

Selective degradation of the mutant protein responsible for most cystic fibrosis, F508del cystic fibrosis transmembrane conductance regulator (CFTR), is initiated by Hsp27, which associates with the small ubiquitin-like modifier (SUMO) E2, Ubc9. They modify F508del with SUMO-2/3, directing F508del to a SUMO-targeted ubiquitin ligase, RNF4. This work implicates SUMO and RNF4 in quality control of a cytosolic transmembrane protein.
Abstract
Small heat shock proteins (sHsps) bind destabilized proteins during cell stress and disease, but their physiological functions are less clear. We evaluated the impact of Hsp27, an sHsp expressed in airway epithelial cells, on the common protein misfolding mutant that is responsible for most cystic fibrosis. F508del cystic fibrosis transmembrane conductance regulator (CFTR), a well-studied protein that is subject to cytosolic quality control, selectively associated with Hsp27, whose overexpression preferentially targeted mutant CFTR to proteasomal degradation. Hsp27 interacted physically with Ubc9, the small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, implying that F508del SUMOylation leads to its sHsp-mediated degradation. Enhancing or disabling the SUMO pathway increased or blocked Hsp27’s ability to degrade mutant CFTR. Hsp27 promoted selective SUMOylation of F508del NBD1 in vitro and of full-length F508del CFTR in vivo, which preferred endogenous SUMO-2/3 paralogues that form poly-chains. The SUMO-targeted ubiquitin ligase (STUbL) RNF4 recognizes poly-SUMO chains to facilitate nuclear protein degradation. RNF4 overexpression elicited F508del degradation, whereas Hsp27 knockdown blocked RNF4’s impact on mutant CFTR. Similarly, the ability of Hsp27 to degrade F508del CFTR was lost during overexpression of dominant-negative RNF4. These findings link sHsp-mediated F508del CFTR degradation to its SUMOylation and to STUbL-mediated targeting to the ubiquitin–proteasome system and thereby implicate this pathway in the disposal of an integral membrane protein.
INTRODUCTION
Successful folding and assembly is a prerequisite for protein exit from the endoplasmic reticulum (ER), whereas the retention of proteins at quality control (QC) checkpoints generally results in their ubiquitylation and degradation by the 26S proteasome, a process denoted as ER-associated degradation (ERAD; Kostova and Wolf, 2003; McCracken and Brodsky, 2003). Even at early stages of biogenesis, proteins begin to encounter a series of QC events that monitor proper protein folding and domain assembly (Ellgaard and Helenius, 2003). ERQC assures that only competent proteins arrive at their appropriate cellular destinations, since the accumulation of aberrant proteins leads to cell stress and the formation of toxic protein aggregates. The selection of proteins for ERAD may be mediated by molecular chaperones, whose bipolar properties facilitate protein folding or degradation, depending on the conformational competency of the target protein (Fewell et al., 2001; Ellgaard and Helenius, 2003).
A prominent ERAD substrate that is subject to cytosolic QC is the cystic fibrosis transmembrane conductance regulator (CFTR), the basis of the cAMP/protein kinase A–induced, anion conductance at the apical membranes of fluid-secreting epithelial cells, including those of the airways, pancreas, and intestines (Pilewski and Frizzell, 1999). Similar to other ATP-binding cassette family members, CFTR (ABCC7) has a modular, multidomain structure, composed of two membrane-spanning domains (MSD1 and MSD2, each composed of six transmembrane segments) and two cytoplasmic nucleotide-binding domains (NBD1 and NBD2). CFTR also contains a central regulatory (R) domain, the primary site of protein kinase–mediated anion channel regulation. The common cystic fibrosis (CF) disease mutant, F508del CFTR (Riordan et al., 1989), omits a phenylalanine from NBD1, defining a class of mutations having defective biogenesis and essentially complete ERAD (Cheng et al., 1990). CFTR's complex folding pattern is reflected in the fact that more than half of the wild-type protein is also degraded in most cells.
CFTR folding is facilitated by an ER-based core chaperone machinery that includes Hsp70 (Yang et al., 1993; Rubenstein and Zeitlin, 2000; Choo-Kang and Zeitlin, 2001), Hsp90 (Loo et al., 1998; Youker et al., 2004), the Hsp40 cochaperones (Meacham et al., 1999; Farinha et al., 2002; Zhang et al., 2002, 2006; Alberti et al., 2004), and calnexin (Pind et al., 1994; Okiyoneda et al., 2004; Farinha and Amaral, 2005). These interactions have been shown to decrease NBD1 aggregation in vitro and to assist with productive CFTR folding (Strickland et al., 1997). However, unstable conformations of CFTR remain bound to chaperones. A prolonged association with Hsp70/Hsp90, for example, allows recruitment of the ubiquitin ligase C-terminus of Hsp70-interacting protein (CHIP), resulting in CFTR ubiquitylation and its degradation by the 26S proteasome (Jensen et al., 1995; Ward et al., 1995; Meacham et al., 2001; Sun et al., 2006; Younger et al., 2006).
Conformational differences in wild-type and F508del CFTR can be monitored by comparing their proteolytic cleavage patterns. While mature, wild-type CFTR exhibits protease resistance, reflecting a compact, folded state of the protein, the digestion patterns of immature wild-type and F508del proteins are similar and less protease-resistant, implying more open, unfolded conformations (Zhang et al., 1998; Du et al., 2005). These findings support the concept that ER-retained F508del CFTR achieves an intermediate conformation along the normal CFTR folding pathway, but that its further maturation is kinetically arrested at one or more ERQC checkpoint(s). This concept implies that F508del CFTR can be rescued from ERAD if the limiting QC step(s) can be identified and appropriately manipulated. Indeed, F508del CFTR can be rescued biochemically and functionally by low temperature (Denning et al., 1992), intragenic suppressors (Teem et al., 1996; DeCarvalho et al., 2002), chemical (Welch, 2004) and molecular (Zhang et al., 2001; Wang et al., 2006) chaperone manipulations, or pharmacophores (Pedemonte et al., 2005; Van Goor et al., 2006). Therefore efforts to identify the QC checkpoints that mediate protein folding versus degradation represent potential therapeutic targets for the correction of most CF, and they could provide significant targets in other diseases of protein folding.
As a foreign protein, CFTR is quantitatively degraded in yeast (Zhang et al., 2001). Using a genomic screen in yeast expressing CFTR, we found increased small heat shock protein (sHsp) expression to reflect sHsp involvement in CFTR ERAD (Ahner et al., 2007). Deletion of the two known yeast sHsps completely stabilized CFTR without affecting the degradation of soluble or other integral membrane ERAD substrates. When overexpressed in human embryonic kidney (HEK293) cells, the mammalian sHsp, αA-crystallin, accelerated F508del CFTR degradation without markedly influencing that of wild-type CFTR.
sHsps have been implicated as ATP-independent chaperones that stabilize proteins during cell stress (Haslbeck et al., 2005) until more favorable conditions return (Horwitz, 1992; Jakob et al., 1993; Ehrnsperger et al., 1997; Lee et al., 1997; Haslbeck et al., 1999, 2004; Biswas and Das, 2004). Ten sHsp homologues are expressed in mammals. Model protein studies show that sHsps tend not to interact with either native conformations or completely denatured proteins, but with proteins of an intermediate, foldable conformation, to maintain their solubility (Rajaraman et al., 1996; Treweek et al., 2000; Lindner et al., 2001). Recent studies of mutant CFTR suggest that altered physical properties of F508del NBD1 impair its ability to participate in effective domain–domain assembly. These transitional conformations of the protein must be stabilized to prevent aggregation, and this requirement would be consistent with sHsp interactions. As for other chaperones, however, if mutant CFTR fails to achieve its native conformation, this interaction targets the protein for ERAD.
In this study, we examined the mechanism of sHsp-mediated CFTR degradation in mammalian cells and found that Hsp27 selectively disposed of F508del CFTR via its interaction with a novel pathway for protein modification by the small ubiquitin-like modifier (SUMO), which was followed by its SUMO-dependent degradation by the ubiquitin–proteasome system.
RESULTS
Hsp27 selectively targets F508del CFTR for degradation
As noted above, a role for sHsps in CFTR degradation was implicated in a yeast screen and verified in mammalian cells (Ahner et al., 2007). Our rationale for the further study of Hsp27, one of 10 members of the mammalian sHsp family, was its role in human diseases (Vidyasagar et al., 2012), its expression in airway cell lines and primary cultures of human bronchial epithelia (Supplemental Figure S1A), and its identification as a component of the CFTR interactome (Wang et al., 2006). Therefore we first examined the involvement of Hsp27 in mutant CFTR disposal by evaluating the impact of its overexpression in HEK293 cells on CFTR steady-state levels. HEK cells were cotransfected with vectors expressing wild-type or F508del CFTR, and with Hsp27 or green fluorescent protein (GFP) cDNAs, the latter as a control protein of similar size. As found for αA-crystallin (Ahner et al., 2007), Hsp27 coexpression reduced steady-state F508del expression levels ∼80% without significantly affecting the expression of wild-type CFTR (Figure 1A).
FIGURE 1:

Hsp27 selectively interacts with F508del CFTR and facilitates its degradation. (A) Hsp27 overexpression reduces F508del levels. HEK293 cells were transfected with 1.5 μg CFTR or F508del CFTR and 1.5 μg GFP or Hsp27 per 60-mm dish. Steady-state CFTR protein levels (bands B and C) were quantified from four independent experiments and averaged. CFTR expression levels in cells coexpressing Hsp27 were normalized to values observed in cells coexpressing GFP. Differences in expressed Hsp27 in the panels are primarily due to differences in film exposure times and not due to differences in the amount of cDNA transfected. Error estimates: SEM (*, p = 0.013). (B) Hsp27 overexpression reduces F508del levels in airway cells. CFBE-F508del cells were electroporated with GFP or Hsp27 cDNAs as described in Materials and Methods, and cell lysates were blotted for CFTR; data are representative of three independent experiments showing a mean reduction of 60% relative to control (p = 0.043). (C) Hsp27 knockdown increases F508del levels. HEK293 cells were transfected as described in (A). CFTR protein levels (bands B and C) were quantified from six independent experiments and averaged. CFTR and F508del CFTR expression levels in cells coexpressing the pGeneClip hMGFP shRNA Hsp27 vector were normalized to those obtained from cells coexpressing a scrambled sequence (scram), as shown in the data summary. SEM: *, p = 0.036; **, p = 0.007. (D) Hsp27 accelerates F508del CFTR degradation. HEK293 cells were transfected with F508del CFTR and GFP or Hsp27 cDNAs as in (A). Cycloheximide chase experiments were performed as described in Materials and Methods. F508del CFTR remaining as a function of time was determined from blot densities in five independent experiments and averaged. Values were normalized to that obtained at the beginning of the chase period (t = 0). Closed circles: F508del CFTR plus GFP control; closed triangles: F508del CFTR plus Hsp27. SEM indicated. (E) Hsp27 interacts preferentially with mutant CFTR. HEK293 cells were transfected with 4 μg wild-type or F508del CFTR and 4 μg GFP or Hsp27 per 100-mm dish, and the interaction between CFTR or F508del CFTR and Hsp27 was assessed by CFTR IP, which was followed by Hsp27 IB. Hsp27 blot densities were normalized to CFTR in the IP. Relative binding represents the average of four independent experiments. SEM: *, p = 0.019.
Second, to provide evidence for the involvement of sHps in CFTR biogenesis in a cell type affected by CF, we overexpressed Hsp27 in the bronchial epithelial cell line, CFBE41o–, which has been stably transduced to express F508del CFTR, hereafter called CFBE-F508del (Bebok et al., 2005). In these cells (Figure 1B), Hsp27 also markedly diminished F508del CFTR steady-state expression; a mean reduction of 60% was found in three experiments.
Third, evidence that F508del CFTR is physiologically targeted for degradation by Hsp27 was provided by knockdown experiments in HEK 293 cells. Decreased cellular Hsp27 levels, induced by the coexpression of short hairpin RNA (shRNA) targeting Hsp27, enhanced the steady-state levels of F508del CFTR ∼fourfold and of wild-type CFTR ∼twofold (Figure 1C), without eliciting immature F508del maturation. These data confirm that mutant CFTR is more susceptible to degradation by physiological Hsp27 levels, reflecting Hsp27’s ability to discriminate the mutant and wild-type proteins.
The kinetic impact of Hsp27 on F508del degradation was revealed in cycloheximide chase experiments showing that Hsp27 overexpression accelerated the disappearance of mutant CFTR following inhibition of protein synthesis (Figure 1D). The mean half-life of the mutant was reduced from 45 to 25 min. Similar findings were obtained for the degradation of newly synthesized F508del CFTR in isotopic pulse–chase experiments (unpublished data).
Next we evaluated the ability of Hsp27 to distinguish physically between wild-type and F508del CFTR by IP of CFTR followed by Hsp27 immunoblot (IB). The co-IP data (Figure 1E) revealed that six times more Hsp27 interacted with F508del versus wild-type CFTR when normalized to the amount of CFTR found in the precipitate. To preserve this potentially transient interaction, we preincubated cells (5 h) prior to lysis with the proteasome inhibitor, MG132, which explains the absence of an Hsp27 effect on F508del levels shown in the primary data. The inhibition of Hsp27-induced F508del degradation demonstrates that the action of the sHsp is mediated by the proteasome. Together these data illustrate the ability of sHsps to distinguish the properties of wild-type and mutant CFTR proteins, presumably based on their conformational differences.
SUMOylation modulates CFTR biogenesis
The SUMO modification pathway resembles that for ubiquitin, being made up of activating (E1) and conjugating (E2) enzymes catalyzing thiol-mediated SUMO transfer reactions, as well as SUMO-specific proteases that reverse substrate modification. Prior yeast two-hybrid and co-IP studies have suggested that Hsp27 interacts with Ubc9 (Joanisse et al., 1998; Brunet Simioni et al., 2009), the only known conjugating enzyme (E2) for transfer of SUMO to substrates. We confirmed this interaction by co-IP of Hsp27 and Ubc9 from coexpressing HEK cells (Figure 2A). Based on these data, and reports of the involvement of the SUMO pathway in protein degradation (Geoffroy and Hay, 2009), SUMO modification of CFTR became a candidate to mediate the action of Hsp27 on F508del degradation.
FIGURE 2:

Hsp27 action links to the SUMO pathway. (A) Hsp27 interacts with the SUMO E2, Ubc9. HEK 293 cells were transfected with the indicated plasmids, 4 μg each per 100-mm dish. The interaction between Hsp27 and myc-Ubc9 was assessed by IP with anti-Hsp27 or anti-myc, followed by IP of myc or Hsp27, respectively, as indicated. (B) SUMOylation pathway components mimic the effect of Hsp27 on CFTR expression. HEK293 cells were cotransfected with CFTR or F508del CFTR plus GFP, myc-Ubc9, or Flag-Senp1, as in Figure 1A. CFTR protein levels were quantified from independent experiments for wild-type (n = 6) and F508del (n = 8). CFTR expression levels (bands B and C) were normalized to values obtained from cells coexpressing GFP. Error estimates are SEM: *, p = 0.024; **, p = 0.028; ***, p = 0.013; vs. GFP controls. (C) Senp1 expression increases F508del expression in airway cells. CFBE-F508del cells were electroporated with GFP or Senp1, which was followed by IB for CFTR. The data are representative of three independent experiments. The fold increase in F508del CFTR expression over control averaged 2.6 ± 0.4 (p = 0.01). (D) Ubc9 accelerates F508del CFTR degradation. HEK293 cells were cotransfected with F508del CFTR and GFP or myc-Ubc9, and mutant CFTR degradation kinetics were determined by cycloheximide chase, as in Figure 1D. Gel densities were normalized to those obtained at the beginning of the chase period in each of five experiments and averaged. Closed circles: F508del CFTR plus GFP; closed triangles: F508del CFTR plus Ubc9. Error estimates are SEM.
As for Hsp27, we coexpressed wild-type or F508del CFTR with Ubc9 or the SUMO protease, Senp1, to examine the impact of SUMO pathway components on steady-state CFTR expression levels. Both Ubc9 and Senp1 were found to be expressed in CFBE cells and in the bronchial epithelial cell line, Calu-3 (unpublished data). As shown in Figure 2B, Ubc9 overexpression selectively lowered F508del CFTR expression levels ∼60%, replicating the action of Hsp27, while Senp1 increased the expression of both wild-type and mutant CFTR more than twofold. To exclude the possibility that this effect is specific for HEK293 cells, we overexpressed Senp1 in CFBE-F508del airway cells and found that the protease increased F508del CFTR expression 2.6-fold (Figure 2C). To verify that the Ubc9 effect is due to its involvement in ER-associated CFTR degradation, we performed cycloheximide chase experiments. Ubc9 overexpression enhanced the rate of F508del CFTR degradation (Figure 2D), reducing its half-life from 44 to 27 min, similar to the data obtained for Hsp27 (Figure 1C). Isotopic pulse–chase experiments replicated these findings and the selectivity of Ubc9 for F508del versus wild-type CFTR degradation (unpublished data).
As in the ubiquitin pathway, the first and essential step in SUMOylation is activation of SUMO by its activating (E1) enzyme, which is composed of an isoform heterodimer, SAE1/SAE2. Expression of the adenoviral protein, GAM1, leads to the degradation of SAE1, which prevents or diminishes target protein SUMOylation (Boggio et al., 2007). When the SUMO pathway was disabled via expression of GAM1, steady-state wild-type and F508del CFTR levels increased by two- and threefold, respectively (Figure 3A). While manipulations of Hsp27 or SUMO pathway components primarily impacted F508del, the knockdown of Hsp27 or SAE1, or Senp1 overexpression, also increased wild-type CFTR expression, although to a lesser degree. These results may reflect a contribution of Hsp27/SUMO to the ∼60% of wild-type CFTR that is degraded in the ER. The parallel increases in immature and mature wild-type CFTR observed in these experiments suggest a primary effect on band B in the ER that then leads to an increased band C. Similar to its action in HEK293 cells, the expression of GAM1 in CFBE-F508del bronchial epithelial cells augmented mutant CFTR expression 2.3-fold (Figure 3B). These findings parallel those obtained from Hsp27 knockdown (Figure 1C) or Senp1 overexpression (Figure 2, B and C), and they further implicate components mediating SUMO conjugation and proteolysis in the biogenesis of CFTR.
FIGURE 3:

Functional linkage between the Hsp27 and SUMO pathways. (A) Disabling the SUMO pathway increases CFTR expression. HEK293 cells were cotransfected with wild-type or F508del CFTR and empty vector (control) or GAM1 cDNAs. GAM1 efficacy was assessed from SAE1 IB. Bottom panel, quantification of CFTR gel densities, normalized to vector controls and averaged: CFTR, n = 6; F508del, n = 5; SEM: *, p = 0.017, **, p = 0.010. (B) Knockdown of the SUMO E1, SAE1, increases F508del CFTR expression in airway cells. CFBE-F508del cells were transduced with empty vector or GAM1 cDNA followed by CFTR IB. The data are representative of three experiments in which SAE1 knockdown increased F508del CFTR expression 2.1 ± 0.3-fold (p = 0.03). (C) SUMO E1 knockdown obviates the impact of Hsp27 on F508del expression. HEK293 cells were transfected with F508del CFTR and GFP or Hsp27 and GAM1 or empty vector DNAs, as indicated. CFTR protein levels were quantified from three independent experiments and normalized to those observed in the GFP–empty vector control. SEM: *, p = 0.04.
Hsp27 is linked functionally to the SUMO pathway
Because Hsp27 and SUMOylation modulate CFTR biogenesis in a similar manner, we determined whether Hsp27 utilizes SUMO modification to exert its effect on mutant CFTR expression. First, we asked whether Hsp27-induced F508del degradation requires an intact SUMOylation pathway. As illustrated in Figure 3C, the ability of Hsp27 to reduce mutant CFTR level was impaired following GAM1-induced knockdown of SAE1. These data link the pathway for SUMOylation of F508del CFTR to its Hsp27-induced degradation in the ER.
To determine whether SUMOylation of CFTR could be detected in vitro using purified components, we incubated purified wild-type or F508del NBD1 protein with ATP, the SUMO E1 activation (SAE1/SAE2) and E2 conjugation (Ubc9) enzymes, plus SUMO. As illustrated in Figure 4A, NBD1 SUMOylation was detected as the shift in molecular mass detected by an NBD1 antibody. Additional control experiments demonstrated that SUMO modification of the NBD required addition of the individual E1, E2, and SUMO components, as well as ATP, and these requirements are reflected by their combined omission in Figure 4A, which yielded no SUMO modification product. Moreover, SUMOylation was favored for the F508del mutant versus wild-type NBD1, such that the mutant protein was selected for modification, even by this simple system, likely due to conformational differences that expose one or more sites for SUMO modification. Prior studies indicate that the conformational defect in F508del NBD1, detected as thermal instability, correlates with the ubiquitylation of this domain in vitro (Rabeh et al., 2012), and preferential mutant NBD1 SUMOylation likely has a similar basis.
FIGURE 4:
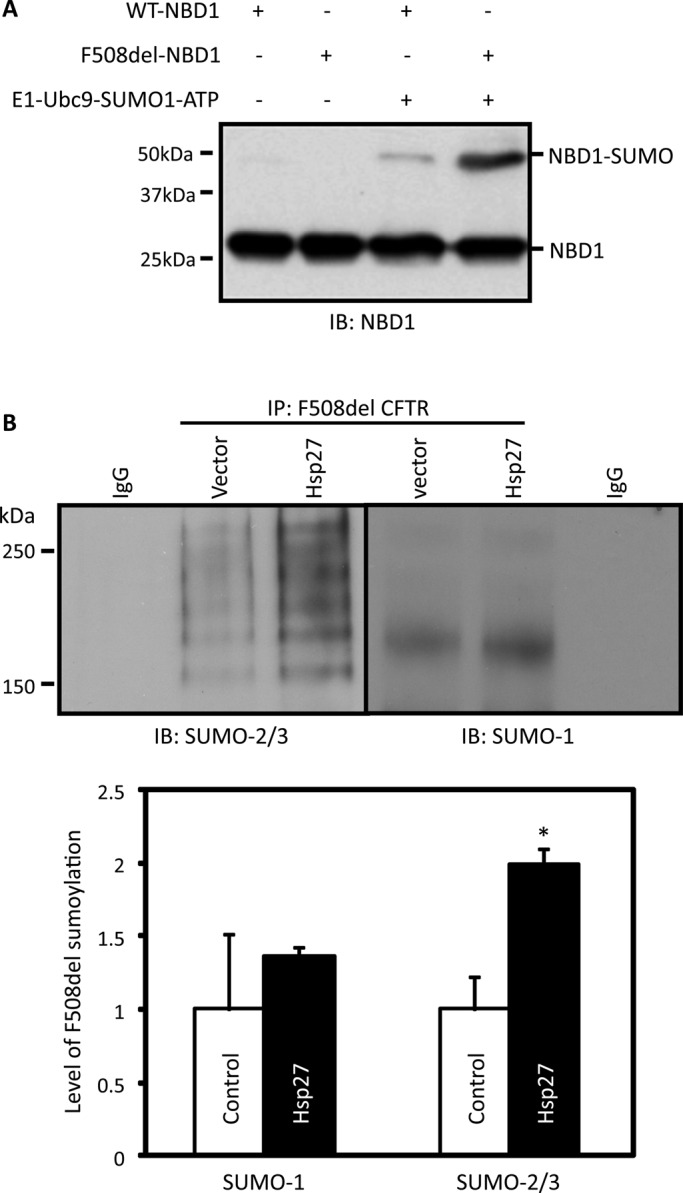
Functional linkage between the Hsp27 and SUMO pathways. (A) F508del NBD1 is preferentially modified by SUMO in vitro. Purified NBD1 proteins containing a single suppressor mutation (1S, F494N) were incubated with or without purified components for 1 h at 27°C as illustrated and described in Materials and Methods. The mixture was then resolved by SDS–PAGE and blotted with anti-NBD1. The thermal stabilities of the wild-type and mutant NBD1-1S proteins show that the 1S mutation marginally improves the F508del NBD1 conformational defect, while improving the solubility of the domain sufficiently to permit in vitro studies (Rabeh et al., 2012). The data are representative of four independent experiments. (B) Hsp27 promotes SUMOylation of F508del CFTR, which prefers SUMO-2/3. HEK293 cells were transfected with F508del CFTR and empty vector or Hsp27 DNAs, as described in Figure 1E. Modification of F508del CFTR by endogenous SUMO paralogues was assessed by CFTR IP followed by SUMO-1 or SUMO-2/3 IB. An irrelevant immunoglobulin G served as a control. SUMO density values were normalized to CFTR in the IPs, and mean values from three experiments are provided in the bottom panel.
Next we assayed the SUMOylation of full-length F508del CFTR in vivo to determine whether Hsp27 overexpression facilitated this process. Mutant CFTR was IPed from HEK293 cells coexpressing Hsp27 or empty vector, and the precipitate was blotted for SUMO-1 or SUMO-2/3 to assess CFTR modification by endogenous SUMO paralogues. While SUMO-1 has only 50% homology to the SUMO-2 and SUMO-3 paralogues, the latter differ by only four amino acids, and the paralogues cannot be distinguished immunologically. As shown in Figure 4B, SUMO bands were observed in the F508del precipitate above its characteristic molecular mass (∼140 kDa), and the expression of Hsp27 increased their intensity, particularly for modification by SUMO-2/3, which increased twofold on average; indicated by the mean data of Figure 4B. SUMO-2 and SUMO-3 contain a consensus SUMOylation motif, which allows them to form poly-chains (Tatham et al., 2001), and the laddering pattern observed for SUMO-2/3 modification is consistent with the concept, but does not prove, that Hsp27 facilitates the poly-SUMOylation of F508del CFTR. It is also possible that the laddering observed reflects, in part, F508del that is both SUMOylated and ubiquitylated, although a similar laddering pattern was observed in response to in vitro SUMOylation with SUMO-2 or SUMO-3 (unpublished data). These NBD1 and full-length CFTR SUMOylation data argue for a direct effect of this modification, rather than modification(s) of other proteins that may indirectly alter the fate of mutant CFTR.
RNF4 mediates SUMO-dependent CFTR degradation
Hsp27 has been implicated in ubiquitin-mediated degradation of a subset of proteins in response to cell stress (Lanneau et al., 2008), but the mechanism of this action is not well defined. The concept of a mechanism for proteolytic regulation of proteins modified by poly-SUMO signals has emerged from the finding that SUMO-2/3 conjugates accumulate when mammalian cells are exposed to proteasome inhibitors (Uzunova et al., 2007). Subsequently SUMO-targeted ubiquitin ligases (STUbLs) were found to represent a relatively new class of ubiquitin E3s that capture sumoylated proteins for degradation by the ubiquitin proteasome system (Tatham et al., 2008). Studies of STUbLs have focused on a small set of proteins that are involved in nuclear regulatory events (Perry et al., 2008).
The possibility that a STUbL mediates the ubiquitylation of SUMOylated CFTR led us to ask whether Hsp27, in addition to promoting F508del SUMOylation (Figure 4B), also facilitates its ubiquitylation. Therefore F508del was IPed from HEK293 cells; this was followed by IB of the precipitate for ubiquitin; the data are summarized in Figure 5A. To promote the accumulation of ubiquitylated proteins, we treated the cells with MG132 (5 h) before lysates were harvested. Hsp27 coexpression increased the ubiquitin signal associated with mutant CFTR by 75% over the vector control. In parallel experiments, the coexpression of CHIP, a recognized Hsp70-linked ubiquitin E3 that targets mutant CFTR (Meacham et al., 2001), increased F508del ubiquitylation 50% in the same time period (unpublished data). These findings are consistent with the concept that Hsp27-linked mutant CFTR degradation is mediated by both SUMOylation and ubiquitylation, as anticipated for the involvement of a SUMO-targeted ubiquitin ligase.
FIGURE 5:

A SUMO-targeted ubiquitin ligase links F508del to the proteasome. (A) Hsp27 promotes F508del ubiquitylation. HEK293 cells were transfected with F508del CFTR and Hsp27 or empty vector, and CFTR modification was assessed by CFTR IP followed by ubiquitin IB. The bottom panel provides mean values for total ubiquitin density from five experiments, which indicate a 75% increase in CFTR ubiquitylation with Hsp27 coexpression. SEM: *, p = 0.004. (B) Expression of RNF4 preferentially decreases F508del expression. HEK293 cells were transfected with wild-type or F508del CFTR and either empty vector or Flag-RNF4 cDNAs. The bottom panel provides steady-state CFTR protein levels quantified from four independent experiments averaged and normalized to their respective controls. SEM: *, p = 0.04; **, p = 0.001. (C) Hsp27 knockdown blocks the ability of RNF4 to reduce F508del CFTR expression. HEK293 cells were transfected with either Hsp27 or scrambled shRNAs, and after 48 h, these cells were transfected with F508del CFTR and either RNF4 or empty vector (control) cDNAs. After 24 h, steady-state F508del protein levels were determined by IB. Mean data from three experiments are summarized in the bottom panel. (D) Dominant-negative RNF4 blocks the ability of Hsp27 to reduce F508del CFTR levels. HEK293 cells were transfected with F508del CFTR and Hsp27 or vector control and RNF4, DN-RNF4s (CS1-CS3), or control DNAs. The DN-RNF4s harbor cysteine-to-serine mutations within the RING domain that mediate thioester ubiquitin transfer, as noted in Materials and Methods. After 24 h, F508del protein levels were determined by IB; the data are representative of two independent experiments in which three DN constructs produced similar results.
The RING-finger ubiquitin E3 ligase, RNF4 (Ring finger protein 4), was the first mammalian STUbL identified (Tatham et al., 2008). It contains four tandem SUMO-interacting motifs (SIMs) at its N-terminus, and therewith recognizes poly-sumoylated targets for ubiquitylation and degradation by the 26S proteasome. Therefore we asked whether RNF4 overexpression would elicit CFTR degradation in coexpressing HEK293 cells. As shown in Figure 5B, RNF4 reduced wild-type and F508del CFTR expression by 40 and 70%, respectively. Because RNF4 interacts preferentially with SUMO poly-chains on its targets (Tatham et al., 2008), these data fit with the finding that CFTR is modified primarily by the SUMO paralogues that are capable of poly-chain formation (Figure 4B).
We then explored the interdependence between RNF4 and Hsp27 in knockdown and overexpression experiments. First, we examined the ability of RNF4 to reduce F508del CFTR expression levels in HEK293 cells with or without reduced Hsp27 expression. As shown in Figure 5C, RNF4 reduced F508del expression ∼70% in cells expressing the scrambled shRNA, as noted above (Figure 5B). However, in cells having reduced Hsp27 expression, F508del expression was enhanced, as found for Hsp27 knockdown (Figure 1C), and the ability of RNF4 to dispose of mutant CFTR was lost. For reasons that are not apparent, the knockdown of Hsp27 was greater in cells coexpressing RNF4, which could account for the ∼50% increase in F508del expression observed under these conditions. It is also possible that a combination of Hsp27 knockdown plus RNF4 overexpression produces off-target effects that increase mutant CFTR expression. Nevertheless, RNF4 required physiological expression levels of Hsp27 to induce mutant CFTR degradation.
Second, we determined whether the ability of Hsp27 to reduce F508del CFTR expression requires functional RNF4. Dominant-negative (DN) expression constructs of RNF4 were kindly provided by Ronald T. Hay (University of Dundee); they were created by introducing paired serine mutations in RING domain catalytic cysteines at the sites identified in Materials and Methods. Whereas the expression of wild-type RNF4 or Hsp27 normally reduced F508del expression, Hsp27 could not reduce F508del levels during the coexpression of three different dominant-negative RNF4 constructs. Wild-type RNF4 normally reduced F508del expression, but these catalytic mutants of RNF4 increased mutant CFTR expression levels. The increase in F508del expression observed during dominant-negative RNF4 expression presumably reflects the interaction of sumoylated F508del with the SIMs of mutant RNF4, which cannot evoke CFTR ubiquitylation, resulting in protection of mutant CFTR from degradation.
Finally, we disabled the SUMO pathway with GAM1, previously shown to increase F508del expression (Figure 3, A and B) and to block the ability of Hsp27 to reduce F508del CFTR expression (Figure 3C). GAM1 also blocked the ability of RNF4 to decrease the expression of mutant CFTR (Figure S2), reflecting the dependence of RNF4 on a functional SUMO pathway. Together these findings demonstrate that the actions of Hsp27 and RNF4 are interdependent and linked by SUMOylation of mutant CFTR.
DISCUSSION
This study implicates a novel SUMO-dependent proteolytic mechanism in the degradation of an extensively studied ERAD substrate, F508del CFTR. We identified a previously unrecognized role for Hsp27 in selectively binding to F508del CFTR and, via its physical interaction with the SUMO E2, Ubc9, facilitating SUMO transfer to the mutant protein. The ability of Hsp27 to promote F508del CFTR degradation required a functional SUMOylation pathway. The preferential modification of F508del CFTR by SUMO-2/3 leads to its recognition by the SUMO-targeted ubiquitin E3 ligase, RNF4, ubiquitylation of the mutant protein, and its proteasomal degradation. Whereas elements of this pathway have been implicated in nuclear regulatory events, these findings provide the first evidence for sHsp-mediated SUMO modification of a nonnuclear ERAD substrate and its transfer to the ubiquitin proteasome system by a STUbL.
sHsps target mutant CFTR degradation
Support for the conclusion that Hsp27 facilitates CFTR degradation via SUMO modification stems from experiments that enhanced or disabled the expression of SUMOylation pathway components. First, Ubc9 interacted with Hsp27, and like the sHsp, its overexpression selectively reduced steady-state levels of F508del CFTR, as well as the half-life of the mutant protein (Figure 2, B and D). Second, reducing the capacity for F508del SUMOylation by overexpression of the SUMO protease, Senp1, increased F508del expression level (Figure 2, B and C), as did knockdown of the SUMO E1, SAE1 (Figure 3, A and B). These results mimic the effect of Hsp27 knockdown (Figure 1B), and they highlight the physiological significance of this pathway in mutant protein degradation. While these maneuvers increased the expression of immature CFTR, they did not, unfortunately, elicit its maturation. Third, knockdown of the SUMO E1 blocked the ability of Hsp27 to reduce F508del CFTR levels (Figure 3C). Fourth, SUMO modification of F508del NBD1 was preferred over that of wild-type CFTR in vitro (Figure 4A), and Hsp27 enhanced the SUMOylation of full-length F508del CFTR in vivo (Figure 4B). Many of these findings were replicated in the airway cell line, CFBE-F508del, which stably expresses the mutant protein, indicating that the operation of this pathway is independent of cellular background.
Although recent reports provide evidence that αB-crystallin and Hsp27 can promote nuclear protein ubiquitylation and degradation (den Engelsman et al., 2003, 2004; Lin et al., 2006; Parcellier et al., 2006; Wu et al., 2009), the mechanism of these actions has not emerged. We found that the Hsp27-mediated degradation of F508del CFTR was associated with increased SUMOylation of the mutant protein (Figure 4B). Similarly, Hsp27 was recently shown to facilitate the SUMO modification of HSF-1 (Brunet Simioni et al., 2009), blocking its function as a nuclear regulator of transcription.
The mechanism by which Hsp27 assists in the transfer of SUMO to mutant CFTR remains to be determined. Because Hsp27 interacts physically with Ubc9, the sHsp may serve as a bridging or scaffolding factor that recruits the SUMO E2 to its substrate, similar to the mechanism by which many RING domain E3 ubiquitin ligases connect their substrates with a ubiquitin-conjugating E2 enzyme, providing for substrate targeting (Deshaies and Joazeiro, 2009). The sHsp, αB-crystallin, plays a similar role in the recruitment of FBX4 to the FBX4-SCF ubiquitin E3 complex, (Lin et al., 2006). It is less likely that Hsp27 exhibits E3 activity, as observed for the Pias family of SUMO E3s, which facilitate SUMO transfer by aligning the Ubc9-SUMO thioester for the ligation reaction, but do not directly interact with substrate (Reverter and Lima, 2005). Rather, the selective co-IP of Hsp27 or αA-crystallin with F508del CFTR (Figure 2B; Ahner et al., 2007) is more consistent with sHsp-mediated recruitment of Ubc9 to the mutant protein.
RNF4 connects SUMOylated CFTR to the proteasome
Until recently, protein modification by ubiquitin and SUMO were thought to have different consequences for protein fate. However, evidence is accumulating from studies in yeast and mammalian cells that SUMO modification can facilitate protein degradation, as reviewed in Geoffroy and Hay (2009). We provide evidence of increased ubiquitylation of F508del CFTR as a consequence of Hsp27 overexpression (Figure 5A). Our data suggest that sHsp-induced F508del CFTR ubiquitylation results from the action of a SUMO-targeted ubiquitin ligase. First, overexpression of the mammalian STUbL, RNF4, mimicked the actions of Hsp27 and Ubc9 in preferentially reducing the expression levels of F508del CFTR (Figure 5B). Second, our in vivo assessment of mutant CFTR SUMOylation indicated its modification by endogenous SUMO-2 or SUMO-3, the nearly identical paralogues that can form SUMO poly-chains (Figure 4B). Increasing evidence indicates that the SUMO and ubiquitin modification pathways are connected by STUbLs, which recognize sumoylated target proteins. As noted, RING-domain E3 ligases often exhibit substrate-targeting motifs; however, RNF4 is distinct in possessing four tandem SIMs within its N-terminus (Perry et al., 2008), with which it targets poly-SUMOylated substrates for ubiquitylation (Tatham et al., 2008). Third, disabling the CFTR SUMOylation pathway via Hsp27 knockdown (Figure 5C) or by reduced SUMO E1 expression (Figure S2) blocked the ability of RNF4 to degrade mutant CFTR. Similarly, catalytically disabled RNF4 blocked Hsp27-induced F508del degradation (Figure 5D). Thus, conditions that reduce the capacity for F508del SUMOylation compromise RNF4-mediated mutant CFTR degradation. Prior studies of the mammalian STUbLs, RNF4 and VHL, have focused entirely on their roles in transcriptional regulation or protein translocation between cytoplasm and nucleus. Our findings provide the first indication that an sHsp induces SUMO-2/3 modification of a transmembrane protein, which links mutant CFTR via RNF4 to the ubiquitin–proteasome system.
Role of sHsps in the selection of mutant CFTR
As noted in the Introduction, the F508del mutation results in near-complete ERAD of the common CFTR mutant due to its inability to achieve a conformation competent for ER exit. Prior work suggests that CFTR domains, such as NBD1, obtain their folded conformations cotranslationally (Kleizen et al., 2005); however, the compact, protease-resistant, structure of wild-type CFTR forms posttranslationally and requires ~30 min for completion of domain assembly (Du et al., 2005). Recent findings indicate that the thermodynamic and kinetic instability of F508del NBD1 are indicative of folding defects in the isolated domain that impair its ability to participate in cooperative domain assembly steps that generate the native structure of export-competent wild-type CFTR (Mendoza et al., 2012; Rabeh et al., 2012). Thermodynamic differences in the folding pathways of wild-type and mutant NBD1 have been modeled as free-energy transitions from the unfolded state (U) to a transition state (T) before the native (N) state is achieved. These states reflect different NBD1 conformations, having different thermal stabilities, in which a more open conformation exposes sites for ubiquitylation (Lukacs and Verkman, 2012), and likely SUMOylation, as reflected by the enhanced SUMO modification of F508del versus wild-type NBD1 observed in vitro (Figure 4A).
The stabilization of a transitional or intermediate conformation of NBD1 by Hsp27, and by Hsp27-mediated NBD SUMOylation, is consistent with model protein studies, showing that sHsps have the ability to distinguish between native and partially denatured proteins, based on small differences in their free energies of unfolding (McHaourab et al., 2002; Koteiche and McHaourab, 2003; Sathish et al., 2004; Shashidharamurthy et al., 2005). In addition, SUMO fusion has the ability to stabilize and enhance the solubility of overexpressed proteins (Peroutka et al., 2011). These properties of sHsps and SUMO modification would be in agreement with the ability of Hsp27 to interact with partially unfolded conformations of F508del or wild-type CFTR to reduce their tendency for aggregation. For the wild-type protein, this interaction is expected to be transient, but the inability of mutant CFTR to obtain the native conformation would lead to a more prolonged interaction with the sHsp, its modification by SUMO, and ERAD. This model is conceptually similar to the preferential interaction of the chaperones Hsp70 and Csp, which target the ubiquitin E3, CHIP, to a complex with F508del CFTR (Meacham et al., 2001; Schmidt et al., 2009). The extent to which the Hsp27/Ubc9/SUMO/RNF4 QC pathway contributes also to the degradation of ∼60% of wild-type CFTR will require further study; however, the findings that knockdown of Hsp27 or the expression of Senp1 or Gam1 increased wild-type CFTR levels may reflect a role for this pathway in determining the fate of the wild-type protein as well.
In summary, we have identified a novel QC mechanism, operative in the cytosolic compartment, which targets the common CFTR mutant F508del for SUMO-dependent ubiquitylation and proteolysis. The targeting of other misfolded proteins by this pathway is under continuing investigation.
MATERIALS AND METHODS
Reagents and antibodies
Monoclonal antibodies targeted CFTR's NBD2 and R domain (#596 and #217, respectively; Cystic Fibrosis Foundation Therapeutics, Bethesda, MD); other monoclonals were to SUMO-1 (Zymed Laboratories, Carlsbad, CA), Ubc9 (Abcam, Cambridge, MA), ubiquitin (Stressgen/Assay Designs, Ann Arbor, MI), the Flag-epitope (Sigma-Aldrich, St. Louis, MO), and the human c-myc (clone 9E10; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Polyclonal antibodies directed at CFTR were a kind gift of D. M. Clarke (University of Toronto, Canada). Antibodies to Hsp27 and Hsp70 were obtained from Stressgen/Assay Designs, SAE1 from Abcam, RNF4 from Sigma-Aldrich, SUMO-2/3 from Enzo Life Sciences (Ann Arbor, MI), and Senp1 from Zymed Laboratories (San Francisco, CA). Horseradish peroxidase–conjugated secondary antibodies, anti-rabbit or anti-mouse, were obtained from Amersham (Piscataway, NJ) and Jackson ImmunoResearch Laboratories (West Grove, PA), respectively.
The Flag-Senp1 plasmid was obtained from Edward T. H. Yeh (MD Anderson Cancer Center, Houston, TX), the Myc-Ubc9 and Myc-Gam1 constructs were obtained from Yin-Yuan Mo (Southern Illinois University School of Medicine, Springfield, IL). Flag-RNF4 and DN Flag-RNF4 constructs (CS1 [C136S/C139S], CS2 [C177S/C180S], CS3 [C136S/C139S/C177S/C180S]) were provided by Ronald T. Hay (University of Dundee, Dundee, UK). The Hsp27 vector was purchased from Origene (Rockville, MD), and GFP was from Clontech (Mountain View, CA). CFTR and F508del CFTR expression vectors were constructed as previously described (Zhang et al., 2002).
Cell culture, transfections, and cell methods
HEK293 cells were cultured in DMEM (Sigma-Aldrich) with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA), 4 mM l-glutamine (Sigma-Aldrich), and penicillin–streptomycin (Gibco-BRL, San Francisco, CA). The human bronchial epithelial cell line, CFBE-F508del, was a gift from J. P. Clancy (University of Alabama at Birmingham, Birmingham, AL). The cells were cultured in modified Earle's medium (Life Technologies, Grand Island, NY) with 10% FBS (Hyclone, Logan, UT), 2 mM l-glutamine (Life Technologies), 50 U/ml penicillin, 50 μg/ml streptomycin (Sigma-Aldrich, St. Louis, MO), and 2 μg/ml puromycin (Invitrogen, Carlsbad, CA). Calu-3 cells (human bronchial epithelial cell line) were cultured in DMEM/F-12 (Sigma-Aldrich) with 15% FBS (Invitrogen), 4 mM l-glutamine (Sigma-Aldrich), and 1% penicillin–streptomycin (Gibco-BRL). Primary human bronchial epithelial cells were received from J. Pilewski (University of Pittsburgh, CF Research Center Cell Culture Core, Pittsburgh, PA). All cell lines were grown in a humidified chamber with 5% CO2 at 37°C.
For protein overexpression, HEK293 cells were transiently transfected with the indicated expression plasmids, aided by Lipofectamine 2000 (Invitrogen). After 24 h, the cells were lysed in 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, and 1% Triton X-100, and equal amounts of protein were resolved on SDS–PAGE and subjected to Western blot analysis. Cells were subjected to cycloheximide chase or isotopic pulse–chase analysis as previously described (Zhang et al., 2002; Ahner et al., 2007). For cycloheximide chases, the culture medium was changed to one containing 100 μg/ml cycloheximide (freshly diluted from a 100 mg/ml stock in dimethyl sulfoxide) and 10 μg/ml brefeldin A (freshly diluted from a 10 mg/ml stock in ethanol). The cells were lysed at indicated time points, and the cell lysates were assayed for CFTR by IB. Bands in images of digitized films were quantitated using ImageJ software (http://rsb.info.nih.gov/ij). For CFTR SUMO or ubiquitin modification experiments, the cultures were treated with 50 μM MG-132 for 5 h before cell lysis. For the detection of in vivo SUMOylation, 20 mM N-ethylmaleimide (NEM) was added to the lysis buffer.
Protein expression in CFBE cells was induced using either electroporation or transfection. For electroporation, cells were trypsinized and resuspended in 600 μl of BTXpress Solution (BTX Harvard Apparatus, Holliston, MA). Cells were placed in an Eppendorf tube and DNA was added; cells were then transferred into a 2-mm gap cuvette (BTX Harvard Apparatus) and electroporated with a BTX ECM 830 using a single pulse at 145 V for 25 ms. Controls using GFP or empty vector DNA were included. Cuvette contents were transferred to a six-well plate, and after cells adhered, media was removed and replaced. On day 1, cells were harvested into RIPA buffer containing a protease inhibitor tablet and sonicated, and protein assays were performed. Alternatively, CFBE cells were transiently transfected with 5 μg of the indicated expression plasmids with 15 μl Lipofectamine 2000 according to the manufacturer's instructions. Cells were lysed on either day 2 or transfected a second time on day 2 and harvested on day 4.
shRNA
For Hsp27 knockdown, a vector expressing an shRNA against Hsp27 was constructed using the GeneClip U1 Hairpin Cloning Systems from Promega (Madison, WI). GACCAAGGATGGCGTGGTG (Arrigo et al., 2005) or the scrambled sequence GAGTGCTGACGAGTGCGAG was cloned into the pGeneClip hMGFP vector according to the manufacturer's instructions. Twenty micrograms of the pGeneClip hMGFP shRNA Hsp27 vector or the scrambled sequence was transfected into HEK293 cells with Lipofectamine 2000 (Invitrogen) per 100-mm dish. After 24 h, cells were trypsinized and divided among four 60-mm dishes. After 24 h, cells in each dish were transfected with 4 μg of the pGeneClip hMGFP Hsp27 shRNA or the scrambled sequence vector and any indicated overexpression constructs. Cells were lysed after a further 24 h, and equal amounts of protein were resolved on SDS–PAGE and subjected to Western blot analysis. Data were quantified and analyzed using ImageJ software, and statistical analyses were performed as outlined above.
CFTR and sHsp co-IPs
A total of 500 μg of HEK293 cell lysate was added to buffer A (13.3 mM HEPES, pH 7.0, 85 mM potassium glutamate, 3.3 mM NaCl, 4.7 mM MgSO4, 13.3 mM ethylene glycol tetraacetic acid, 2.2 mM CaCl2 [vol/vol: 2/1]) and incubated with 1 μg of CFTR antibody M3A7 (Cell Signaling Technologies, Boston, MA) and CFTR C-terminal antibody (R&D Systems, Minneapolis, MN) for 2 h. The immunocomplex was isolated by incubation with protein A and G agarose (Invitrogen) beads for 2 h. Precipitates were isolated and washed three times with buffer A plus 0.25% NP-40, and the proteins were resolved on SDS–PAGE and subjected to Western blot analysis.
In vitro SUMOylation assay
The in vitro assay was performed using reagents purchased from Enzo Life Sciences (Ann Arbor, MI) together with wild-type or F508del human NBD1 variants that contained a single solubilizing mutation, F494N, as previously described (Rabeh et al., 2012). In brief, 15 ng of purified NBD1 was incubated in SUMOylation buffer with a reaction mixture containing recombinant E1 (0.4 μM), Ubc9 (4 μM), SUMO (3 μM), and Mg-ATP (2 mM), with or without purified recombinant human Hsp27 protein (15 ng), for 1 h at 27˚C. After the reaction was terminated with SDS sample buffer containing 2-mercaptoethanol, reaction products were fractionated on 12% SDS–PAGE, and the gel shift resulting from SUMO modification was detected by immunoblotting using anti-NBD1 (#660; Cystic Fibrosis Foundation Therapeutics).
In vivo ubiquitylation and SUMOylation assays
For CFTR SUMOylation, a total of 1 mg of HEK293 cell lysate, prepared using lysis buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1% NP-40, pH 7.5, and 20 mM NEM), was incubated with 1 μg of #217 CFTR R domain and 596 NBD2 antibodies (Cystic Fibrosis Foundation Therapeutics) overnight. Immunocomplexes were isolated by incubation with protein A agarose beads (Invitrogen) for 4 h. Precipitates were isolated and washed three times with lysis buffer, and the proteins were resolved on 5% SDS–PAGE and subjected to Western blot analysis for SUMO.
For the ubiquitylation assay, HEK293 cells were transfected in 60-mm dishes and treated with 50 μM MG132 for 5 h prior to cell lysis. Cells were then lysed in RIPA buffer (Cell Signaling Technology, Danvers, MA) supplemented with 0.1% SDS, Complete Protease Inhibitor Cocktail (Roche, Indianapolis, IN), 2 mM phenylmethylsulfonyl fluoride, and 20 mM NEM. CFTR was IPed from 500 μg HEK293 lysate with mouse antibodies (#217 and 596; CFFT, Bethesda, MD) cross-linked to Protein A/G Plus Agarose beads (Pierce, Rockford, IL). Ubiquitin in the immunoprecipitate was detected by IB using rabbit anti-ubiquitin (SPA-200; Enzo Life Sciences, Ann Arbor, MI).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK68196 and DK72506) and the Cystic Fibrosis Foundation (FRIZZE05XX0) to R.A.F. and from Cystic Fibrosis Canada, the Canadian Institute of Health Research, and the Canadian Foundation for Innovation–Research Infrastructure and Research Support to G.L.L., who holds a Canada Research Chair.
Abbreviations used:
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHIP
C-terminus of Hsp70-interacting protein
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum–associated degradation
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HEK
human embryonic kidney
- IB
immunoblot
- IP
immunoprecipitation
- NBD
nucleotide binding domain
- NEM
N-ethylmaleimide
- QC
quality control
- R domain
regulatory domain
- RNF4
Ring finger protein 4
- shRNA
short hairpin RNA
- sHsp
small heat shock protein
- SIMs
SUMO-interacting motifs
- STUbLs
SUMO-targeted ubiquitin ligases
- SUMO
small ubiquitin-like modifier
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0678 on November 14, 2012.
REFERENCES
- Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL. Small heat-shock proteins select ΔF508-CFTR for endoplasmic reticulum-associated degradation. Mol Biol Cell. 2007;18:806–814. doi: 10.1091/mbc.E06-05-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Bohse K, Arndt V, Schmitz A, Hohfeld J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. 2004;15:4003–4010. doi: 10.1091/mbc.E04-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo AP, Firdaus WJ, Mellier G, Moulin M, Paul C, Diaz-latoud C, Kretz-remy C. Cytotoxic effects induced by oxidative stress in cultured mammalian cells and protection provided by Hsp27 expression. Methods. 2005;35:126–138. doi: 10.1016/j.ymeth.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, Sorscher EJ, Clancy JP. Failure of cAMP agonists to activate rescued ΔF508 CFTR in CFBE410− airway epithelial monolayers. J Physiol. 2005;569:601–615. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Das KP. Role of ATP on the interaction of α-crystallin with its substrates and its implications for the molecular chaperone function. J Biol Chem. 2004;279:42648–42657. doi: 10.1074/jbc.M404444200. [DOI] [PubMed] [Google Scholar]
- Boggio R, Passafaro A, Chiocca S. Targeting SUMO E1 to ubiquitin ligases: a viral strategy to counteract sumoylation. J Biol Chem. 2007;282:15376–15382. doi: 10.1074/jbc.M700889200. [DOI] [PubMed] [Google Scholar]
- Brunet Simioni M, De Thonel A, Hammann A, Joly AL, Bossis G, Fourmaux E, Bouchot A, Landry J, Piechaczyk M, Garrido C. Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene. 2009;28:3332–3344. doi: 10.1038/onc.2009.188. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes ΔF508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol. 2001;281:L58–68. doi: 10.1152/ajplung.2001.281.1.L58. [DOI] [PubMed] [Google Scholar]
- DeCarvalho AC, Gansheroff LJ, Teem JL. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ΔF508. J Biol Chem. 2002;277:35896–35905. doi: 10.1074/jbc.M205644200. [DOI] [PubMed] [Google Scholar]
- den Engelsman J, Bennink EJ, Doerwald L, Onnekink C, Wunderink L, Andley UP, Kato K, de Jong WW, Boelens WC. Mimicking phosphorylation of the small heat-shock protein αB-crystallin recruits the F-box protein FBX4 to nuclear SC35 speckles. Eur J Biochem. 2004;271:4195–4203. doi: 10.1111/j.1432-1033.2004.04359.x. [DOI] [PubMed] [Google Scholar]
- den Engelsman J, Keijsers V, de Jong WW, Boelens WC. The small heat-shock protein αB-crystallin promotes FBX4-dependent ubiquitination. J Biol Chem. 2003;278:4699–4704. doi: 10.1074/jbc.M211403200. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Du K, Sharma M, Lukacs GL. The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- Ehrnsperger M, Graber S, Gaestel M, Buchner J. Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997;16:221–229. doi: 10.1093/emboj/16.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25:5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Nogueira P, Mendes F, Penque D, Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Annu Rev Genet. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- Geoffroy MC, Hay RT. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat Rev. 2009;10:564–568. doi: 10.1038/nrm2707. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Braun N, Stromer T, Richter B, Model N, Weinkauf S, Buchner J. Hsp42 is the general small heat shock protein in the cytosol of Saccharomyces cerevisiae. EMBO J. 2004;23:638–649. doi: 10.1038/sj.emboj.7600080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Walke S, Stromer T, Ehrnsperger M, White HE, Chen S, Saibil HR, Buchner J. Hsp26: a temperature-regulated chaperone. EMBO J. 1999;18:6744–6751. doi: 10.1093/emboj/18.23.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Joanisse DR, Inaguma Y, Tanguay RM. Cloning and developmental expression of a nuclear ubiquitin-conjugating enzyme (DmUbc9) that interacts with small heat shock proteins in Drosophila melanogaster. Biochem Biophys Res Commun. 1998;244:102–109. doi: 10.1006/bbrc.1998.8214. [DOI] [PubMed] [Google Scholar]
- Kleizen B, van Vlijmen T, de Jonge HR, Braakman I. Folding of CFTR is predominantly cotranslational. Mol Cell. 2005;20:277–287. doi: 10.1016/j.molcel.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koteiche HA, McHaourab HS. Mechanism of chaperone function in small heat-shock proteins. Phosphorylation-induced activation of two-mode binding in αB-crystallin. J Biol Chem. 2003;278:10361–10367. doi: 10.1074/jbc.M211851200. [DOI] [PubMed] [Google Scholar]
- Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. 2008;12:743–761. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GJ, Roseman AM, Saibil HR, Vierling E. A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 1997;16:659–671. doi: 10.1093/emboj/16.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-αB crystallin) complex. Mol Cell. 2006;24:355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner RA, Treweek TM, Carver JA. The molecular chaperone α-crystallin is in kinetic competition with aggregation to stabilize a monomeric molten-globule form of α-lactalbumin. Biochem J. 2001;354:79–87. doi: 10.1042/0264-6021:3540079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken AA, Brodsky JL. Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD) Bioessays. 2003;25:868–877. doi: 10.1002/bies.10320. [DOI] [PubMed] [Google Scholar]
- McHaourab HS, Dodson EK, Koteiche HA. Mechanism of chaperone function in small heat shock proteins. Two-mode binding of the excited states of T4 lysozyme mutants by αA-crystallin. J Biol Chem. 2002;277:40557–40566. doi: 10.1074/jbc.M206250200. [DOI] [PubMed] [Google Scholar]
- Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA, Thomas PJ. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–174. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Harada K, Takeya M, Yamahira K, Wada I, Shuto T, Suico MA, Hashimoto Y, Kai H. ΔF508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol Biol Cell. 2004;15:563–574. doi: 10.1091/mbc.E03-06-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcellier A, et al. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 2006;20:1179–1181. doi: 10.1096/fj.05-4184fje. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroutka RJ, III, Orcutt SJ, Strickler JE, Butt TR. SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol Biol. 2011;705:15–30. doi: 10.1007/978-1-61737-967-3_2. [DOI] [PubMed] [Google Scholar]
- Perry JJ, Tainer JA, Boddy MN. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem Sci. 2008;33:201–208. doi: 10.1016/j.tibs.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79:S215–255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- Pind S, Riordan JR, Williams DB. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994;269:12784–12788. [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A, Lukacs GL. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman K, Raman B, Rao CM. Molten-globule state of carbonic anhydrase binds to the chaperone-like α-crystallin. J Biol Chem. 1996;271:27595–27600. doi: 10.1074/jbc.271.44.27595. [DOI] [PubMed] [Google Scholar]
- Reverter D, Lima CD. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature. 2005;435:687–692. doi: 10.1038/nature03588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Sathish HA, Koteiche HA, McHaourab HS. Binding of destabilized βB2-crystallin mutants to α-crystallin: the role of a folding intermediate. J Biol Chem. 2004;279:16425–16432. doi: 10.1074/jbc.M313402200. [DOI] [PubMed] [Google Scholar]
- Schmidt BZ, Watts RJ, Aridor M, Frizzell RA. Cysteine string protein promotes proteasomal degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) by increasing its interaction with the C terminus of Hsp70-interacting protein and promoting CFTR ubiquitylation. J Biol Chem. 2009;284:4168–4178. doi: 10.1074/jbc.M806485200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidharamurthy R, Koteiche HA, Dong J, McHaourab HS. Mechanism of chaperone function in small heat shock proteins: dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. J Biol Chem. 2005;280:5281–5289. doi: 10.1074/jbc.M407236200. [DOI] [PubMed] [Google Scholar]
- Strickland E, Qu BH, Millen L, Thomas PJ. The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1997;272:25421–25424. doi: 10.1074/jbc.272.41.25421. [DOI] [PubMed] [Google Scholar]
- Sun F, Zhang R, Gong X, Geng X, Drain PF, Frizzell RA. Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J Biol Chem. 2006;281:36856–36863. doi: 10.1074/jbc.M607085200. [DOI] [PubMed] [Google Scholar]
- Tatham MH, Geoffroy MC, Shen L, Plechanovova A, Hattersley N, Jaffray EG, Palvimo JJ, Hay RT. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol. 2008;10:538–546. doi: 10.1038/ncb1716. [DOI] [PubMed] [Google Scholar]
- Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, Hay RT. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 2001;276:35368–35374. doi: 10.1074/jbc.M104214200. [DOI] [PubMed] [Google Scholar]
- Teem JL, Carson MR, Welsh MJ. Mutation of R555 in CFTR-delta F508 enhances function and partially corrects defective processing. Receptors Channels. 1996;4:63–72. [PubMed] [Google Scholar]
- Treweek TM, Lindner RA, Mariani M, Carver JA. The small heat-shock chaperone protein, α-crystallin, does not recognize stable molten globule states of cytosolic proteins. Biochim Biophys Acta. 2000;1481:175–188. doi: 10.1016/s0167-4838(00)00109-6. [DOI] [PubMed] [Google Scholar]
- Uzunova K, et al. Ubiquitin-dependent proteolytic control of SUMO conjugates. J Biol Chem. 2007;282:34167–34175. doi: 10.1074/jbc.M706505200. [DOI] [PubMed] [Google Scholar]
- Van Goor F, et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Vidyasagar A, Wilson NA, Djamali A. Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair. 2012;5:7. doi: 10.1186/1755-1536-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Welch WJ. Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol. 2004;15:31–38. doi: 10.1016/j.semcdb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Wu Y, Liu J, Zhang Z, Huang H, Shen J, Zhang S, Jiang Y, Luo L, Yin Z. HSP27 regulates IL-1 stimulated IKK activation through interacting with TRAF6 and affecting its ubiquitination. Cell Signal. 2009;21:143–150. doi: 10.1016/j.cellsig.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Zhang F, Kartner N, Lukacs GL. Limited proteolysis as a probe for arrested conformational maturation of delta F508 CFTR. Nat Struct Biol. 1998;5:180–183. doi: 10.1038/nsb0398-180. [DOI] [PubMed] [Google Scholar]
- Zhang H, Peters KW, Sun F, Marino CR, Lang J, Burgoyne RD, Frizzell RA. Cysteine string protein interacts with and modulates the maturation of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2002;277:28948–28958. doi: 10.1074/jbc.M111706200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Schmidt BZ, Sun F, Condliffe SB, Butterworth MB, Youker RT, Brodsky JL, Aridor M, Frizzell RA. Cysteine string protein monitors late steps in CFTR biogenesis. J Biol Chem. 2006;281:11312–11321. doi: 10.1074/jbc.M512013200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.