). Hydrolysis of GTP then drives the rapid disassembly of the SRP•FtsY complex, allowing the two proteins to be recycled for additional rounds of targeting (Shan and Walter, 2005).
The SRP RNA is a ubiquitous and indispensable component of the SRP. The Escherichia coli 4.5S RNA contains the universally conserved domain IV of eukaryotic SRP RNA, which forms a hairpin structure capped by a highly conserved GGAA tetraloop (Batey et al., 2000). The SRP RNA binds with picomolar affinity to the SRP54 (or Ffh) M domain in the vicinity of the signal sequence–binding site (Batey et al., 2001). It also regulates the interaction between the Ffh and FtsY GTPases during protein targeting. The tetraloop of the SRP RNA mediates a key electrostatic interaction with FtsY, which accelerates the stable association between the SRP and FtsY GTPases by a factor of 200–3000 (Shen et al., 2011). This stimulation occurs only in the presence of RNC bearing correct signal sequences, or stimulatory detergents and signal peptides that partially mimic the effect of RNC, ensuring that the recognition of cargo is tightly coupled to its membrane delivery during protein targeting (Siu et al., 2007; Bradshaw et al., 2009; Zhang et al., 2010; Shen et al., 2011). In addition, the SRP RNA also activates GTP hydrolysis in the SRP•FtsY complex ∼10-fold, whereas the cargo negatively regulates this GTPase activation (Peluso et al., 2001; Zhang et al., 2010). Thus there is extensive molecular communication between the cargo, the SRP RNA, and the GTPases throughout different stages of protein targeting. However, the precise molecular mechanism that allows information to be propagated from the signal sequence–binding site in the M domain to the SRP RNA and the GTPases remains to be defined.
Flanking the signal sequence–binding site is an evolutionarily conserved flexible region, the fingerloop, which forms a “flap” over the hydrophobic binding groove. In the absence of a signal sequence, the M domain can adopt a “closed” conformation in which the fingerloop inserts several of its hydrophobic residues into the signal sequence–binding site; it has been proposed that this conformation stabilizes the hydrophobic signal sequence–binding pocket in the free SRP (Rosendal et al., 2003). The fingerloop has also been crystallized in an “open” conformation, in which it folds back from the signal sequence–binding pocket (Keenan et al., 1998). In a recent crystal structure of the M domain in complex with a signal peptide, several residues of the fingerloop directly interact with the hydrophobic signal peptide (Janda et al., 2010). These observations have led to the suggestion that the fingerloop forms a flexible “lid” that closes down on the signal sequence–binding groove upon cargo binding to the SRP, providing additional hydrophobic contacts with the hydrophobic signal peptide (Keenan et al., 1998, 2001; Rosendal et al., 2003). In addition, the flexibility of the fingerloop, along with the abundance of methionine residues in the M domain, is thought to provide an adaptable binding site that accommodates a variety of signal sequences (Keenan et al., 2001; Bernstein, 1998; Koch et al., 2003; Halic and Beckmann, 2005). Nevertheless, no direct experiments assessing the contribution of the fingerloop in signal sequence binding have been available, and the role of this highly conserved loop in SRP function has remained unclear.
Given its proximity to the signal sequence–binding site, the fingerloop is in an ideal position to sense information about signal sequences; the conformational plasticity of the fingerloop also makes it a good candidate for transmitting this information to the GTPases. In support of this notion, a previous study reported that mutations in the fingerloop disrupt the ability of the SRP RNA to stimulate Ffh•Fts complex assembly (Bradshaw and Walter, 2007; Hainzl et al., 2011). However, these defects could also be explained by the inability of the fingerloop mutants to bind signal sequences. In this work, we defined the roles of the SRP fingerloop on individual molecular steps during the protein-targeting reaction. Our results showed that the fingerloop is not strictly required for high-affinity signal sequence binding, but rather, it helps mediate conformational changes in response to signal sequence binding that propagates to the remainder of Ffh and the 4.5S RNA, thus activating the SRP and FtsY GTPases and facilitating the delivery and unloading of cargo to the translocon.
RESULTS
The fingerloop domain is essential for Ffh function
To characterize the fingerloop (FL) of Ffh in vivo, we first constructed strain SLD108 to facilitate complementation tests (see Materials and Methods). SLD108 is not viable at 42°C, because the sole functional copy of ffh is expressed from a temperature-sensitive plasmid, pFfhTSpc (Table 1). Growth can be restored at 42°C if the strain is also transformed with a plasmid expressing a functional copy of ffh, pBADffhN6x, which expresses ffh under control of the araBAD operator and promoter. To determine the importance of the fingerloop for Ffh function, we also constructed the ffhΔFL allele by deleting a 60–base pair region that encodes the fingerloop (Figure 1A; Zheng and Gierasch, 1997) on pBADffhN6x (see Materials and Methods).
TABLE 1:
Bacterial strains and plasmids.
| Strain or plasmid | Relevant genotype or description | Source or reference |
|---|---|---|
| E. coli strains | ||
| NEB5α | fhuA2Δ(argF-lacZ)U169 phoA glnV44 80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 (general cloning host) | New England Biolabs |
| ECF529 | ΔaraBAD, ΔrhaBAD, ΔaraFGH, ΔaraE, rrnBPI(CTC-AGA)-lacYA177C | Bowers et al., 2004 |
| XLU102 | ECF529, Δbla::frt | This study |
| SLD108 | XLU102, ffh::kan1, pFfhTSpc | This study |
| Plasmids | ||
| pFfhTSpc | pSC101ts, ffh+, spc (SpcR) | Lab collection |
| pBADffh6x | araC, ffh+, bla (AmpR), ColE1 (vector for expressing ffh under ParaBAD control) | This study |
| pBADffhN6x | pBADffh6x (NheI) | This study |
| pBADffhN6xΔFL | pBADffhN6x with fingerloop deleted | This study |
| pBADffhN6xLM-AA | pBADffhN6x with LM→AA mutation | |
| pBADffhN6xPG-AA | pBADffhN6x with PG→AA mutation | |
FIGURE 1:
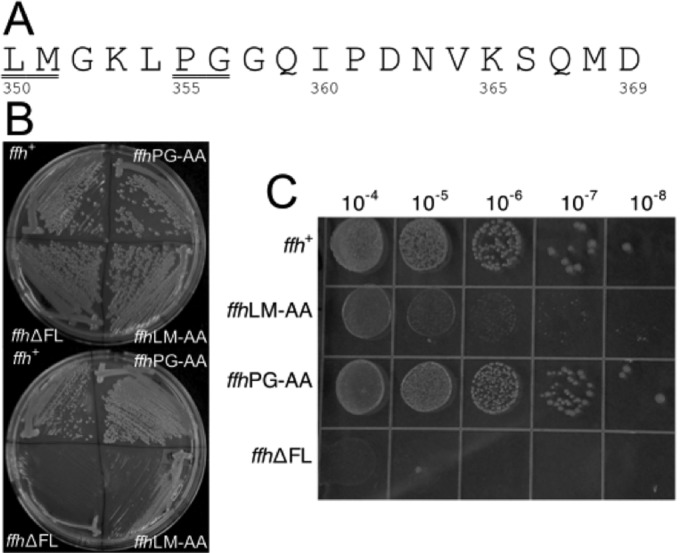
Phenotypes of ffh fingerloop mutants. (A) The amino acid sequence of the fingerloop domain of Ffh deleted in the ffh∆FL allele. Positions of amino acids of the E. coli Ffh protein are shown. The underlined amino acids were converted to alanine in the ffhLM→AA and ffhPG→AA alleles. (B) Plasmids expressing ffh∆FL, ffhPG→AA, ffhLM→AA, and ffh+ alleles were transformed into the temperature-sensitive strain SLD108 and cultured at 30°C (top) or 42°C (bottom) as shown. (C) SLD108 transformants expressing each of the four ffh alleles were spotted onto LB+Amp+l-arabinose plates at the dilutions shown at the top and incubated at 42°C overnight.
In cells carrying pBADffhN6x, we observed growth at both the permissive temperature of 30°C, as well as at 42°C, the nonpermissive temperature for pFfhTSpc replication (Figure 1B). We observed, conveniently, that arabinose was not necessary for this plasmid to complement ffh::kan1 when grown at 42°C, due to leaky expression from the araBAD promoter at the elevated temperature (Figure 1B). Colonies that appeared at 42 °C were retested and confirmed to be spectinomycin sensitive (SpcS), indicating loss of the pFfhTSpc plasmid. Moreover, the SpcS transformants were only able to grow at 30°C when provided with 0.01% l-arabinose (unpublished data). In contrast to the wild-type control, expression of ffhΔFL failed to complement ffh::kan1 in SLD108 (Figure 1B). Consistent with this result, none of the cells recovered from the heavy portion of the streak were SpcS nor were they able to grow at 30°C.
The fingerloop is often unstructured in crystallographic and biochemical studies of the SRP (Zheng and Gierasch, 1997; Cleverley et al., 2001; Doudna and Batey, 2004; Janda et al., 2010). As an alternative approach to identify key features in the fingerloop, we compared the amino acid sequences of this loop from 109 distinct species representing all three domains of life, using multiple sequence alignment (Thompson et al., 1994). These analyses identified two amino acid pairs, Leu350/Met351 and Pro355/Gly356 from E. coli, that are highly conserved (Figure 1A). To assess the importance of these residues, we generated mutant alleles in which each amino acid pair was converted to alanines. When expressed in SLD108, the ffhLM→AA allele complemented only slightly better than ffh∆FL, while expression of ffhPG→AA complemented as well as ffh+ (Figure 1B). Expression levels of all Ffh constructs were consistent across the board (Supplemental Figure S1).
To further test the function of the ffh mutants, we took advantage of the features of SLD108, as described in Materials and Methods, that allow l-arabinose to induce gene expression at levels that directly correlate with its concentration homogeneously throughout the population of cells (Morgan-Kiss et al., 2002). We used this system to determine whether elevated gene expression of the mutant ffh alleles could restore growth to SLD108 at the nonpermissive temperature. As expected, increased expression of ffhΔFL failed to restore viability to SLD108, and only a minor increase in growth was observed when ffhLM→AA was expressed at higher levels. As observed previously, expression of the ffhPG→AA allele supported growth of SLD108 at levels indistinguishable from wild-type ffh (Figure 1C).
Fingerloop is important for SRP-mediated protein targeting
To directly test the effect of the fingerloop mutations on cotranslational protein targeting, we used a well-established in vitro assay that examines the ability of purified SRP and FtsY to target a model SRP substrate, preprolactin (pPL), to ER microsomal membranes (Powers and Walter, 1997; Shan et al., 2007). The efficiency of targeting and translocation can be quantified based on cleavage of pPL signal sequence upon its successful incorporation into the membrane (Figure 2A). Wild-type SRP efficiently targeted pPL, reaching a translocation efficiency of greater than 60% at saturating FtsY concentrations (Figure 2, A and B). Deletion of the fingerloop significantly reduced the targeting efficiency, with only ∼30% successful targeting and translocation at saturating FtsY concentrations (Figure 2, A and B). Further, a much higher FtsY concentration was required to reach saturation for the targeting reaction mediated by SRP(∆FL). The LM→AA mutant also displayed impaired targeting of pPL, but the defect is milder than that of mutant SRP(∆FL) (Figure 2, A and B).
FIGURE 2:
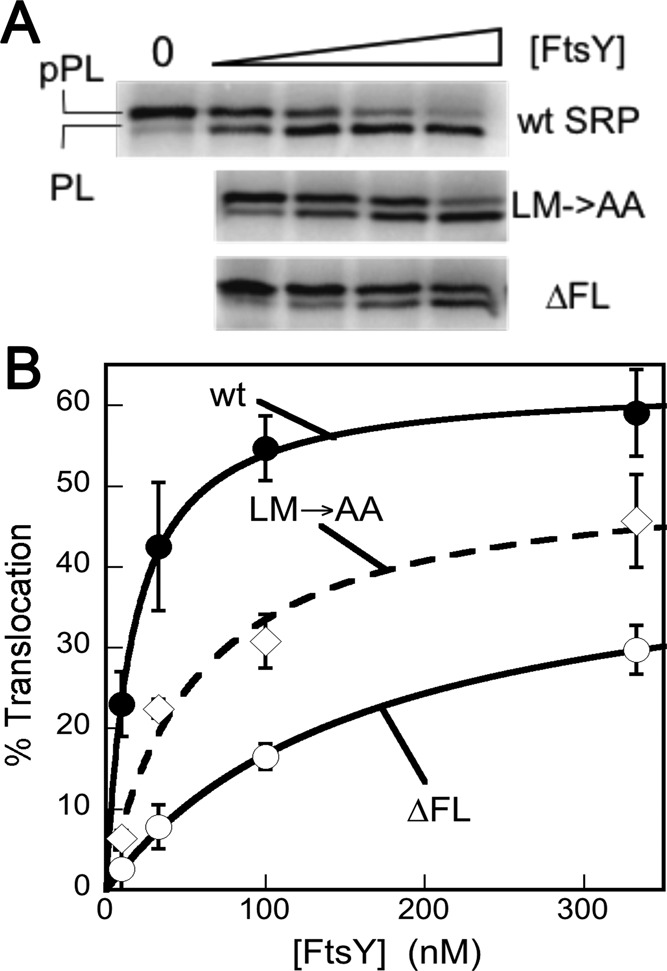
SRP fingerloop mutants are defective in protein targeting and translocation. (A) Cotranslational targeting and translocation of 35S-labeled pPL into ER microsomal membranes by wild-type and mutant SRP. (B) Quantification of the data in (A) for wild-type SRP (●), SRP(LM→AA) (◊), and SRP(∆FL) (◯).
Qualitatively, these results agreed with the in vivo observations, and together they provide direct evidence that the conserved fingerloop plays an important role in cotranslational protein targeting. We note that several factors could contribute to the stronger phenotype of fingerloop mutants in vivo than in vitro. The in vitro assay represents a single round of targeting and translocation, whereas the defects of SRP mutations can accumulate over multiple rounds of targeting in vivo. In addition, the slower translation rate in vitro than in vivo gives the SRP and FtsY a longer window of time to complete the targeting reaction, such that defects in their assembly (see below: The fingerloop facilitates signal sequence–induced stimulation of SRP•FtsY complex assembly) could be masked. Finally, SRP is limiting in vivo and competes among a much larger number of translating ribosomes than in translation extracts; mutational effects on SRP-RNC binding (see below: The fingerloop is not strictly required for high-affinity cargo binding by the SRP) are therefore easily masked in the in vitro targeting assay but could contribute more significantly in vivo.
The fingerloop is not strictly required for high-affinity cargo binding by the SRP
To test whether compromised binding affinity for signal sequences accounts for the defects of fingerloop mutants in protein targeting, we compared the binding affinities of the wild-type and mutant SRPs for RNCs bearing the nascent chain of FtsQ, a bona fide SRP substrate (RNCFtsQ; Zhang et al., 2010). RNCs bearing the nascent chain of firefly luciferase (RNCLuc), which contains no signal sequences, served as a control for the ability of SRP to bind ribosomes translating incorrect substrate proteins. SRP was labeled with fluorescein at Cys-421 near the signal sequence–binding groove, and SRP-RNC binding was monitored as a change in the fluorescence anisotropy of Ffh(C421)-fluorescein (Zhang et al., 2010). Equilibrium titrations based on this anisotropy signal showed that wild-type SRP binds to RNCFtsQ and RNCLuc with equilibrium dissociation constants (Kd) of 1.7 and 128 nM, respectively (Figure 3, A and B, filled circles), reflecting a 102-fold contribution of the signal sequence to cargo binding. Unexpectedly, both the ∆FL and LM→AA mutants of SRP were able to bind tightly to RNCFtsQ, with less than threefold change in the value of Kd (Figure 3, A, open symbols, and C). The binding affinity of SRP for RNCLuc was also not substantially affected by the fingerloop mutations (Figure 3, B, open symbols, and C).
FIGURE 3:

Fingerloop mutants did not exhibit significant defects in cargo binding. (A and B) Equilibrium titrations to measure the binding of wild-type Ffh (●), mutant Ffh(∆FL) (◯), and mutant Ffh(LM→AA) (◊) to RNCs bearing the nascent chain from FtsQ (A) or luciferase (B). The lines are quadratic fits of data to Eq. 1 in Materials and Methods. (C) Summary of the binding affinities from (A) and (B). (D and E) FRET was used to monitor the association (D) and dissociation (E) of wild-type SRP, SRP(∆FL) (●), and SRP(LM→AA) (◊) for binding RNC1A9L, as described in Materials and Methods. The results with wt SRP (dotted line) are from Saraogi, Akopian, and Shan, unpublished data. (F) Summary of the association and dissociation rate constants (kon and koff, respectively) of wild-type SRP, SRP(∆FL), and SRP(LM→AA) for binding RNC. The Kd values were calculated according to Kd = koff/kon.
To directly monitor signal sequence interactions with the Ffh M domain, the binding of wild-type and mutant SRPs to the RNC were measured using a fluorescent nonnatural amino acid, 7-hydroxycoumaryl ethylglycine (Cm), incorporated near an engineered signal sequence, 1A9L, on the nascent chain (Saraogi et al., 2011). Förster resonance energy transfer (FRET) between Cm-labeled RNC1A9L and boron dipyromethene fluorescein (BODIPY-FL) labeled at residue 421 of Ffh M domain reports directly on the docking of the signal sequence into its binding groove (Saraogi et al., 2011). This assay allowed us to measure, in real time, the association and dissociation rate constants of cargo-SRP binding (Figure 3, D and E, respectively). The results showed that mutants SRP(∆FL) and SRP(LM→AA) bind and dissociate from RNC1A9L with rate constants that differ by no more than threefold from wild-type SRP (Figure 3F). The values of Kd calculated from these rate constants is only twofold weaker with mutant SRP(∆FL) and sixfold weaker with mutant SRP(LM→AA) (Figure 3F). These results support conclusions from the anisotropy assay; together they indicate that the fingerloop is not strictly necessary for high-affinity signal sequence binding by the SRP. The modest effects of the fingerloop mutants on SRP-RNC binding could contribute, in part, to the phenotype of fingerloop mutants in vivo. Nevertheless, it would be easily masked in the in vitro targeting reaction, in which the concentration of SRP is >50-fold above the Kd values, even with SRP(LM→AA). Thus the observed defects of fingerloop mutants in the in vitro targeting assay could not be attributed to their reduced ability to bind the RNC and instead must arise from subsequent steps in the SRP pathway.
The fingerloop facilitates signal sequence–induced stimulation of SRP•FtsY complex assembly
To efficiently deliver its substrate proteins to the membrane, SRP must rapidly assemble a stable complex with its receptor FtsY. However, to ensure fidelity of protein targeting, complex assembly between free SRP and FtsY is extremely slow but is substantially accelerated by correct cargoes (Zhang et al., 2010) and, to a lesser extent, by signal peptides or the detergent Nikkol, which mimics the effect of signal peptides (Bradshaw et al., 2009). We therefore asked whether efficient SRP•FtsY complex assembly in response to cargo is affected by deletion or mutation of the fingerloop. To this end, we measured the rate constants for formation of the 5′- guanylylimido-diphosphate (GppNHp)-stabilized complex between SRP and FtsY, using either FRET between N-(7-dimethylamino-4-methylcoumarin-3-yl) (DACM)-labeled SRP(C235) and BODIPY-FL–labeled FtsY(C487) or acrylodan-labeled SRP(C235), which specifically changes fluorescence upon guanosine-5′-triphosphate (GTP)-dependent formation of the stable complex (Zhang et al., 2008, 2009). We determined complex assembly rate constants under three conditions: 1) without any stimulant; 2) in the presence of the signal-peptide mimic Nikkol; and 3) in the presence of RNCFtsQ. In the latter cases, SRP was preincubated with saturating concentrations of Nikkol or RNC based on the information from previous studies (Bradshaw et al., 2009; Zhang et al., 2010) and above to ensure that >98% of SRP was loaded with cargo or the signal peptide mimic, so that effects of the fingerloop mutations on cargo/signal sequence–binding affinities were bypassed.
In the absence of any stimulant, complex assembly for wild-type SRP and the fingerloop mutants were slow and differed by no more than threefold, ranging from 250–610 M−1s−1 (Figure 4, A and C). Consistent with previous results (Bradshaw and Walter, 2007; Bradshaw et al., 2009), stable SRP•FtsY complex assembly was accelerated 50-fold with wild-type SRP in the presence of Nikkol, but this stimulation was abolished with the ∆FL and LM→AA mutations (Figures S2 and 4C). These data support the suggestion that the fingerloop helps mediate the signal peptide–induced stimulation of complex assembly (Bradshaw and Walter, 2007).
FIGURE 4:

The effects of fingerloop mutants on SRP•FtsY complex assembly. (A and B) Measurements of SRP•FtsY complex assembly kinetics of wild-type SRP (●), mutant SRP(∆FL) (◯), and mutant SRP(LM→AA) (♦) without any stimulants (A) and in the presence of RNCFtsQ (B). The lines are linear fits of the data to Eq. 2 in Materials and Methods. (C) Summary of the complex formation rates from (A), (B), and Figure S2.
As previously demonstrated, RNCFtsQ exerts a larger stimulatory effect on the SRP•FtsY complex assembly than signal peptides or Nikkol, accelerating their complex assembly more than 103-fold (Figure 4, B and C; Bradshaw et al., 2009; Zhang et al., 2009; Shen et al., 2011). Intriguingly, RNCFtsQ also provided significant stimulation for the mutants SRP(∆FL) and SRP(LM→AA), increasing their complex assembly rate constants by 360- and 620-fold, respectively (Figure 4, B and C). In contrast to the observations in the presence of Nikkol, the mutants SRP(∆FL) and SRP(LM→AA) exhibited only 10- and 2.5-fold slower complex assembly kinetics in the presence of RNCFtsQ, respectively. Thus the additional presence of the ribosome in a complete cargo partially rescued the defects of the fingerloop mutants in mediating efficient SRP•FtsY complex assembly in response to a signal peptide mimic.
GTP-dependent assembly of stable SRP•FtsY complex comprises two steps: the formation of a transient early intermediate followed by a GTP-dependent rearrangement of this intermediate to a stable closed complex (Zhang et al., 2008, 2009). Using established fluorescence assays and conditions (see Materials and Methods), we further dissected which of these steps was affected by the fingerloop mutations. In the presence of cargo, the early intermediate formed by the SRP(∆FL) and SRP(LM→AA) mutants were two- to threefold weaker compared with the early intermediate formed by wild-type SRP (Figure S3 and Table 2). In addition, this intermediate rearranged to the closed complex two- to fourfold slower with mutant SRP(∆FL) and SRP(LM→AA) than with wild-type SRP (Figure S4 and Table 2). Thus the combination of defects in stabilizing the early intermediate and in mediating the early → closed rearrangement accounted for the overall defect of fingerloop mutants in assembling the stable SRP•FtsY complex.
TABLE 2:
Effects of fingerloop mutations on the equilibrium stability of the early complex in the presence of RNCFtsQ (Kd,early) and on the rate constants for rearrangement of the cargo-SRP•FtsY early complex to the stable closed complex (krearrange).
| SRP | ∆FL | LM→AA | |
|---|---|---|---|
| Kd,early (nM) | 86 (1) | 175 (2) | 236 (4) |
| krearrange (s−1) | 0.6 (1) | 0.18 (0.3) | 0.3 (0.5) |
The rate and equilibrium constants are derived from the data in Figures S3 and S4. The values in parenthesis are relative to that of wild-type SRP.
The fingerloop is important for GTPase activation and cargo unloading
Although the fingerloop mutants exhibited defects in cargo-induced stimulation of SRP•FtsY assembly, in the presence of RNC these defects were modest and insufficient to account for their observed defects in cotranslational protein targeting, especially with the ΔFL mutant. We therefore asked whether additional downstream steps in the SRP pathway were also impaired by these mutations. Previous work has shown that after a stable SRP•FtsY complex is assembled, GTPase activation in this complex is crucial for the successful unloading of cargo from the SRP to the translocation machinery on the target membrane (Shan et al., 2007). We therefore asked whether the fingerloop mutations impaired the ability of the SRP•FtsY complex to activate its GTPase sites.
To this end, we monitored the reciprocally stimulated GTPase reaction between SRP and FtsY (Peluso et al., 2001). In this assay, the observed reaction rates at subsaturating FtsY concentrations are rate-limited by and reflect the assembly of the SRP•FtsY complex, whereas the rate constant at saturating FtsY concentrations (kcat) reports on the GTP hydrolysis rate once a stable complex is formed (Figures 5 and S5). The complex formed by the wild-type SRP hydrolyzed GTP efficiently, with a kcat of 100 min−1 (Figures 5 and S5). For both fingerloop mutants, the observed GTPase rates at subsaturating FtsY concentrations were much slower (Figure S5, A and B, kcat/Km), reflecting their kinetic defects in complex assembly in the presence of the signal peptide mimic Nikkol (Figures S2 and 4C). However, once a stable GTPase complex was formed at saturating FtsY concentrations, mutant SRP(LM→AA) exhibited minimal defects in activated GTP hydrolysis, whereas mutant SRP(∆FL) had a significantly reduced GTPase rate (Figures 5 and S5, kcat), indicating an additional defect of this mutant in undergoing GTPase activation.
FIGURE 5:
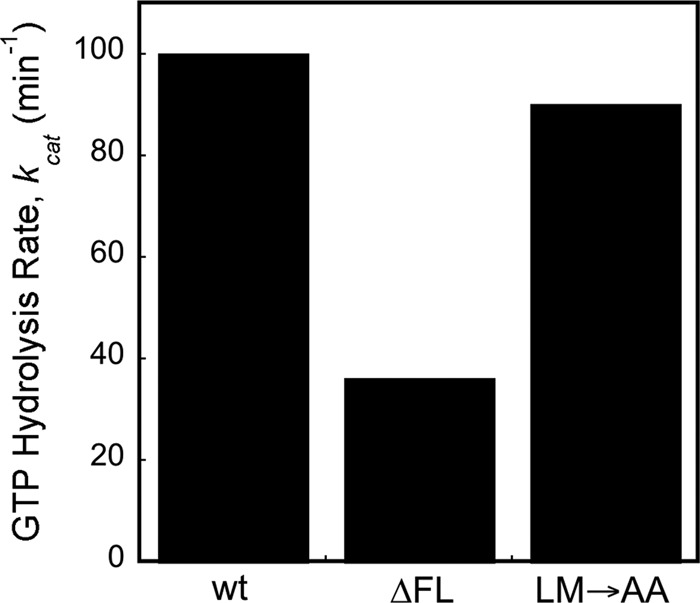
Deletion of the fingerloop results in inefficient GTPase activation. The reciprocally stimulated GTPase reaction between SRP and FtsY was determined for wild-type SRP and mutants SRP(LM→AA) and SRP(∆FL).
Mutant SRPs and FtsYs that specifically block GTPase activation block protein targeting at late stages, when the unloading and transfer of the cargo need to occur (Shan et al., 2007). We therefore asked whether the fingerloop also plays an important role in the timely and efficient transfer of cargo to translocation sites on the target membrane. To address this question, we modified our targeting assay to more specifically isolate this cargo-unloading step (Figure 6A). We generated [35S]methionine-labeled, stalled RNCs bearing the pPL86 nascent chain (RNCpPL86) via in vitro translation. RNCpPL86 was incubated with saturating SRP (wild-type or ∆FL), FtsY, and GTP for sufficient time to allow the formation of a stable RNCpPL86•SRP•FtsY complex, such that the kinetic defect of mutant SRP(ΔFL) in complex assembly was bypassed. Microsomes were then added to trigger the transfer of RNCpPL86 from the targeting complex to translocation sites on the ER membrane, which was monitored at different time points by sedimentation (Figure 6A). This experiment showed that the targeting complex formed by wild-type SRP was able to unload ∼35% of RNCpPL86 to the membrane, and the unloading reaction was complete as early as 15 s (Figure 6B, white bars). In contrast, cargo transfer proceeded much more slowly with mutant SRP(ΔFL), and even after 2 min, <20% of RNCpPL86 stably engaged with the microsomal membrane (Figure 6B, black bars). These results directly demonstrated that the fingerloop plays an important role in the cargo handover event at the last stage of the protein-targeting reaction.
FIGURE 6:

Deletion of the fingerloop impaired unloading of cargo to the ER membrane. (A) Schematic of the experiment to isolate the cargo-unloading process. (B) Percentage of RNCpPL86 stably engaged with the membrane mediated by wild-type SRP and mutant SRP(∆FL).
The fingerloop is crucial for SRP RNA–mediated stimulatory effects
The effects of the fingerloop mutants, especially Ffh(ΔFL), in the GTPase assay above were reminiscent of the effects of removing the SRP RNA (Peluso et al., 2000, 2001; Shen et al., 2011), which accelerates complex assembly between SRP and FtsY and promotes their subsequent GTPase activation. This raises the possibility that the defects of the fingerloop mutants were caused by defective function of the SRP RNA. To test whether this is the case, we measured the GTPase activity for wild-type and mutant Ffh in the absence of the SRP RNA. Under these conditions, both Ffh(∆FL) and Ffh(LM→AA) exhibited kcat and kcat/Km values similar to those of wild-type Ffh (Figure 7, A and D), indicating that the intrinsic ability of Ffh to form a complex with FtsY and to hydrolyze GTP are unaffected by the fingerloop mutations. Thus the defects of the fingerloop mutants in complex assembly and GTPase activation described above likely arise from the inability of the SRP RNA to exert its stimulatory effect on the GTPase interactions.
FIGURE 7:

Effects of fingerloop mutants on complex assembly and GTPase activation are linked to the SRP RNA. (A) Reciprocally stimulated GTPase reaction in the absence of SRP RNA for wild-type Ffh (●) and mutants Ffh(LM→AA) (♦) and Ffh(∆FL) (◯). The lines are fits of data to Eq. 3 in Materials and Methods. Measurement of the disassembly of the SRP•FtsY complex formed by Ffh(∆FL) (B) and Ffh(LM→AA) (C). The lines are single exponential fits of the data, which gave the dissociation rate constants. (D) Summary of the kcat and Km values from the data in (A). (E) Summary of the rate constants obtained from (B) and (C). The equilibrium stability (Kd) of the SRP·FtsY closed complex (wild-type and mutants) were calculated according to Kd = koff/kon, based on rates obtained in (B) and (C) and Figures 4C and S2.
To provide additional evidence for this notion, we tested another unique signature of the action of SRP RNA: its ability to accelerate the disassembly, as well as the assembly, of the Ffh•FtsY complex without perturbing the equilibrium stability of this complex (Peluso et al., 2000; Shen et al., 2011). If the defects exhibited by the fingerloop mutants are associated with defective function of the SRP RNA, then these mutants will phenocopy the effect of SRP RNA deletion and exhibit much slower complex dissociation rates (koff). Using acrylodan-labeled SRP(C235), we measured the dissociation rate constants of the stable SRP•FtsY complex with the fingerloop mutants. The GTPase complex assembled by SRP(∆FL) exhibited a dissociation rate constant 80-fold slower than that of wild-type SRP (Figure 7, B and E), approaching the value observed in the absence of the SRP RNA (Peluso et al., 2000; Shen et al., 2011). Mutant SRP(LM→AA) exhibited a similar, albeit milder, reduction in complex disassembly kinetics (Figure 7, C and E). The equilibrium stability of the SRP•FtsY complex, derived from the complex assembly and disassembly rates, was unaffected by the fingerloop mutants (Figure 7E), analogous to the effects of mutating or removing the SRP RNA. Together, these results strongly suggest that the fingerloop is necessary for SRP RNA to exert its stimulatory effects on the SRP and FtsY GTPases during cotranslational protein targeting.
DISCUSSION
Cotranslational protein targeting by SRP is essential for maintaining the proper localization of proteins in all cells. During this process, recognition of the signal sequence on the translating ribosome must be tightly coupled to its rapid delivery to the target membrane and efficient unloading onto the translocation machinery. This coupling requires that the GTPase domains in the SRP and FtsY actively communicate with spatial and temporal cues from the cargo and the target membrane. In this work, we showed that the universally conserved SRP fingerloop helps convey the information about signal sequence in the M domain of SRP to its NG domain; this facilitates multiple stages of the targeting reaction, including recruitment of the SR, subsequent activation of the GTPases, and the handover of cargo to the translocation machinery in the membrane.
The fingerloop flanks the signal sequence–binding site, forming a flexible flap that could close down on the signal sequence–binding groove upon cargo binding to the SRP (Keenan et al., 1998; Rosendal et al., 2003; Janda et al., 2010; Hainzl et al., 2011). Further, the structural plasticity of the fingerloop, together with the richness of methionine residues in the M domain, has been proposed to provide the conformational flexibility necessary for the SRP to bind diverse signal sequences (Bernstein, 1998; Keenan et al., 1998). Given this, it was surprising to find that mutation or even deletion of the entire fingerloop still resulted in subnanomolar binding affinities of SRP for its substrates. One possible explanation is that the fingerloop exerts a similar effect on both sides of the binding equilibrium: the free Ffh and Ffh bound to the signal sequence. Crystallographic studies showed that in the absence of signal sequences, the fingerloop could insert into the hydrophobic signal sequence–binding groove to stabilize the free Ffh (Figure 8A, SRP; Rosendal et al., 2003). Upon cargo recognition, the interaction of the fingerloop with the binding groove is replaced by interaction with the signal peptide (Figure 8A, RNC•SRP), thereby giving rise to an apparent “isoenergetic” effect on cargo binding affinity. Nevertheless, the results here strongly suggest the fingerloop helps ensure a proper mode of signal-sequence docking in the M domain that is required to induce important conformational changes in the SRP, as manifested by the defects of these mutants in subsequent steps of protein targeting. The observation that the LM→AA mutant binds the RNC more weakly than the ∆FL mutant but is nevertheless more efficient in mediating protein targeting and supporting cell growth further highlights the importance of these downstream steps.
FIGURE 8:

A model for the role of SRP fingerloop (brown) in relaying the information of signal sequence binding to the GTPases and enabling multiple stages of SRP•FtsY interactions during protein targeting is shown in (A). Step 1, the presence of a signal sequence in the M domain is propagated to the NG domain (both in light blue, connected by the flexible linker in dark blue) by the fingerloop, priming the SRP for binding its receptor, FtsY (green), near the SRP RNA's tetraloop end (orange). In step 2, FtsY associates with SRP to form the [RNC·SRP·FtsY]early intermediate. During step 3, [RNC·SRP·FtsY]early rearranges to activate GTP hydrolysis and facilitate the transfer of the RNC to the translocation machinery. Structures or structural models for each complex (PDB IDs [from top to bottom]: 1QZW, 2J28, and 2XKV; Rosendal et al., 2003; Halic et al., 2006; Estrozi et al., 2011) are shown adjacent to the respective SRP/FtsY diagrams. (B) Free-energy profile explaining the different observed effects of fingerloop in the presence of the signal sequence mimic Nikkol (red) or the RNC (black). In both cases, removal of the fingerloop (dashed line) disfavored the conformational change of SRP to an active conformation (ΔΔG) more conducive to complex assembly with FtsY. In the presence of Nikkol, this conformational change is unfavorable, even with wild-type SRP; thus the effect of the fingerloop is fully manifested (ΔΔG = ΔΔG‡Nik). In contrast, when bound to the RNC, the SRP is preorganized into the active conformation; thus, although removal of the fingerloop exerts the same destabilizing effect, the full extent of its defect is masked in the observed complex assembly rates (ΔΔG‡RNC < ΔΔG).
A major effect of fingerloop mutations is that the SRP and FtsY GTPases lose their ability to respond to the signal peptide mimic Nikkol and efficiently assemble a complex with one another. How does the fingerloop exert this effect? Several observations here and from previous work offered a few clues. The fingerloop mutations phenocopied the effects of deleting the SRP RNA on SRP•FtsY assembly, suggesting they abolished the ability of this RNA to accelerate this process. The conserved tetraloop of the SRP RNA provides a tethering interaction that holds FtsY near the SRP GTPase to facilitate their initial encounter (Siu et al., 2007; Shen and Shan, 2010). In this mechanism, the SRP's NG domain must be properly positioned close to the RNA tetraloop; this likely requires a reorientation of the relative position of the M and NG domains from that in the free SRP, which appears to be triggered by the cargo and less effectively by the signal peptide or the signal peptide mimic Nikkol (Figure 8A, step 1; Halic et al., 2006; Bradshaw et al., 2009; Zhang et al., 2009, 2010; Estrozi et al., 2011; Hainzl et al., 2011; Shen et al., 2011). We therefore deem it most likely that the SRP fingerloop helps the SRP to undergo this structural rearrangement in response to signal sequence binding, inducing it into a more active conformation for FtsY recruitment.
This hypothesis is further supported by the observation that mutation of the SRP fingerloop has a much less deleterious effect when the SRP is bound to a complete cargo, RNCFtsQ, than to the less effective signal peptide mimic Nikkol. This intriguing relationship between the SRP fingerloop and the RNC is akin to “synthetic lethality” effects, which suggests that the fingerloop and the RNC play overlapping and redundant roles in inducing a more active conformation of SRP for complex assembly (Figure 8B). The RNC, by interacting with both the M and N domains of the SRP, is highly effective in bringing the SRP's NG domain into close proximity to the RNA tetraloop. Thus, in the presence of RNC, the SRP is predominantly in the active conformation (Figure 8B, black line). This redundancy would compensate for the destabilizing effect of the fingerloop mutations, partially masking their deleterious effect (Figure 8B, ΔΔG‡RNC < ΔΔGFL). In contrast, Nikkol is less effective at inducing SRP molecules into the “active” structure (Figure 8B, red line; Shen et al., 2011). Although mutation of the fingerloop exerts the same destabilizing effect on the active conformation, the full extent of this effect is manifested (Figure 8B, ΔΔG‡Nikkol = ΔΔGFL), as there is no redundancy to buffer the deleterious effect of these mutations.
Once the early RNC•SRP•FtsY complex is formed (Figure 8A, step 2; Zhang et al., 2008, 2009; Estrozi et al., 2011), it undergoes additional conformational changes that are essential for subsequent steps of the pathway and ultimately lead to unloading of the cargo (Figure 8A, step 3; Shan et al., 2007; Ataide et al., 2011) and activation of GTP hydrolysis (Shan et al., 2007; Siu et al., 2007; Zhang et al., 2009). The SRP RNA also facilitates GTPase activation (Peluso et al., 2001; Ataide et al., 2011), and recent work further suggested a potential model for this stimulatory effect: at late stages of the GTPase rearrangements, the Ffh•FtsY NG domain complex could detach from SRP RNA's tetraloop and, instead, interact with the 5′, 3′-distal end of the SRP RNA, where GTP hydrolysis can be stimulated (Figure 8A, step 3; Ataide et al., 2011). In this structure, the NG domain complex would be removed from the signal sequence–binding site and the ribosome exit site, which could represent a conformation more conducive to the release of cargo. Regardless of the model, our observations here that deletion of the fingerloop abolishes SRP RNA–dependent GTPase activation and also impairs the unloading of cargo strongly suggest that the fingerloop is also intimately involved in late conformational rearrangements of SRP that complete the protein-targeting cycle (Figure 8A, step 3).
Together, the results presented here demonstrate that the SRP fingerloop contributes to multiple steps in the targeting reaction beyond initial signal sequence binding, including the recruitment of the SR, subsequent GTPase activation, and cargo unloading. In light of the recent structural work, it is intriguing to note that all of these molecular steps require global reorganization of the relative position of the M and NG domains of the SRP, during which the linker connecting its G and M domains undergoes major restructuring (Figure 8A). Because of its proximity to the signal sequence–binding site, we speculate that the fingerloop is the first structural element that senses signal sequences and changes conformation. Through the remainder of the M domain, this information is amplified and leads to the restructuring of the M-G domain linker, thus inducing more global rearrangements of the SRP in both the early and late stages of the protein-targeting reaction.
MATERIALS AND METHODS
Materials
The strains and plasmids used in this study are shown in Table 1. All antibiotics and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). Restriction enzymes used for cloning were obtained from New England Biolabs (Ipswich, MA) and Fermentas Life Sciences (Glen Burnie, MD). Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA). Antibiotics were used at the following concentrations: ampicillin (Amp), 100 μg/ml; kanamycin (Kan), 30 μg/ml; spectinomycin (Spc), 100 μg/ml.
Ffh, FtsY, and 4.5S RNA were expressed and purified using established protocols (Peluso et al. 2001). Single cysteine mutations were constructed using the QuikChange mutagenesis protocol (Stratagene, Agilent, Santa Clara, CA) and were purified using the same procedures as wild-type protein. The fluorescent dyes fluorescein, BODIPY-FL, DACM, and acrylodan were purchased from Invitrogen (Carlsbad, CA). RNCs were prepared and purified as previously described (Schaffitzel and Ban, 2007; Saraogi et al., 2011). Single cysteine mutants of Ffh and FtsY were labeled using maleimide chemistry and purified as previously described (Zhang et al. 2008). Labeling efficiency was usually >95%.
Plasmid constructions
All plasmids are derivatives of pBADffh6x, a plasmid that expresses an allele of ffh that expresses a hexahistidine epitope tag at the carboxy terminus of Ffh (Table 1). This plasmid was made by PCR amplification of ffh from E. coli genomic DNA using primers ffhN.S (ACCATGGTTGATAATTTAACCGATCGTTTGTCGC) and ffhC-AS (TCAATGGTGATGGTGATGATGACCGGTACG). The primers were designed to introduce an NcoI restriction site (shown in bold in primer ffhN-F) to the PCR product. The amplification product was introduced to pBAD-topo (Invitrogen), such that the 3′ end of ffh was fused in-frame with a hexahistidine coding sequence. The resulting plasmid was subsequently digested with NcoI and religated yielding pBADffh6x.
This plasmid was further modified using site-directed mutagenesis to introduce an NheI restriction site at the start of the fingerloop coding region, yielding pBADffhN6x. No amino acid substitutions resulted from this change. The fingerloop region of Ffh, corresponding to amino acids 350–369 (Figure 1A), was deleted in plasmid pBADffhN6xΔFL. This plasmid was made by PCR amplification of ffh using primers ffhNheI-FL.S (ATGGCTAGCAAAGTGCTGGTGCGTATGGAAGCC) and ffhNhe-FL.AS (CCCCCAGGCTTCCCTGGTCC). The PCR product was digested with NheI (site shown in bold) and BlpI (site contained within the PCR product), and the gel-purified DNA was ligated into pBADffhN6x digested with the same enzymes.
Two additional ffh alleles were also constructed by site-directed mutagenesis of pBADffhN6x (ffhLM→AA and ffhPG→AA; Figure 1A). The relevant region of each plasmid construct was confirmed by DNA sequencing (DNA Facility, Iowa State University, Ames, IA). Expression of ffh from all plasmids was confirmed by using the InVision His-Tag In-Gel Stain (Invitrogen), which was used to detect the hexahistidine epitope tag at the carboxy terminus of Ffh (Figure S1).
Strain constructions
To characterize function of the fingerloop mutants in vivo, we constructed SLD108. This strain is deleted for genes whose products are necessary for arabinose transport (araFGH, araE) and utilization (araBAD). In addition, SLD108 expresses a mutant LacY permease that allows homogenous uptake of arabinose throughout the population, such that the heterogeneity of gene expression of genes under araC control is eliminated (Morgan-Kiss et al., 2002). For construction of SLD108, ECF529 (Bowers et al., 2004) was first modified by lambda Red homologous recombination to inactivate bla (AmpR) encoded on the chromosome of this strain and replacing it with a KanR gene cassette. For this, primers bla-KD4.S (ATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTtgtgtaggctggagctgcttc) and bla-KD4.AS (TTACCAATGCTTAATCAGTGAGGCACCTATCTCAGCGATCTGTCTAcatatgaatatcctccttag) were used to amplify a PCR product using pKD4 as a template (Datsenko and Wanner, 2000). Sequences in uppercase designate the portions of the primers with homology to bla, and sequences in lowercase are homologous to pKD4 (Datsenko and Wanner, 2000). The gel-purified PCR product was electroporated into ECF529 transformed with pSIM5, as previously described (Datta et al., 2006), and KanR, AmpS recombinants were identified.
The KanR cassette was subsequently deleted using Flp-mediated site-specific recombination, as previously described (Datsenko and Wanner, 2000). For complete construction of SLD108, the resulting KanS (sensitive) strain was subsequently transformed with pFfhTSpc expressing ffh+ from a temperature-sensitive replicon (Phillips, 1999) and imparting SpcR (spectinomycin resistant), and the ffh::kan1 allele (Phillips and Silhavy, 1992) was introduced by P1 transduction, as previously described (Peterson and Phillips, 2008).
For complementation tests, plasmids expressing the different ffh alleles were transformed into SLD108. AmpR colonies were restreaked on LB+Amp agar plates and incubated at 30°C and 42°C. Where indicated, dilutions of saturated cultures were spotted onto LB+Amp plates containing 0%, 0.01%, and 0.02% of l-arabinose and incubated overnight at 42°C.
Fluorescence measurements
Fluorescence measurements were carried out on a FluoroLog-3-22 spectrofluorometer (Jobin-Yvon, Edison, NJ) in assay buffer (50 mM KHEPES, pH 7.5, 150 mM KOAc, 10 mM Mg(OAc)2, 2 mM dithiothreitol [DTT], 10% glycerol, with or without 0.01% Nikkol). The buffer also contained 100–200 μM GppNHp, a nonhydrolyzable GTP analogue. All reactions were carried out at 25ºC, unless otherwise stated.
The binding affinities of SRP for RNCs or ribosomes were determined using two methods. In the first approach, fluorescence anisotropy measurements were carried out with 5–10 nM of fluorescein-labeled Ffh(C421) and various concentrations of RNCFtsQ or RNCLuc. Observed anisotropy values (A) are fitted to Eq. 1,
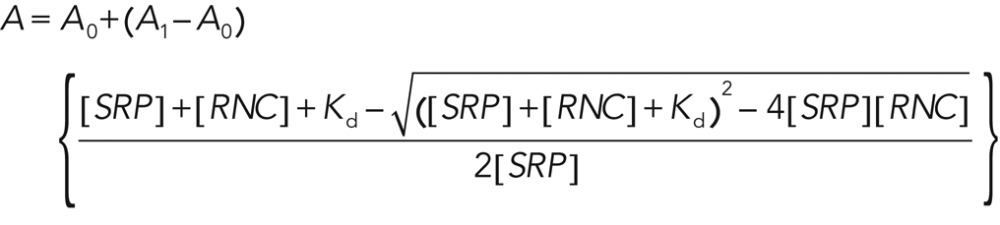 (1) (1)
|
in which A0 is the anisotropy value of free SRP, A1 is the anisotropy value when SRP is bound to cargo, and Kd is the equilibrium dissociation constant of SRP for the RNC (Zhang et al. 2010). In a second approach, the binding of SRP to the RNC was determined using FRET between 7-hydroxycoumarin–labeled RNC1A9L and BODIPY-labeled SRP (Saraogi et al., 2011). Time courses for SRP-RNC assembly (kon) and disassembly (koff) were determined using either a FluoroLog-3-22 spectrofluorometer (Jobin-Yvon) or an SF-2004 stopped-flow apparatus (KinTek, Pittsford, NY). For determining SRP-RNC assembly rates, 20 nM RNC1A9L was mixed with various concentrations of SRP. Linear fits (Eq. 2) of the observed rate constants for SRP-RNC binding (kobsd) was plotted as a function of SRP concentration to give the second-order association rate constant, kon:
|
|
For determining SRP-RNC disassociation rate constants, 20 nM RNC1A9L was preincubated with saturating amounts of labeled SRP. The preformed RNC•SRP complex was then chased with >10-fold excess unlabeled SRP. Exponential fits to the time course give the dissociation rate constant.
Association rate constants for SRP–FtsY complex formation were determined using two different assays (Zhang et al., 2009): 1) FRET between donor (DACM)- and acceptor (BODIPY-FL)-labeled SRP(C235) and FtsY(C487), respectively; or 2) change in the fluorescence of SRP(C235) labeled with acrylodan, an environmentally sensitive dye. In all cases, saturating concentrations of RNCs (50- or 100-fold above the respective Kd value) were used to ensure that SRP was bound with cargo. Complex assembly was initiated by mixing SRP with various amounts of FtsY in the presence of 100 μM GppNHp, and the time course of fluorescence change was monitored using a FluoroLog-3-22 spectrofluorometer (Jobin-Yvon) or an SF-2004 stopped-flow apparatus (KinTek). The data were fitted to Eq. 2, except that the term [SRP] was replaced by [FtsY].
Equilibrium titrations of the early intermediate were carried out using FRET, as described previously (Zhang et al., 2008). Rate constants for rearrangement of the early intermediate to the stable complex were measured using Ffh-C235 labeled with acrylodan. An RNC•SRP•FtsY early intermediate was preformed in the presence of saturating SRP/Ffh and FtsY with respect to the Kd value of the early intermediate. The reaction was initiated by mixing 500 mM GppNHp with the early intermediate. The time course of fluorescence change was fitted to single-exponential functions to give the rearrangement rate constants. For experiments concerning SRP or Ffh loaded with different RNCs, concentrations 50- to 100-fold above their respective Kd for Ffh were used to ensure > 90% occupancy of SRP by the cargo.
GTPase assay
All GTPase assays were performed at 25°C in assay buffer (50 mM KHEPES, pH 7.5, 150 mM KOAc, 10 mM Mg(OAc)2, 2 mM DTT, 0.01% Nikkol, and 10% glycerol). GTP hydrolysis reactions were followed and analyzed as previously described (Peluso et al., 2001). In general, reciprocally stimulated GTPase reactions between SRP and FtsY were determined in reactions containing 100–500 nM wild-type or mutant Ffh, 200–1000 nM 4.5S RNA (where applicable), 100 μM GTP, doped with γ-32P-GTP (MP Biomedicals, Solon, OH), and various concentrations of FtsY. The concentration dependence of the observed rate constant (kobsd) is fitted to Eq. 3, in which kcat is the rate constant at saturating FtsY concentrations and Km is the concentration of FtsY that gives half the maximal rate:
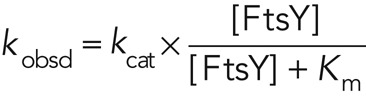 (3) (3)
|
Translocation assay
The protein-targeting efficiency of wild-type Ffh and mutants ∆FL and LM→AA was determined by a cotranslational translocation assay using pPL as a substrate, as described previously (Shan et al., 2007). Reactions were carried out using 333 nM SRP, various concentrations of FtsY, and 2 eq of trypsin-digested, salt-washed ER microsomal membranes (TKRM).
Cargo-unloading assay
The targeting of RNCpPL86 was performed with slight modifications of the procedures used by Wilson et al. (1988). Stalled RNCs bearing pPL86 were generated by in vitro translation using wheat germ translation extract (Promega Corporation, Madison, WI) in the presence of 35[S]methionine (MP Biomedicals). The stalled RNCpPL86 was incubated with saturating SRP or SRP(∆FL), FtsY, and GTP (to final concentrations of 250 nM, 1 μM, and 3 mM, respectively) for 30 min at room temperature to ensure that the ternary complex, RNCpPL86•SRP/SRP(∆FL)•FtsY, was formed. Cargo unloading was initiated by addition of 2 eq of TKRM at 25°C. The reaction was stopped at different time points (15, 30, 60, 90, and 120 s) by flash-freezing with liquid nitrogen. The samples were thawed on ice and immediately layered onto a sucrose cushion (0.5 M sucrose, 50 mM KHEPES, pH 7.5, 150 mM KOAc, 10 mM Mg(OAc)2, and 2 mM DTT). Membrane-bound RNCs were sedimented by centrifugation at 55,000 rpm at 4°C for 3 min (TLA100, Optima TLX Ultracentrifuge; Beckman Coulter, Brea, CA). The reaction was analyzed by 15% SDS–PAGE and autoradiography.
Supplementary Material
Acknowledgments
We thank members of the Shan group for helpful comments on the manuscript. This work was supported by National Institutes of Health (NIH) grant GM078024, the Beckman Young Investigator Award, the David and Lucile Packard Fellowship in Science and Engineering, and the Henry Dreyfus Teacher-Scholar Award to S.-O.S. G.J.P. was supported by NIH grant R01 GM069628. A.R.A. was supported by NIH/NRSA training grant 5T32GM07616 and by the Betty and Gordon Moore Foundation.
Abbreviations used:
- Amp
ampicillin
- BODIPY-F
boron dipyromethene fluorescein
- Cm
7-hydroxycoumaryl ethylglycine
- DACM
N-(7-dimethylamino-4-methylcoumarin-3-yl)
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- FRET
Förster resonance energy transfer
- GppNHp
5′-guanylylimido-diphosphate
- GTP
guanosine-5′-triphosphate
- Kan
kanamycin
- pPL
preprolactin
- RNC
ribosome nascent chain complex
- Spc
spectinomycin
- SR
SRP receptor
- SRP
signal recognition particle
- TKRM
trypsin-digested, salt-washed ER microsomal membranes
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0434) on November 7, 2012.
*Present address: Department of Medicine, Division of Infectious Diseases, Vanderbilt University, Nashville, TN 37232.
REFERENCES
- Ataide SF, Schmitz N, Shen K, Ke A, Shan SO, Doudna JA, Ban N. The crystal structure of the signal recognition particle in complex with its receptor. Science. 2011;331:881–886. doi: 10.1126/science.1196473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey RT, Rambo RP, Lucast L, Rha B, Doudna JA. Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science. 2000;287:1232–1239. doi: 10.1126/science.287.5456.1232. [DOI] [PubMed] [Google Scholar]
- Batey RT, Sagar MB, Doudna JA. Structural and energetic analysis of RNA recognition by a universally conserved protein from the signal recognition particle. J Mol Biol. 2001;307:229–246. doi: 10.1006/jmbi.2000.4454. [DOI] [PubMed] [Google Scholar]
- Bernstein HD. Protein targeting: getting into the groove. Curr Biol. 1998;8:R715–R718. doi: 10.1016/s0960-9822(98)70456-7. [DOI] [PubMed] [Google Scholar]
- Bowers LM, Lapoint K, Anthony L, Pluciennik A, Filutowicz M. Bacterial expression system with tightly regulated gene expression and plasmid copy number. Gene. 2004;340:11–18. doi: 10.1016/j.gene.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Bradshaw N, Neher SB, Booth DS, Walter P. Signal sequences activate the catalytic switch of SRP RNA. Science. 2009;323:127–130. doi: 10.1126/science.1165971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw N, Walter P. The signal recognition particle (SRP) RNA links conformational changes in the SRP to protein targeting. Mol Biol Cell. 2007;18:2728–2734. doi: 10.1091/mbc.E07-02-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleverley RM, Zheng N, Gierasch LM. The cost of exposing a hydrophobic loop and implications for the functional role of 4.5 S RNA in the Escherichia coli signal recognition particle. J Biol Chem. 2001;276:19327–19331. doi: 10.1074/jbc.M011130200. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Constantino N, Court DL. A set of recombineering plasmids for gram-negative bacteria. Gene. 2006;379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Doudna JA, Batey RT. Structural insights into the signal recognition particle. Annu Rev Biochem. 2004;73:539–557. doi: 10.1146/annurev.biochem.73.011303.074048. [DOI] [PubMed] [Google Scholar]
- Egea PF, Shan SO, Napetschnig J, Savage DF, Walter P, Stroud RM. Substrate twinning activates the signal recognition particle and its receptor. Nature. 2004;427:215–221. doi: 10.1038/nature02250. [DOI] [PubMed] [Google Scholar]
- Estrozi LF, Boehringer D, Shan SO, Ban N, Schaffitzel C. Cryo-EM structure of the E. coli translating ribosome in complex with SRP and its receptor. Nat Struct Mol Biol. 2011;18:88–90. doi: 10.1038/nsmb.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM. Heterodimeric GTPase core of the SRP targeting complex. Science. 2004;303:373–377. doi: 10.1126/science.1090827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freymann DM, Keenan RJ, Stroud RM, Walter P. Structure of the conserved GTPase domain of the signal recognition particle. Nature. 1997;385:361–364. doi: 10.1038/385361a0. [DOI] [PubMed] [Google Scholar]
- Gilmore R, Blobel G, Walter P. Protein translocation across the endoplasmic reticulum. I. Detection in the microsomal membrane of a receptor for the signal recognition particle. J Cell Biol. 1982a;95:463–469. doi: 10.1083/jcb.95.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore R, Walter P, Blobel G. Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J Cell Biol. 1982b;95:470–477. doi: 10.1083/jcb.95.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainzl T, Huang S, Merilainen G, Brannstrom K, Sauer-Eriksson AE. Structural basis of signal-sequence recognition by the signal recognition particle. Nat Struct Mol Biol. 2011;18:389–391. doi: 10.1038/nsmb.1994. [DOI] [PubMed] [Google Scholar]
- Halic M, Becker T, Pool MR, Spahn CM, Grassucci RA, Frank J, Beckmann R. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427:808–814. doi: 10.1038/nature02342. [DOI] [PubMed] [Google Scholar]
- Halic M, Beckmann R. The signal recognition particle and its interactions during protein targeting. Curr Opin Struct Biol. 2005;15:116–125. doi: 10.1016/j.sbi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Halic M, Blau M, Becker T, Mielke T, Pool MR, Wild K, Sinning I, Beckmann R. Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature. 2006;444:507–511. doi: 10.1038/nature05326. [DOI] [PubMed] [Google Scholar]
- Janda CY, Li J, Oubridge C, Hernandez H, Robinson CV, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–510. doi: 10.1038/nature08870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan RJ, Freymann DM, Stroud RM, Walter P. The signal recognition particle. Annu Rev Biochem. 2001;70:755–775. doi: 10.1146/annurev.biochem.70.1.755. [DOI] [PubMed] [Google Scholar]
- Keenan RJ, Freymann DM, Walter P, Stroud RM. Crystal structure of the signal sequence binding subunit of the signal recognition particle. Cell. 1998;94:181–191. doi: 10.1016/s0092-8674(00)81418-x. [DOI] [PubMed] [Google Scholar]
- Koch HG, Moser M, Muller M. Signal recognition particle-dependent protein targeting, universal to all kingdoms of life. Rev Physiol Biochem Pharmacol. 2003;146:55–94. doi: 10.1007/s10254-002-0002-9. [DOI] [PubMed] [Google Scholar]
- Montoya G, Svensson C, Luirink J, Sinning I. Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature. 1997;385:365–368. doi: 10.1038/385365a0. [DOI] [PubMed] [Google Scholar]
- Morgan-Kiss RM, Wadler C, Cronan JE., Jr. Long-term and homogeneous regulation of the Escherichia coli araBAD promoter by use of a lactose transporter of relaxed specificity. Proc Natl Acad Sci USA. 2002;99:7373–7377. doi: 10.1073/pnas.122227599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso P, Herschlag D, Nock S, Freymann DM, Johnson AE, Walter P. Role of 4.5S RNA in assembly of the bacterial signal recognition particle with its receptor. Science. 2000;288:1640–1643. doi: 10.1126/science.288.5471.1640. [DOI] [PubMed] [Google Scholar]
- Peluso P, Shan SO, Nock S, Herschlag D, Walter P. Role of SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry. 2001;40:15224–15233. doi: 10.1021/bi011639y. [DOI] [PubMed] [Google Scholar]
- Peterson JM, Phillips GJ. Characterization of conserved bases in 4.5S RNA of Escherichia coli by construction of new F′ factors. J Bacteriol. 2008;190:7709–7718. doi: 10.1128/JB.00995-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GJ. New cloning vectors with temperature sensitive replication. Plasmid. 1999;41:78–81. doi: 10.1006/plas.1998.1380. [DOI] [PubMed] [Google Scholar]
- Phillips GJ, Silhavy TJ. The E. coli ffh gene is necessary for viability and efficient protein export. Nature. 1992;359:744–746. doi: 10.1038/359744a0. [DOI] [PubMed] [Google Scholar]
- Pool MR, Stumm J, Fulga TA, Sinning I, Dobberstein B. Distinct modes of signal recognition particle interaction with the ribosome. Science. 2002;297:1345–1348. doi: 10.1126/science.1072366. [DOI] [PubMed] [Google Scholar]
- Powers T, Walter P. Co-translational protein targeting catalyzed by the Escherichia coli signal recognition particle and its receptor. EMBO J. 1997;16:4880–4886. doi: 10.1093/emboj/16.16.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendal KR, Wild K, Montoya G, Sinning I. Crystal structure of the complete core of archaeal signal recognition particle and implications for interdomain communication. Proc Natl Acad Sci USA. 2003;100:14701–14706. doi: 10.1073/pnas.2436132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraogi I, Zhang D, Chandrasekaran S, Shan SO. Site-specific fluorescent labeling of nascent proteins on the translating ribosome. J Am Chem Soc. 2011;133:14936–14939. doi: 10.1021/ja206626g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffitzel C, Ban N. Generation of ribosome nascent chain complexes for structural and functional studies. J Struct Biol. 2007;158:463–471. doi: 10.1016/j.jsb.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Schaffitzel C, Oswald M, Berger I, Ishikawa T, Abrahams JP, Koerten HK, Koning RI, Ban N. Structure of the E. coli signal recognition particle bound to a translating ribosome. Nature. 2006;444:503–506. doi: 10.1038/nature05182. [DOI] [PubMed] [Google Scholar]
- Shan SO, Chandrasekar S, Walter P. Conformational changes in the GTPase modules of the signal reception particle and its receptor drive initiation of protein translocation. J Cell Biol. 2007;178:611–620. doi: 10.1083/jcb.200702018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan SO, Walter P. Co-translational protein targeting by the signal recognition particle. FEBS Lett. 2005;579:921–926. doi: 10.1016/j.febslet.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Shen K, Shan SO. Transient tether between the SRP RNA and SRP receptor ensures efficient cargo delivery during cotranslational protein targeting. Proc Natl Acad Sci USA. 2010;107:7698–7703. doi: 10.1073/pnas.1002968107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Zhang X, Shan SO. Synergistic actions between the SRP RNA and translating ribosome allow efficient delivery of the correct cargos during cotranslational protein targeting. RNA. 2011;17:892–902. doi: 10.1261/rna.2610411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu FY, Spanggord RJ, Doudna JA. SRP RNA provides the physiologically essential GTPase activation function in cotranslational protein targeting. RNA. 2007;13:240–250. doi: 10.1261/rna.135407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Johnson AE. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu Rev Cell Biol. 1994;10:87–119. doi: 10.1146/annurev.cb.10.110194.000511. [DOI] [PubMed] [Google Scholar]
- Wilson C, Connoly T, Morison T, Gilmore R. Integration of membrane proteins into the endoplasmic reticulum requires GTP. J Cell Biol. 1988;107:67–77. doi: 10.1083/jcb.107.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kung S, Shan SO. Demonstration of a multistep mechanism for assembly of the SRP x SRP receptor complex: implications for the catalytic role of SRP RNA. J Mol Biol. 2008;381:581–593. doi: 10.1016/j.jmb.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Rashid R, Wang K, Shan SO. Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science. 2010;328:757–760. doi: 10.1126/science.1186743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Schaffitzel C, Ban N, Shan SO. Multiple conformational switches in a GTPase complex control co-translational protein targeting. Proc Natl Acad Sci USA. 2009;106:1754–1759. doi: 10.1073/pnas.0808573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Gierasch LM. Domain interactions in E. coli SRP: stabilization of M domain by RNA is required for effective signal sequence modulation of NG domain. Mol Cell. 1997;1:79–87. doi: 10.1016/s1097-2765(00)80009-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.