

Abstract
Respiratory syncytial virus (RSV) causes substantial morbidity and life-threatening lower respiratory tract disease in infants, young children and the elderly. Understanding the host response to RSV infection is critical for developing disease-intervention approaches. The role of microRNAs (miRNAs) in post-transcriptional regulation of host genes responding to RSV infection is not well understood. In this study, it was shown that RSV infection of a human alveolar epithelial cell line (A549) induced five miRNAs (let-7f, miR-24, miR-337-3p, miR-26b and miR-520a-5p) and repressed two miRNAs (miR-198 and miR-595), and showed that RSV G protein triggered let-7f expression. Luciferase–untranslated region reporters and miRNA mimics and inhibitors validated the predicted targets, which included cell-cycle genes (CCND1, DYRK2 and ELF4), a chemokine gene (CCL7) and the suppressor of cytokine signalling 3 gene (SOCS3). Modulating let-7 family miRNA levels with miRNA mimics and inhibitors affected RSV replication, indicating that RSV modulates host miRNA expression to affect the outcome of the antiviral host response, and this was mediated in part through RSV G protein expression.
Introduction
Respiratory syncytial virus (RSV) is an important paediatric and geriatric challenge causing substantial hospitalizations, clinic visits and >14 000 deaths per annum (CDCP, 2008). RSV is a prototype of the genus Paramyxovirus with a 15 kb negative-sense ssRNA genome encoding 11 proteins (NS1, NS2, N, P, M, SH, G, F, M2-1, M2-2 and L). Despite 60 years of intense efforts towards an RSV vaccine, there is a lack of effective prophylactic and therapeutic intervention, mainly due to a poor understanding of the host–virus interface. Whilst recent antiviral efforts have begun to target host pathways to inhibit virus replication (Li et al., 2009; Meliopoulos et al., 2012; Panda et al., 2011) and RNA interference approaches using small interfering RNAs (siRNAs) to target RSV have shown success at reducing virus replication in a mouse model (Alvarez et al., 2009) and are currently in phase II clinical trials (Alvarez et al., 2009), mitigating the host immune response that results in bronchiolitis remains a challenge.
Among the 11 RSV proteins, the non-structural proteins (NS1/2) cooperatively inhibit activation and nuclear translocation of interferon (IFN) regulatory factor 3 (Bossert et al., 2003; Spann et al., 2005), and mediate inhibition of cytokine production by proteasome-mediated degradation of the signal transducer and activator of transcription factor 2 (Elliott et al., 2007; Lo et al., 2005), whilst the surface proteins F and G are involved in attachment and entry, along with nucleolin (Tayyari et al., 2011). The RSV G protein also interacts with Toll-like receptors (Kurt-Jones et al., 2000; Murawski et al., 2009; Oshansky et al., 2009a), and negatively affects type I IFN (Moore et al., 2008; Tripp et al., 1999) and cytokine and chemokine expression (Tripp et al., 2000a), in part by induction of suppressor of cytokine signalling (SOCS) proteins in normal human bronchoepithelial cells and mouse lung epithelial cells (Moore et al., 2008; Oshansky et al., 2009a). In addition, a highly conserved CX3C chemokine motif in the RSV G protein mimics fractalkine, modulating fractalkine-mediated immune responses (Tripp et al., 2001).
RSV infection in vitro and in vivo induces early, middle and late host genome-wide gene transcription (Janssen et al., 2007; Martínez et al., 2007); however, these increases are not directly reflected in the host proteome (Munday et al., 2010) and this is not completely understood. RSV infection causes G1/S arrest in A549 cells (Gibbs et al., 2009) and HEp-2 cells (Mohapatra et al., 2009), and a G2/M cell-cycle arrest in primary human bronchial epithelial cells via induction of transforming growth factor β1 and a reduction in p53, both in vitro and in vivo. RSV infection also induces stress granule formation via a protein kinase R-dependent pathway in HEp-2 cells, leading to increased virus replication in cytosolic viral inclusion bodies (Lindquist et al., 2010, 2011). Components of stress granules are shared with processing bodies, which are sites for accumulation of 22 nt RNAs called microRNAs (miRNAs) (Fabian et al., 2010; Selbach et al., 2008), which regulate gene expression post-transcriptionally by binding to a complementary sequence in the 3′UTR of a target gene(s) via a ‘seed’ region (nt 2–7) in the miRNA, and cause transcript degradation or a block in translation (Fabian et al., 2010; Selbach et al., 2008). Consequently, miRNA deregulation linked to virus infection and replication can affect global gene expression. Recently, infection of normal tracheal epithelial cells with a recombinant GFP-expressing RSV (rgRSV) was shown to repress 24 miRNAs to varying extents (from −0.5-fold to −2.9-fold relative to mock-infected cells) and to induce two miRNAs (Othumpangat et al., 2012). Of the miRNAs discovered, six were predicted to govern the neurotrophin nerve growth factor (NGF) gene, and miR-221 was shown to regulate NGF. As bronchial epithelia represent a mixed population of cells and miRNA expression can vary considerably among cell types, we investigated the deregulation of miRNA expression following RSV infection of A549 cells, a type II respiratory epithelial model. In this model, microarray data validated by quantitative real-time PCR (qPCR) showed that a different set of miRNAs (let-7f, miR-337, miR-520a, miR-24, miR-26b, miR-198 and miR-595) was deregulated following RSV infection. We determined that the RSV G protein modified let-7f expression and showed that let-7 miRNAs regulated several key host genes induced during RSV infection. Modulation of let-7f miRNA also regulated virus replication, a feature not attributable to the induction of antiviral cytokines alone. These studies suggest that RSV G protein-induced let-7 miRNA expression regulates host genes during RSV infection to modulate virus replication.
Results
RSV infection deregulates host miRNA expression
To evaluate RSV deregulation of host miRNA expression, A549 cells were infected with recombinant wild-type RSV (6340WT; m.o.i. of 1) or were mock treated in triplicate, and expression of all known mature miRNAs was determined at 24 h post-infection (p.i.) by microarray analysis. The miRNAs let-7, let-7a, let-7f and miR-337 were significantly (P≤0.01) induced ≥1.5-fold among replicates, whilst miR-224 showed consistent repression of at least 1.5-fold among replicates (Table S1, available in JGV Online). The miRNAs miR-24, miR-26b, miR-29a, miR-320a and miR-520a-5p (miR-520a) were also induced ≥1.5-fold, whilst miR-198, miR-224 and miR-595 were repressed by at least 1.5-fold (Table S1). qPCR performed using miRNA-specific oligonucleotides validated approximate inductions of twofold for miRNAs let-7f and miR-337, 1.7-fold for miR-520a and miR-24 (Fig. 1) and fourfold for miR-26b (Fig. 1). Although multiple miRNAs were induced, we focused on let-7 miRNAs, as the induction of this family was consistent among replicates and let-7f showed the highest induction among various let-7 members.
Fig. 1.
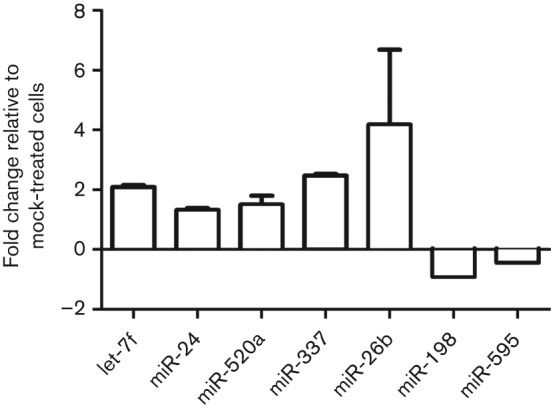
6340WT infection deregulates host miRNA expression. A549 cells were infected or mock infected with RSV 6340WT virus at an m.o.i. of 1 for 24 h. The data represent the mean qPCR fold change±sem of let-7f (let-7f), miR-337-3p (miR-337), miR-520a-5p (miR-520a), miR-24, miR-26b, miR-198 and miR-595 from three independent experiments relative to mock-infected cells, with values >1.0 considered to be upregulation and values below 1.0 considered to be downregulation.
RSV G protein elicits miRNA expression
RSV G protein expression has been shown to modify cytokine and chemokine expression and to induce SOCS1- and SOCS3-negative regulation of type I IFNs (Moore et al., 2008; Oshansky et al., 2009a, b; Tripp et al., 1999, 2000a, 2001). To determine whether RSV G protein affected expression of the validated miRNAs (Fig. 1), A549 cells were infected (m.o.i. of 1) with recombinant RSV (6340WT) or with a recombinant RSV mutant virus lacking the G gene (RSVΔG), and expression of let-7f, miR-337, miR-520a, miR-26b and miR-24 was determined at 24 h p.i. Both 6340WT and RSVΔG replicated to similar levels over the short period of infection; however, in the absence of the G protein gene (RSVΔG), expression of let-7f was significantly (P<0.001) lower, whilst levels of miR-337 and miR-24 were significantly (P<0.05) upregulated (Table 1 and Fig. 2a). Expression of miR-520a was decreased slightly in RSVΔG-infected cells, but this change was not statistically significant, and miR-26b expression remained unchanged compared with 6340WT infection. These results indicated that RSV G protein expression was associated with let-7f induction but repressed miR-24 and miR-337 expression. To confirm that RSV G protein induced let-7f (Fig. 2a), A549 cells were treated with purified RSV G protein (1.0 µg ml−1) (Oshansky et al., 2009a), RSV F protein (1.0 µg ml−1) or infected (m.o.i. of 1) with 6340WT or 6340ΔG virus (Fig. 2b). A549 cells treated with purified RSV G protein showed a remarkable and significant (P<0.01) induction of let-7f relative to mock-treated cells, supporting a role for RSV G protein-mediated induction of miRNA let-7f. RSV F treatment did not change let-7f expression, confirming that RSV G is the major inducer for let-7f.
Table 1. Expression levels of miRNAs in A549 cells following infection with 6340WT or RSVΔG virus.
ns, Not significant. All experiments were carried out in duplicate.
| miRNA | 6340WT | RSVΔG | P value | ||
| Mean | sem | Mean | sem | ||
| let-7f | 2.0900 | 0.060 | 0.1655 | 0.079 | <0.0001 |
| miR-24 | 1.310 | 0.070 | 1.890 | 0.084 | 0.0125 |
| miR-337-3p | 2.45 | 0.0141 | 3.47 | 0.296 | 0.013 |
| miR-520a-5p | 1.505 | 0.3464 | 1.196 | 0.316 | ns |
| miR-26b | 3.376 | 4.22866 | 2.923 | 3.60 | ns |
Fig. 2.
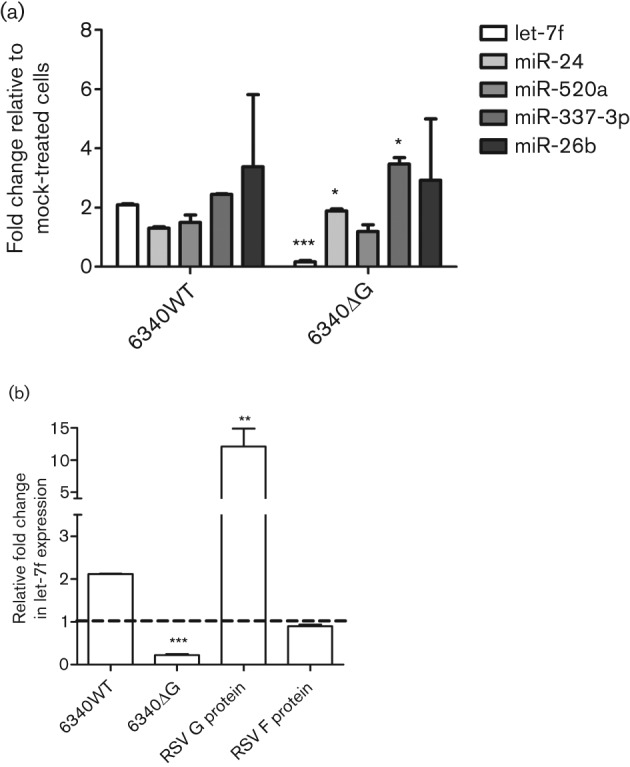
RSV G protein regulates miRNA expression during infection. (a) Expression of let-7f, miR-24, miR-520a, miR-26b and miR-337 was measured in A549 cells infected with 6340WT or 6340ΔG virus at 24 h p.i. (b) Expression of let-7f in cells treated with purified RSV G protein (1.0 µg ml−1) or RSV F protein (1.0 µg ml−1) and at 24 h after infection with 6340WT or 6340ΔG virus. Data represent mean fold changes in copy number±sem from three independent experiments relative to mock-infected cells. Statistical significance is indicated: ***P<0.001, **P<0.01, *P<0.05.
let-7f regulates numerous host genes responding to RSV infection
As RSV infection modified let-7 miRNA expression (Fig. 1), and RSV G protein expression considerably affected let-7f (Fig. 2), we focused on the role of let-7f in the host response. To identify let-7f targets regulated by base pairing between the miRNA ‘seed’ region and the 3′UTR of the gene (Friedman et al., 2009), potential let-7f targets based on published genes linked to RSV infection and those predicted to be let-7f targets were mined using multiple computational algorithms (TargetScan, miRbase and PicTar). These potential targets were then mapped against a dataset of genes known to be deregulated at different times after RSV infection based on published microarray data (Fjaerli et al., 2007; Janssen et al., 2007; Martínez et al., 2007; Wu et al., 2011). From this analysis, 102 genes were identified as significant let-7f targets, 117 genes were found to be deregulated during RSV infection and 27 genes overlapped these two areas (Fig. 3a). The 27 genes were expressed at various time points (≤6 h, 6–12 h, and ≥12 h p.i.) after RSV infection of A549 cells, and were probably also regulated by other let-7 miRNAs, because of the 100 % identity in the let-7 ‘seed’ sequences (Fig. 3b) (Fjaerli et al., 2007; Huang et al., 2008; Martínez et al., 2007). Regulation by let-7f of a subset of these genes was validated with let-7f inhibitors/mimics (Fig. 3c). Commercial let-7f and miR-24 inhibitor and mimics used in this study consistently prevented or increased the incorporation of the miRNA guide strand into the RNA-induced silencing complex (RISC) complex via proprietary design (Fig. 3c) (Vermeulen et al., 2007). Different concentrations of let-7f inhibitors were tested, where 25 nM let-7f inhibitor reduced native let-7f levels by ≥85 % in 24 h and were not cytotoxic (Alamar Blue reduction assay; AbD SeroTec). Therefore, 25 nM was used in all transfection assays. It is important to note that, whilst the let-7f inhibitors were miRNA specific and were able to distinguish between different members of the let-7 family, let-7 mimics affected the native levels of all let-7 family members because the let-7 seed sites were identical across all let-7 miRNAs (Fig. 3b). Thus, for genuine let-7f targets, it would be expected that inhibitors and mimics would increase and decrease Luc expression, respectively, relative to non-targeting controls, although not to the same extent. Fold changes in Luc expression were calculated using the formula described in Methods. Moreover, based on findings from a previous study in human cells (Johnson et al., 2007), the magnitude of differences in Luc expression would be expected to be modest compared with those of the controls.
Fig. 3.

Luciferase (Luc)–UTR assays used to validate predicted let-7f gene targets. RSV G protein induced let-7f and other miRNAs regulate multiple genes during RSV infection. (a) Venn diagram depicting the overlap between predicted let-7f gene targets and genes deregulated during RSV infection. Genes that were examined further are shown in bold. SOCS3, Suppressor of cytokine signalling 3; CCND1, cyclin D1; SMOX, spermine oxidase; HOXA1, homeobox A1 transcription factor; TNFAIP3, tumour necrosis factor α-induced protein 3; ELF4, E74-like factor 4; DYRK2, dual-specificity tyrosine phosphorylation regulated kinase 2; CCL7, chemokine (C-C motif) ligand 7; PLAUR, plasminogen activator, urokinase receptor; VLDLR, very low density lipoprotein receptor; GLRX3, glutaredoxin 3; SERPING, serpin peptidase inhibitor, clade G. The dashed line indicates the baseline value. (b) Seed sequence (nt 2–8) conservation (boxed area) among miRNAs of the let-7 family. (c) Sequence of let-7f and miR-24 inhibitor and mimic sequences. The nature of chemical modifications (N0–16) on inhibitors and mimics are proprietary and not known. (d) Sequence alignment of various gene 3′UTRs and let-7f and miR-24. Numbers correspond to nucleotides in the 3′UTR. (e) let-7f regulates multiple genes during RSV infection. Luc–3′UTRs of putative let-7f targets were co-transfected into A549 cells with pSEAP2-Control (transfection control) and inhibitors or mimics for let-7f and/or miR-24. Data represent mean fold change±sem in Luc values [measured in relative light units (RLU)] from three independent experiments between inhibitor- and mimic-transfected cells relative to a non-target control (NTC) inhibitor or mimic. Statistical significance is indicated for all transfections represented in (e) and (f): ***P<0.001; **P<0.01; *P<0.05. (f) Cooperative activity of let-7f and miR-24 on DYRK2–Luc expression. A549 cells were transfected with DYRK2-pMLC plasmid and let-7f /miR-24 inhibitor/mimic alone or with DYRK2-pMLC plasmid and equimolar concentrations of let-7f+miR-24 inhibitor/mimic together with pSEAP2-Control plasmid as a transfection control. Data represent the fold change in Luc expression±sem from two independent experiments with the dashed line indicating the baseline value.
The 3′UTRs for 12 (SOCS3, CCND1, SMOX, HOXA1, TNFAIP3, ELF4, DYRK2, CCL7, PLAUR, VLDLR, GLRX3 and SERPING1) of the 27 genes representing early, middle and late times after RSV infection (Fjaerli et al., 2007; Huang et al., 2008; Martínez et al., 2007) were cloned into a pMetLucControl (pMLC) plasmid that constitutively expresses secreted Metridia longa Luc from a cytomegalovirus promoter. Plasmid mixes of gene-specific luc–UTR constructs and transfection control plasmid [pSEAP2-Control, expressing secreted alkaline phosphatase (SEAP)] were co-transfected into A549 cells along with let-7f miRNA inhibitors or mimics (Dharmacon, Thermo Fisher), or with miRNA inhibitor negative control or mimic negative control that targeted Caenorhabditis elegans miR-67, and which have been validated not to affect the expression of any human gene (Dharmacon, Thermo Fisher).
Differences in Luc expression between let7-f inhibitor- and mimic-transfected cells were statistically (P<0.01) significant for five (SOCS3, CCND1, ELF4, DYRK2 and CCL7) of the 12 genes examined relative to the negative controls (Fig. 3e). All these genes exhibited a strong match between the let-7f seed site and the 3′UTR of the target gene (Fig. 3d). Both let-7f and miR-24 were predicted to regulate the DYRK2 gene (Fig. 3d). As miRNAs function as a molecular rheostat to fine-tune gene expression and may act cooperatively with other miRNAs (Asirvatham et al., 2009), we investigated whether combined miRNA inhibition of let-7f and miR-24 further increased Luc expression. Accordingly, cells were transfected with DYRK2-pMLC plasmid and either transfected with let-7f or miR-24 inhibitor alone or co-transfected with DYRK2-pMLC plasmid and equimolar amounts of let-7f and miR-24 inhibitors or mimics. Concomitant inhibition of let-7f and miR-24 resulted in a significant (P<0.05) increase in Luc expression relative to cells transfected with let-7f or miR-24 inhibitor alone (Fig. 3f), suggesting that let-7f also acts cooperatively to regulate gene expression.
let-7f regulates its targets via the RISC pathway
Gene transcripts regulated by miRNAs are processed by RISC complexes, which contain the Ago2 protein as a conserved core component (Fabian et al., 2010). Precipitating RISC using an anti-Ago2 mAb has been shown to significantly enrich for miRNA-regulated transcripts (Dölken et al., 2010). To validate let-7f-regulated transcripts, RISC-associated mRNA and target transcripts were precipitated from 6340WT-infected (m.o.i. of 1) or mock-infected cells using a mAb against Ago2 (a component of the RISC) or anti-bromodeoxyuridine (BrdU) control mAb. The CCND1 gene, which is induced early during RSV infection (Martínez et al., 2007), was enriched in anti-Ago2-precipitated RNA from RSV-infected cells but not in anti-BrdU-precipitated RNA from RSV-infected cells (Fig. 4a), or from similarly treated mock-infected Vero cell RNA precipitated with anti-Ago2 or anti-BrdU mAb (Fig. 4a). This showed that the CCND1 transcript was associated with the RISC in RSV-infected cells, presumably for miRNA-mediated translational repression. As the RSV G protein induced let-7f (Fig. 2b), and let-7f regulated CCND1 expression (Fig. 3e), it was expected that there would be differential enrichment of let-7f transcripts in RISC-associated RNA from 6340WT-infected cells compared with 6340ΔG-infected cells. qPCR analysis of RISC-associated mRNA precipitated from 6340WT-infected cells using anti-Ago2 mAb, but not using anti-BrdU mAb, showed an approximately threefold let-7f enrichment compared with RNA from 6340ΔG-infected cells (P = 0.0005) (Fig. 4b). These results supported the findings showing RSV G protein induction of let-7f expression (Fig. 2b) and its involvement in regulating CCND1 expression via the RISC pathway (Figs 3e and 4a).
Fig. 4.
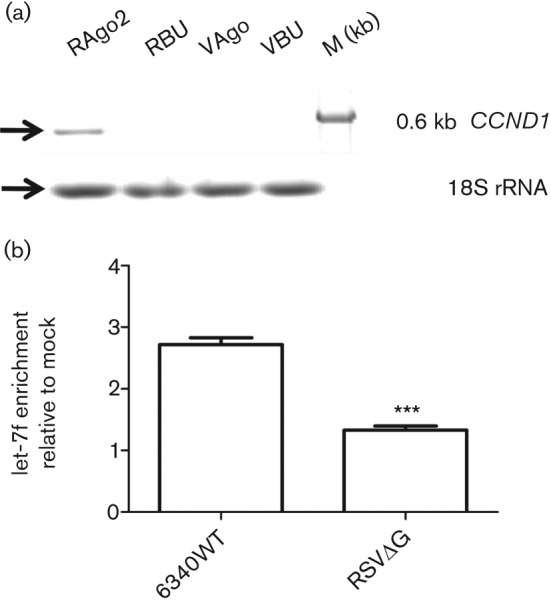
RISC complexes from RSV 6340WT-infected cells are enriched for CCND1 and let-7f transcripts. (a) RISC-associated RNA from mock-, 6340WT- and 6340ΔG-infected cells were assayed for CCND1 by PCR. CCND1 UTR amplicons (0.6 kb) were amplified as described in Methods in two independent experiments. 18S rRNA was used as a loading control. RAgo, Anti-Ago2-precipitated RNA from RSV-infected cells; RBU, anti-BrdU-precipitated RNA from RSV-infected cells; VAgo, mock-infected Vero cell RNA precipitated with anti-Ago2; VBU, mock-infected Vero cell RNA precipitated anti-BrdU. (b) Enrichment of let-7f in RISC immunoprecipitated RNA from 6340WT- and RSVΔG-infected cells was assayed by qPCR and normalized to that of mock-infected cells from two independent experiments. Results are shown as means±sem, and Student’s t-test was used to measure the statistical significance of the data: ***P<0.001.
RSV replication is modulated by let-7f and miR-24
To determine whether let7-f affected RSV replication, A549 cells were transfected with let-7f miRNA inhibitor or mimic, or with controls, for 24 h and the cells were incubated for a further 48 h, assayed for cytotoxicity and subsequently infected with rgRSV expressing GFP at an m.o.i. of 0.5. Transfection with let-7f miRNA inhibitors, mimics or controls was not cytotoxic. rgRSV has been shown to replicate with a similar titre and time course as wild-type RSV in untreated A549 cells (Hallak et al., 2000) At an m.o.i. of 0.5, rgRSV-infected A549 cells showed a peak GFP fluorescence at day 3 p.i. and hence were processed at this time point for RSV plaque assays on Vero E6 cells using an anti-RSV F-based plaque assay. The RSV plaque assays showed that inhibitor transfections reduced rgRSV plaque numbers, comparable to the results for positive-control siRNA (Fig. 5). A qPCR for RSV M gene copy numbers as well as GFP measurements also showed similar trends (data not shown). These data showed that miRNA inhibition can modulate RSV replication. As we did not observe statistically significant differences in antiviral cytokine expression after inhibitor/mimic transfection (unpublished observations), the effect on virus replication was probably due to a global deregulation of host gene expression. This is supported by a previous study showing that let-7 mimic transfections affected global gene expression profiles, deregulating 629 genes across multiple cellular pathways (Johnson et al., 2007). We also analysed the RSV genome for potential seed sites for the above miRNAs using blast. The results (matrix = BLOSUM62, E value cut-off = 10.0) were filtered to identify hits in the miRNA seed site (nt 2–8). let-7f and miR-24 did not show any significant homology in the seed site with any region in the RSV genome in both sense and anti-sense orientations, ruling out a direct inhibition of virus replication by these miRNAs. This supports our hypothesis that modulation of virus replication in inhibitor-transfected cells is probably effected by modulating cellular pathways. These data suggest that RSV-modulated miRNAs have a pro-viral role and that modulation of these miRNAs affects virus replication by affecting multiple cellular pathways.
Fig. 5.
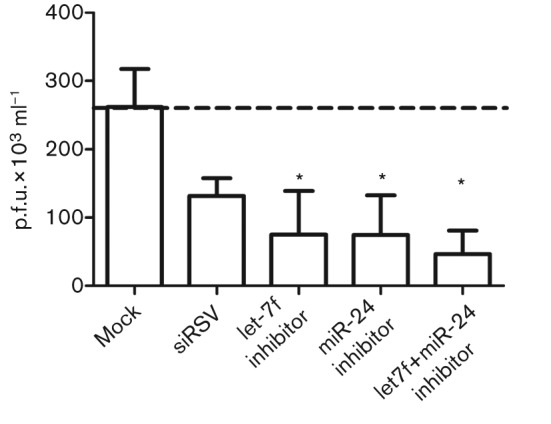
Modulation of miRNA levels deregulates virus replication. A549 cells were mock transfected or transfected in two independent experiments with inhibitors of let-7f and miR-24 separately and together (let-7f+miR-24), followed by infection with rgRSV at an m.o.i. of 0.5. The number of RSV p.f.u. was measured at day 3 p.i. using an anti-RSV F-based plaque assay relative to mock-infected cells. siRNA against the RSV N gene (siRSV) was used as a silencing control. The dashed line indicates basal p.f.u. levels in the mock-transfected control.
Discussion
RSV is an important paediatric challenge, and understanding the miRNA regulated RSV–host interface is critical for vaccine development. As miRNA expression is regulated by multiple mechanisms such as Toll-like receptor recognition of pathogen-associated molecular patterns (Taganov et al., 2006), extra- or intracellular signalling (O’Connell et al., 2007; Taganov et al., 2006), processing of IFN-stimulated gene transcripts (Berezikov et al., 2007; Morlando et al., 2008), direct viral induction of miRNA promoters (Taganov et al., 2006) and as an off-target effect of viral inhibition of cellular processes, a central theme of this study was to determine the miRNAs that were deregulated following RSV infection in an established in vitro model of RSV infection, and to determine the effect of modulating these miRNAs on virus replication.
We identified a set of miRNAs that were deregulated (five induced and two repressed) during RSV infection of A549 cells where let-7f expression was induced most abundantly following RSV infection and was found to be regulated in part by RSV G protein. Treatment with purified RSV G protein enhanced let-7f expression and this was not observed following RSV F treatment. This is the first report of an RSV gene product regulating the expression of a host miRNA. let-7f showed maximum expression among differentially expressed let-7 miRNAs in A549 cells (copies per cell: let-7a, ~200; let-7b and let-7c, ~100; let-7e, ~50; let-7f, ~750; let-7g, ~80; let-7i, ~25; Johnson et al., 2007), and our data are consistent with this study. The fold changes reported in this study are similar to a recently published study (Othumpangat et al., 2012) that showed miR-221 deregulation in normal tracheal epithelial cells following rgRSV infection. miR-221 was postulated to be a major regulator of NGF, and miR-221 upregulation reduced NGF expression and virus replication.
To compare our findings with the above study, we performed an extensive analysis of miRNAs targeting NGF using ten different algorithms (DIANAmT, miRanda, miRDB, miRWalk, RNAhybrid, PICTAR4, PICTAR5, PITA, RNA22 and Targetscan) and failed to find any significant seed match between miR-221 and the NGF promoter and the 3′- or 5′UTR. Additionally, blast analysis of NGF mRNA (GenBank accession no. NM_002506.2) versus miR-221-5p (miRNA base accession no. MIMAT0004568) or miR-221-3p (miRNA base accession no. MIMAT0000278) failed to show any hits between the miR-221 seed site and the NGF-coding region, suggesting that the reported miR-221 regulation of NGF is an off-target effect of miR-221 transfection. In contrast, NGF treatment has been shown to induce miR-221/miR-222 via the ERK1/2-mediated pathway in culture (Terasawa et al., 2009), and RSV infection induces NGF expression (Othumpangat et al., 2009). miR-221 is a known negative regulator of the tumour suppressor genes PTEN (Zhang et al., 2010b), Bim (Terasawa et al., 2009) and PUMA (Zhang et al., 2010a) and transcription factor Foxo3a (Hamada et al., 2012). Hence, treatment with pre-miR-221 could be hypothesized to reduce the activity of these tumour suppressors, enhance apoptosis and reduce viral titres. It is important to note that Othumpangat et al. (2012) also identified miR-574 (repressed −0.5-fold relative to mock-infected cells) as a regulator of NGF, although its impact on virus replication was not studied. Differences between our findings and those above probably reflect the different cell types and viruses examined, as miRNA expression profiles vary considerably among cell types (Johnson et al., 2007; Landgraf et al., 2007).
let-7f gene targets were identified using a meta-analysis of computationally predicted let-7f targets and published microarray data on RSV-deregulated host genes. Of the genes predicted to be let-7f targets, 12 were tested using Luc–UTR assays and five genes (CCND1, SOCS3, ELF4, DYRK2 and CCL7) showed modest but statistically significant differences following let-7f inhibitor and mimic treatment relative to non-targeting controls. The findings also showed that let-7f and CCND1 transcripts co-localized in RISCs in RSV-infected but not in mock-infected cells using Ago2 immunoprecipitation, and were selectively enriched in 6340WT- versus RSVΔG-infected A549 cells, further supporting observations on the role of the RSV G protein. As let-7 miRNAs have 100 % sequence identity in their seed site, the results suggested that the let-7f target genes identified may also be regulated by other let-7 miRNAs. Inhibition of let-7f alone or in combination with miRNA miR-24 led to a significant reduction in rgRSV viral titres as measured by plaque assays. Lack of any significant homology between these miRNAs and the RSV genome and the negligible effects of these miRNAs on cytokine expression (unpublished observations) suggest that the observed reduction in viral titres is probably due to gene target modulation by let-7f.
CCND1 and ELF4 are important in cell-cycle regulation, affecting the G1/S phase transition, whilst ELF4 and DYRK2 inhibit p53-mediated induction of apoptosis (Maddika & Chen, 2009; Taira et al., 2007; Taura et al., 2011). CCND1, as a complex with CDK4/6, promotes G1/S phase transition, whilst DYRK2 regulates apoptosis via p53 phosphorylation (Taira et al., 2007). ELF4 has been shown to be induced in RSV-infected cells at 12 h p.i., leading to expression of the E3 ubiquitin ligase Mdm2, which ubiquitinates p53 and targets it for proteasome-mediated degradation (Maddika & Chen, 2009). As previous studies have shown that RSV infection arrests cells in G1 (Johnson et al., 2007; Mohapatra et al., 2009), our findings of let-7 inhibition of CCND1, DYRK2 and ELF4 translation suggest that let-7-mediated gene regulation is one of the mechanisms employed by RSV (Groskreutz et al., 2007; Mohapatra et al., 2009).
The results of this study also showed that let-7f regulates CCL7/MCP3 and SOCS3, two genes involved in the antiviral cytokine response. We have previously shown that RSV G protein-mediates inhibition of chemokine mRNA expression by bronchoalveolar leukocytes responding to RSV infection (Tripp et al., 2000a) and induces interleukin-8 (IL-8) (Tripp et al., 2000a), and in this study let-7f was shown to regulate ELF4, a known inducer of IL-8 (Hedvat et al., 2004). Thus, RSV G protein expression is linked to let-7f deregulation and downstream modulation of IL-8 expression, which has been associated with RSV disease pathogenesis (Johnson & Graham, 2004). The RSV G protein is a well-documented immunomodulatory glycoprotein that is produced as both a membrane-bound and a soluble form (Roberts et al., 1994) and is implicated in the induction of substance P, a neurokinin that mediates inflammation and enhanced pulmonary disease in RSV-infected BALB/c mice (Tripp et al., 2000b). Importantly, RSV G protein expression has been linked to Th2-type cytokine skewing in the immune response to RSV infection in mice (Becker, 2006; Tripp et al., 2000a, b; Varga et al., 2000) and to inhibition of early chemokine mRNA expression (Tripp et al., 2000a) via a highly conserved CX3C chemokine motif located in the central conserved cysteine-rich region (Harcourt et al., 2006; Li et al., 2006; Tripp et al., 2001). The previous findings that RSV G protein inhibits type I IFNs through induction of SOCS1 and SOCS3 expression (Moore et al., 2008; Oshansky et al., 2009a) are consistent with the findings in this study showing RSV G protein induction of let-7f and governance of the SOCS3 gene. It appears in this context that a consequence of RSV G protein expression is induction of SOCS3-negative regulation of type I IFNs, a process that would facilitate virus replication. However, this pro-viral function attributed to the RSV G protein appears to be balanced by the host response, where G protein also induces let-7f expression, which upregulated SOCS3 expression.
An outcome of let-7f regulation of host genes seems to be delayed viral clearance. The data from Fig. 5 clearly showed that inhibition of let-7 and/or miR-24 affected virus replication significantly. These results suggest that host miRNAs may have a role in regulating virus replication similar to other RNA and DNA viruses (Jopling et al., 2006; Lagos et al. 2010; Roberts et al., 2011; Triboulet et al., 2007), either by affecting cellular pathways or by directly regulating viral transcription and/or translation. Although we analysed host mRNAs in this study, we did not analyse the effect of these miRNAs on viral gene transcription or translation, and this could be an additional mechanism employed by the virus to control viral gene expression, similar to related viruses such as influenza. These findings provide a better understanding of the mechanisms that contribute to the host response to infection and disease pathogenesis, and move the field closer to the development of safe and effective RSV disease-intervention strategies. Further studies are needed to elucidate the spectra of potential mechanisms of miRNA activation associated with RSV infection in order to understand the kinetics of miRNA deregulation, the role of other viral proteins in miRNA deregulation and the global impact of these miRNAs on virus replication by other RNA viruses.
Methods
Cell culture and viruses.
Mycoplasma-free virus stocks of recombinant wild-type RSV strain A2 (6340WT) and RSV lacking only the G protein gene (RSVΔG; a kind gift of Dr Mark Peeples, Center for Vaccines & Immunity, The Research Institute at Nationwide Children’s Hospital, OH, USA) were expanded in Vero E6 cells (ATCC CCL-81) and maintained in Dulbecco’s modified essential medium (DMEM; Hyclone) supplemented with 5 % heat-inactivated FBS (Hyclone), as described previously (Oshansky et al., 2009a). A549 cells (ATCC CCL-185) grown in DMEM supplemented with 5 % serum as above were used for all infections. A549 cells were infected at an m.o.i. of 0.5 or 1, as described previously (Oshansky et al., 2009a).
qPCR for validation of miRNA microarray data.
miRNA DNA amplicons were generated from DNase-treated A549 RNA using an Ncode miRNA cDNA synthesis kit (Invitrogen) following the manufacturer’s instructions. Amplicons were quantified, and five tenfold cDNA dilutions in triplicate and equal amounts of 1 : 10-diluted cDNA from experimental samples in triplicate were used in a real-time qPCR assay on a Mx3000/Mx3005P instrument followed by dissociation curve analysis. Sequences of forward primers were based on mature miRNA sequences from miRbase (Table S2). Amplification was carried out using the following program: initial incubation at 52 °C for 2 min and 95 °C for 10 min, and 40 cycles of 15 s denaturation at 95 °C, 1 min annealing at 57 °C and 1 min extension at 68 °C, followed by dissociation curve analysis. Only standards/replicates showing an amplification efficiency between 90 and 110 % with an R2 value >0.985, and a slope of between −3.1 and −3.4 were used for copy number calculations. Fold changes in copy number were calculated and compared for statistical significance using Student’s t-test from duplicate experiments.
Computational analysis of miRNA targets.
Predicted host gene targets for the differentially expressed miRNAs were computationally mined from miRbase (Griffiths-Jones et al., 2008), TargetScan (Lewis et al., 2005) and PicTar (Krek et al., 2005). Consensus target genes predicted by the three algorithms were compared with host genes identified previously to be affected by RSV infection (Martínez et al., 2007) to narrow the number of genes that might be potential targets for RSV-deregulated miRNAs. The 3′UTR sequence of the genes of interest was amplified from A549 cDNA using oligonucleotides with a NotI site (GGCCGC) in the forward primer and an XbaI/SpeI site (TCTAGA/ACTAGT) in the reverse primer using LongAmp Taq (New England Biolabs) under the following conditions: initial denaturation at 94 °C for 30s and 30 cycles of 94 °C for 10 s, 55 °C for 30 s and 65 °C for 30s, with a final extension at 65 °C for 10 min. Correct-sized amplicons were cloned into a pMetLucControl (pMLC) plasmid (Clontech). The oligonucleotide sequences used for UTR cloning are shown in Table S3. Plasmids were verified by restriction digestion and sequencing.
Luc–UTR reporter plasmid design and Luc reporter assays.
All transfections were carried out in triplicate in at least three independent experiments. A549 cells (2×104 per well) were transfected for 18 h using Lipofectamine 2000 with 200 ng gene-specific luc–UTR reporter plasmid (pMLC-UTR), 20 ng transfection control plasmid pSEAP2-Control (Clontech) and 25/50 nM specific or non-targeting miRNA mimics/inhibitor (NTC) to C. elegans miR-67 (Thermo Fisher) following the manufacturer’s instructions. The sequences of the miRNA inhibitors and mimics are given in Fig. 3(c). Luc and SEAP expression for each transfection was measured at 24 and 48 h post-transfection using a Ready-to-Glow kit (Clontech) following the manufacturer’s protocol. Fold changes in Luc expression were calculated using the formula: fold change in inhibitor or mimic = (Luctest/SEAPtest)/(LucNTC/SEAPNTC).
The data presented are means±sem from three independent experiments. Statistical significance was determined using Student’s t-test using GraphPad Prism version 5.0.
RISC immunoprecipitation assays.
Cell lysates were prepared at 24 h p.i. using cell lysis buffer [25 mM Tris/HCl (pH 7.5), 0.5 % NP-40, 150 mM KCl, 1 mM NaF, 2 mM EDTA, 0.5 mM DTT; all from Sigma] and a protease inhibitor tablet (Roche), and then used for RISC immunoprecipitation assays as described previously (Dölken et al., 2010). Briefly, protein G–Sepharose beads (50 µl; Thermo Scientific) were incubated with rat anti-Ago2 hybridoma (a kind gift from Drs Juergen Haas, University of Edinburgh Medical School, Edinburgh, UK, and Gunther Meister, Universität Regensburg, Regensburg, Germany) or with BrdU mAb or FBS-free DMEM (controls) overnight at 4°C with mixing followed by two washes with cell lysis buffer to remove non-specifically bound material. Cell lysate from mock-treated or 6340WT-infected cells was added to the beads and incubated overnight at 4 °C to allow binding of the anti-Ago2–protein G complex with RISC complexes. The beads were washed five times in immunoprecipitation wash buffer [50 mM Tris/HCl (pH 7.5), 300 mM NaCl, 0.01 % NP-40, 5 mM MgCl2; all from Sigma], followed by one wash with ice-cold PBS to remove the detergent. RISC-associated RNA was extracted from the beads using Qiazol (Qiagen) and the RNA isolated following the miRNAeasy protocol described for qPCR.
miRNA transfections and virus replication and plaque assays.
All transfections were carried out as described above. Transfected cells were infected with mycoplasma-free rgRSV (m.o.i. of 0.5) for 2 h in serum-free DMEM, followed by a change to complete medium containing 5 % FBS. Cell lysates were prepared at 3 days p.i., sonicated and centrifuged at 300 g at 4 °C for 5 min. Tenfold dilutions of supernatant were made in serum-free DMEM on ice and 200 µl per well was added to 24-well plates containing 2×105 Vero E6 cells per well in quadruplicate for 2 h followed by the addition of 1 ml 2 % carboxymethylcellulose. Plates were incubated for 6 days at 37 °C with 5 % CO2 and 95 % humidity, fixed with acetone : methanol (60 : 40) for 10 min at 4 °C and stained for RSV F protein using mAb 131-2A (produced in house). Plaques were detected with goat anti-mouse whole IgG coupled to alkaline phosphatase and developed using nitro blue tetrazolium (Thermo Fisher). Statistical analysis was carried out using a two-tailed Student’s t-test.
Acknowledgements
The authors wish to thank Drs Juergen Haas and Meister Gunther for their kind gift of the mAb against Ago2.
Footnotes
Three supplementary tables are available with the online version of this paper.
References
- Alvarez R., Elbashir S., Borland T., Toudjarska I., Hadwiger P., John M., Roehl I., Morskaya S. S., Martinello R. & other authors (2009). RNA interference-mediated silencing of the respiratory syncytial virus nucleocapsid defines a potent antiviral strategy. Antimicrob Agents Chemother 53, 3952–3962 10.1128/AAC.00014-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asirvatham A. J., Magner W. J., Tomasi T. B. (2009). miRNA regulation of cytokine genes. Cytokine 45, 58–69 10.1016/j.cyto.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker Y. (2006). Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy – a review. Virus Genes 33, 235–252 [DOI] [PubMed] [Google Scholar]
- Berezikov E., Chung W.-J., Willis J., Cuppen E., Lai E. C. (2007). Mammalian mirtron genes. Mol Cell 28, 328–336 10.1016/j.molcel.2007.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert B., Marozin S., Conzelmann K. K. (2003). Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol 77, 8661–8668 10.1128/JVI.77.16.8661-8668.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDCP (2008). Respiratory syncytial virus activity – United States, July 2007–December 2008. MMWR Morb Mortal Wkly Rep 57, 1355–1358 [PubMed] [Google Scholar]
- Dölken L., Malterer G., Erhard F., Kothe S., Friedel C. C., Suffert G., Marcinowski L., Motsch N., Barth S., Beitzinger M. (2010). Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 7, 324–334 10.1016/j.chom.2010.03.008 [DOI] [PubMed] [Google Scholar]
- Elliott J., Lynch O. T., Suessmuth Y., Qian P., Boyd C. R., Burrows J. F., Buick R., Stevenson N. J., Touzelet O. & other authors (2007). Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin–cullin E3 ligase. J Virol 81, 3428–3436 10.1128/JVI.02303-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. R., Sonenberg N., Filipowicz W. (2010). Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79, 351–379 10.1146/annurev-biochem-060308-103103 [DOI] [PubMed] [Google Scholar]
- Fjaerli H. O., Bukholm G., Skjaeret C., Holden M., Nakstad B. (2007). Cord blood gene expression in infants hospitalized with respiratory syncytial virus bronchiolitis. J Infect Dis 196, 394–404 10.1086/519168 [DOI] [PubMed] [Google Scholar]
- Friedman R. C., Farh K. K., Burge C. B., Bartel D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105 10.1101/gr.082701.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J. D., Ornoff D. M., Igo H. A., Zeng J. Y., Imani F. (2009). Cell cycle arrest by transforming growth factor β1 enhances replication of respiratory syncytial virus in lung epithelial cells. J Virol 83, 12424–12431 10.1128/JVI.00806-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Saini H. K., van Dongen S., Enright A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res 36 (Database issue), D154–D158 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groskreutz D. J., Monick M. M., Yarovinsky T. O., Powers L. S., Quelle D. E., Varga S. M., Look D. C., Hunninghake G. W. (2007). Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J Immunol 179, 2741–2747 [DOI] [PubMed] [Google Scholar]
- Hallak L. K., Spillmann D., Collins P. L., Peeples M. E. (2000). Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J Virol 74, 10508–10513 10.1128/JVI.74.22.10508-10513.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada N., Fujita Y., Kojima T., Kitamoto A., Akao Y., Nozawa Y., Ito M. (2012). MicroRNA expression profiling of NGF-treated PC12 cells revealed a critical role for miR-221 in neuronal differentiation. Neurochem Int 60, 743–750 10.1016/j.neuint.2012.03.010 [DOI] [PubMed] [Google Scholar]
- Harcourt J., Alvarez R., Jones L. P., Henderson C., Anderson L. J., Tripp R. A. (2006). Respiratory syncytial virus G protein and G protein CX3C motif adversely affect CX3CR1+ T cell responses. J Immunol 176, 1600–1608 [DOI] [PubMed] [Google Scholar]
- Hedvat C. V., Yao J., Sokolic R. A., Nimer S. D. (2004). Myeloid ELF1-like factor is a potent activator of interleukin-8 expression in hematopoietic cells. J Biol Chem 279, 6395–6400 10.1074/jbc.M307524200 [DOI] [PubMed] [Google Scholar]
- Huang Y.-C., Li Z., Hyseni X., Schmitt M., Devlin R. B., Karoly E. D., Soukup J. M. (2008). Identification of gene biomarkers for respiratory syncytial virus infection in a bronchial epithelial cell line. Genomic Med 2, 113–125 10.1007/s11568-009-9080-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen R., Pennings J., Hodemaekers H., Buisman A., van Oosten M., de Rond L., Oztürk K., Dormans J., Kimman T., Hoebee B. (2007). Host transcription profiles upon primary respiratory syncytial virus infection. J Virol 81, 5958–5967 10.1128/JVI.02220-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. R., Graham B. S. (2004). Contribution of respiratory syncytial virus G antigenicity to vaccine-enhanced illness and the implications for severe disease during primary respiratory syncytial virus infection. Pediatr Infect Dis J 23 (Suppl.), S46–S57 10.1097/01.inf.0000108192.94692.d2 [DOI] [PubMed] [Google Scholar]
- Johnson C. D., Esquela-Kerscher A., Stefani G., Byrom M., Kelnar K., Ovcharenko D., Wilson M., Wang X., Shelton J. & other authors (2007). The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res 67, 7713–7722 10.1158/0008-5472.CAN-07-1083 [DOI] [PubMed] [Google Scholar]
- Jopling C. L., Norman K. L., Sarnow P. (2006). Positive and negative modulation of viral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb Symp Quant Biol 71, 369–376 10.1101/sqb.2006.71.022 [DOI] [PubMed] [Google Scholar]
- Krek A., Grün D., Poy M. N., Wolf R., Rosenberg L., Epstein E. J., MacMenamin P., da Piedade I., Gunsalus K. C. & other authors (2005). Combinatorial microRNA target predictions. Nat Genet 37, 495–500 10.1038/ng1536 [DOI] [PubMed] [Google Scholar]
- Kurt-Jones E. A., Popova L., Kwinn L., Haynes L. M., Jones L. P., Tripp R. A., Walsh E. E., Freeman M. W., Golenbock D. T. & other authors (2000). Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 1, 398–401 10.1038/80833 [DOI] [PubMed] [Google Scholar]
- Lagos D., Pollara G., Henderson S., Gratrix F., Fabani M., Milne R. S., Gotch F., Boshoff C. (2010). miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat Cell Biol 12, 513–519 10.1038/ncb2054 [DOI] [PubMed] [Google Scholar]
- Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A. O. & other authors (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414 10.1016/j.cell.2007.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B. P., Burge C. B., Bartel D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- Li X.-Q., Fu Z. F., Alvarez R., Henderson C., Tripp R. A. (2006). Respiratory syncytial virus (RSV) infects neuronal cells and processes that innervate the lung by a process involving RSV G protein. J Virol 80, 537–540 10.1128/JVI.80.1.537-540.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Brass A. L., Ng A., Hu Z., Xavier R. J., Liang T. J., Elledge S. J. (2009). A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A 106, 16410–16415 10.1073/pnas.0907439106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist M. E., Lifland A. W., Utley T. J., Santangelo P. J., Crowe J. E., Jr (2010). Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol 84, 12274–12284 10.1128/JVI.00260-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist M. E., Mainou B. A., Dermody T. S., Crowe J. E., Jr (2011). Activation of protein kinase R is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology 413, 103–110 10.1016/j.virol.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M. S., Brazas R. M., Holtzman M. J. (2005). Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol 79, 9315–9319 10.1128/JVI.79.14.9315-9319.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S., Chen J. (2009). Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat Cell Biol 11, 409–419 10.1038/ncb1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez I., Lombardía L., García-Barreno B., Domínguez O., Melero J. A. (2007). Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J Gen Virol 88, 570–581 10.1099/vir.0.82187-0 [DOI] [PubMed] [Google Scholar]
- Meliopoulos V. A., Andersen L. E., Birrer K. F., Simpson K. J., Lowenthal J. W., Bean A. G., Stambas J., Stewart C. R., Tompkins S. M. & other authors (2012). Host gene targets for novel influenza therapies elucidated by high-throughput RNA interference screens. FASEB J 26, 1372–1386 10.1096/fj.11-193466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra S., Park S. J., Boyapalle S., Pastey M. K., Graham B. S., Blanck G. (2009). Human respiratory syncytial virus reduces the number of cells in S-phase and increases GADD153 expression in HEp-2 cells. Acta Virol 53, 207–211 10.4149/av_2009_03_207 [DOI] [PubMed] [Google Scholar]
- Moore E. C., Barber J., Tripp R. A. (2008). Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol J 5, 116 10.1186/1743-422X-5-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlando M., Ballarino M., Gromak N., Pagano F., Bozzoni I., Proudfoot N. J. (2008). Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 15, 902–909 10.1038/nsmb.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munday D. C., Emmott E., Surtees R., Lardeau C. H., Wu W., Duprex W. P., Dove B. K., Barr J. N., Hiscox J. A. (2010). Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus. Mol Cell Proteomics 9, 2438–2459 10.1074/mcp.M110.001859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murawski M. R., Bowen G. N., Cerny A. M., Anderson L. J., Haynes L. M., Tripp R. A., Kurt-Jones E. A., Finberg R. W. (2009). Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol 83, 1492–1500 10.1128/JVI.00671-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell R. M., Taganov K. D., Boldin M. P., Cheng G., Baltimore D. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A 104, 1604–1609 10.1073/pnas.0610731104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshansky C. M., Krunkosky T. M., Barber J., Jones L. P., Tripp R. A. (2009a). Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a Toll-like receptor pathway. Viral Immunol 22, 147–161 10.1089/vim.2008.0098 [DOI] [PubMed] [Google Scholar]
- Oshansky C. M., Zhang W., Moore E., Tripp R. A. (2009b). The host response and molecular pathogenesis associated with respiratory syncytial virus infection. Future Microbiol 4, 279–297 10.2217/fmb.09.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othumpangat S., Gibson L. F., Samsell L., Piedimonte G. (2009). NGF is an essential survival factor for bronchial epithelial cells during respiratory syncytial virus infection. PLoS ONE 4, e6444 10.1371/journal.pone.0006444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othumpangat S., Walton C., Piedimonte G. (2012). MicroRNA-221 modulates RSV replication in human bronchial epithelium by targeting NGF expression. PLoS ONE 7, e30030 10.1371/journal.pone.0030030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D., Das A., Dinh P. X., Subramaniam S., Nayak D., Barrows N. J., Pearson J. L., Thompson J., Kelly D. L. & other authors (2011). RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc Natl Acad Sci U S A 108, 19036–19041 10.1073/pnas.1113643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. R., Lichtenstein D., Ball L. A., Wertz G. W. (1994). The membrane-associated and secreted forms of the respiratory syncytial virus attachment glycoprotein G are synthesized from alternative initiation codons. J Virol 68, 4538–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. P., Lewis A. P., Jopling C. L. (2011). miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 39, 7716–7729 10.1093/nar/gkr426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 10.1038/nature07228 [DOI] [PubMed] [Google Scholar]
- Spann K. M., Tran K. C., Collins P. L. (2005). Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J Virol 79, 5353–5362 10.1128/JVI.79.9.5353-5362.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov K. D., Boldin M. P., Chang K.-J., Baltimore D. (2006). NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 103, 12481–12486 10.1073/pnas.0605298103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira N., Nihira K., Yamaguchi T., Miki Y., Yoshida K. (2007). DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 25, 725–738 10.1016/j.molcel.2007.02.007 [DOI] [PubMed] [Google Scholar]
- Taura M., Suico M. A., Fukuda R., Koga T., Shuto T., Sato T., Morino-Koga S., Okada S., Kai H. (2011). MEF/ELF4 transactivation by E2F1 is inhibited by p53. Nucleic Acids Res 39, 76–88 10.1093/nar/gkq762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayyari F., Marchant D., Moraes T. J., Duan W., Mastrangelo P., Hegele R. G. (2011). Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med 17, 1132–1135 10.1038/nm.2444 [DOI] [PubMed] [Google Scholar]
- Terasawa K., Ichimura A., Sato F., Shimizu K., Tsujimoto G. (2009). Sustained activation of ERK1/2 by NGF induces microRNA-221 and 222 in PC12 cells. FEBS J 276, 3269–3276 10.1111/j.1742-4658.2009.07041.x [DOI] [PubMed] [Google Scholar]
- Triboulet R., Mari B., Lin Y.-L., Chable-Bessia C., Bennasser Y., Lebrigand K., Cardinaud B., Maurin T., Barbry P. & other authors (2007). Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 315, 1579–1582 10.1126/science.1136319 [DOI] [PubMed] [Google Scholar]
- Tripp R. A., Moore D., Jones L., Sullender W., Winter J., Anderson L. J. (1999). Respiratory syncytial virus G and/or SH protein alters Th1 cytokines, natural killer cells, and neutrophils responding to pulmonary infection in BALB/c mice. J Virol 73, 7099–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp R. A., Jones L., Anderson L. J. (2000a). Respiratory syncytial virus G and/or SH glycoproteins modify CC and CXC chemokine mRNA expression in the BALB/c mouse. J Virol 74, 6227–6229 10.1128/JVI.74.13.6227-6229.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp R. A., Moore D., Winter J., Anderson L. J. (2000b). Respiratory syncytial virus infection and G and/or SH protein expression contribute to substance P, which mediates inflammation and enhanced pulmonary disease in BALB/c mice. J Virol 74, 1614–1622 10.1128/JVI.74.4.1614-1622.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp R. A., Jones L. P., Haynes L. M., Zheng H., Murphy P. M., Anderson L. J. (2001). CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat Immunol 2, 732–738 10.1038/90675 [DOI] [PubMed] [Google Scholar]
- Varga S. M., Wissinger E. L., Braciale T. J. (2000). The attachment (G) glycoprotein of respiratory syncytial virus contains a single immunodominant epitope that elicits both Th1 and Th2 CD4+ T cell responses. J Immunol 165, 6487–6495 [DOI] [PubMed] [Google Scholar]
- Vermeulen A., Robertson B., Dalby A. B., Marshall W. S., Karpilow J., Leake D., Khvorova A., Baskerville S. (2007). Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA 13, 723–730 10.1261/rna.448107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Munday D. C., Howell G., Platt G., Barr J. N., Hiscox J. A. (2011). Characterization of the interaction between human respiratory syncytial virus and the cell cycle in continuous cell culture and primary human airway epithelial cells. J Virol 85, 10300–10309 10.1128/JVI.05164-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Zhang J., Zhang A., Wang Y., Han L., You Y., Pu P., Kang C. (2010a). PUMA is a novel target of miR-221/222 in human epithelial cancers. Int J Oncol 37, 1621–1626 [DOI] [PubMed] [Google Scholar]
- Zhang J., Han L., Ge Y., Zhou X., Zhang A., Zhang C., Zhong Y., You Y., Pu P., Kang C. (2010b). miR-221/222 promote malignant progression of glioma through activation of the Akt pathway. Int J Oncol 36, 913–920 [DOI] [PubMed] [Google Scholar]