

Abstract
The RecG enzyme, a superfamily 2 helicase, is present in nearly all bacteria. Here we report for the first time that the recG gene is also present in the genomes of most vascular plants as well as in green algae, but is not found in other eukaryotes or archaea. The precise function of RecG is poorly understood, although ample evidence shows that it plays critical roles in DNA repair, recombination and replication. We further demonstrate that Mycobacterium tuberculosis RecG (RecGMtb) DNA binding activity had a broad substrate specificity, whereas it only unwound branched-DNA substrates such as Holliday junctions (HJs), replication forks, D-loops and R-loops, with a strong preference for the HJ as a helicase substrate. In addition, RecGMtb preferentially bound relatively long (≥40 nt) ssDNA, exhibiting a higher affinity for the homopolymeric nucleotides poly(dT), poly(dG) and poly(dC) than for poly(dA). RecGMtb helicase activity was supported by hydrolysis of ATP or dATP in the presence of Mg2+, Mn2+, Cu2+ or Fe2+. Like its Escherichia coli orthologue, RecGMtb is also a strictly DNA-dependent ATPase.
Introduction
Mycobacterium tuberculosis, the aetiological agent of the re-emerging disease tuberculosis (TB), remains a global health threat, killing at least 1.5 million individuals every year (WHO, 2011). The emergence of extensively and extremely drug-resistant M. tuberculosis strains, coupled with the HIV/AIDS pandemic, has exacerbated the risk of TB resurgence, underlining the urgent need to develop interventions that halt the spread of this disease. M. tuberculosis, an intracellular human pathogen, successfully combats many host cell defence mechanisms, including genotoxic stress, using efficient DNA repair pathways that help to maintain its genome integrity, in spite of accumulating DNA damage during infection (Ambur et al., 2009; Dos Vultos et al., 2009; Gorna et al., 2010; Mizrahi & Andersen, 1998; Stallings & Glickman, 2010). It is believed that these DNA repair pathways promote M. tuberculosis survival and increase its pathogenicity and virulence (Gorna et al., 2010; Warner, 2010). Thus, in-depth characterization of the mechanisms that protect the M. tuberculosis genome and promote its virulence and/or capacity to develop drug resistance may lead to novel therapeutic targets and attenuate the increasing risk of global resurgence of TB.
Escherichia coli RecG, a 76 kDa monomeric helicase with a particular affinity for branched-DNA substrates such as replication forks, Holliday junctions (HJs), and D- and R-loops, has been shown to play roles in DNA repair, recombination and replication (Lloyd & Sharples, 1993; McGlynn et al., 1997; McGlynn & Lloyd, 2000, 2001; Rudolph et al., 2010b; Vincent et al., 1996; Whitby & Lloyd, 1998). E. coli RecG is widely believed to promote regression of stalled replication forks, when fork progression is blocked by lesions in the DNA template strand, thereby facilitating repair or bypass of the lesion (McGlynn & Lloyd, 2001, 2002). In vitro studies suggest that RecG actively unwinds stalled replication forks, generating a four-way junction product that resembles an HJ (McGlynn & Lloyd, 2000, 2001; McGlynn et al., 2001), and also promotes HJ branch migration (Müller & West, 1994; Whitby et al., 1994). Structural characterization of a complex between Thermotoga maritima RecG and a forked-DNA substrate has revealed the mechanism by which RecG recognizes junctions (Singleton et al., 2001). E. coli RecG inhibits inappropriate DNA amplification and aberrant chromosome segregation in cells exposed to UV irradiation (Rudolph et al., 2009a, b), and is also essential in cells lacking 3′ ssDNA exonucleases to counteract PriA helicase-mediated DNA re-replication (Rudolph et al., 2010a). Furthermore, a recent study has revealed that RecG promotes resolution of intermolecular recombination intermediates that are poorly recognized/resolved by RuvABC (Fonville et al., 2010).
Strains with mutations in recG have been shown to exhibit complex and variable phenotypes, including transformation deficiency in Neisseria meningitidis (Sun et al., 2005), growth defects and reduced radio-resistance in Deinococcus radiodurans (Wu et al., 2009), and sensitivity to UV irradiation and oxidative stress in Pseudomonas aeruginosa (Ochsner et al., 2000). recG mutation has also been suggested to be responsible for the susceptibility of Staphylococcus aureus to quinolone (Niga et al., 1997) and of E. coli to bleomycin, metronidazole and ciprofloxacin (Kosa et al., 2004; Tamae et al., 2008).
In this study, we have characterized the recombinant RecG enzyme from M. tuberculosis H37Rv (RecGMtb) for its DNA binding, unwinding and ATPase activities in order to delineate its potential roles in the DNA metabolism of M. tuberculosis.
Methods
Cloning the M. tuberculosis recG gene.
M. tuberculosis genomic DNA was isolated from a M. tuberculosis H37Rv (ATCC 25618) culture. The M. tuberculosis recG (Rv2973c) gene was PCR-amplified using a forward primer (5′-CGCATATGGCGTCGTTAAGCGATCGGCTC-3′) and reverse primer (5′-CGCTCGAGTCATGACTTATCTAAGTATTCGATGC-3′) which contained NdeI and XhoI restriction sites, respectively (underlined). The PCR product was digested with NdeI and XhoI and ligated into a similarly digested pET28b(+) vector (Novagen). The resulting construct, pET28b–recG with an N-terminal His6-tag, was then transformed into E. coli ER2566 (New England Biolabs; NEB). The nucleotide sequence of the construct was verified by DNA sequencing (ABI).
Overexpression and purification of the RecGMtb protein.
The recombinant RecGMtb protein was purified to homogeneity as follows. E. coli ER2566 harbouring pET28b–recG was inoculated into Luria–Bertani broth (Difco) supplemented with 50 µg kanamycin ml−1, 2.5 mM betaine hydrochloride and 0.5 M sorbitol, and grown at 37 °C to OD600 ~0.4. The culture was then transferred to 18 °C and induced at OD600 ~0.6 with 0.5 mM IPTG. After overnight growth, cells were harvested, lysed and purified using a nickel-nitrilotriacetic acid agarose column as described in the QIAexpressionist protocol for native purification of His6-tagged proteins from E. coli (Qiagen, 2003). β-Mercaptoethanol (5 mM) was added to the lysis, wash and elution buffers as indicated in the protocol. After elution from the column, the eluates containing RecGMtb were pooled and dialysed overnight against buffer comprising 50 mM NaH2PO4 (pH 7.5), 300 mM NaCl and 1 mM DTT. The N-terminal His6-tag was cleaved off using biotinylated thrombin (Novagen) following the manufacturer’s protocol. Further purification was carried out on a HiTrap Q HP column (GE Healthcare) after the buffer had been exchanged with 20 mM Bistris (pH 7.2), 100 mM NaCl and 1 mM DTT. Fractions containing pure RecGMtb were pooled and dialysed against storage buffer [20 mM Bistris (pH 7.5), 300 mM NaCl, 1 mM DTT, 20 % glycerol (w/v)] and stored at −80 °C until use. RecGMtb protein concentration was determined using the Bradford method (Bio-Rad) using BSA as standard.
Model DNA substrate preparation for DNA binding, unwinding and ATPase assays.
DNA substrates were prepared essentially as described by Brosh et al. (2006) with some modifications. Briefly, oligonucleotides were 5′ end-labelled using [γ-32P]ATP (PerkinElmer) and T4 PNK enzyme (NEB) for 1 h at 37 °C, followed by enzyme inactivation at 65 °C for 20 min. Unincorporated ATPs were removed using illustra MicroSpin G-25 columns (GE Healthcare). Labelled and unlabelled complementary oligonucleotides were mixed at a molar ratio of 1 : 2.5 in annealing buffer [40 mM Tris/HCl (pH 8.0), 50 mM NaCl], denatured at 95 °C for 5 min, and allowed to cool to room temperature for about 3 h. The annealed products were resolved on an 8 % non-denaturing polyacrylamide gel. The bands containing the completely annealed substrates were excised and DNA was eluted into buffer comprising 10 mM Tris/HCl (pH 8.0) and 0.5 mM EDTA by incubating overnight at 4 °C. The concentrations of the eluted DNA substrates were estimated as described by Brosh et al. (2006) and are given in moles of substrate molecules. For ATPase assays, DNA cofactors employed were prepared by annealing equimolar concentrations of complementary strands. Proper annealing of the prepared DNA cofactors was verified by resolving on a non-denaturing 10 % polyacrylamide gel and staining with SYBR Safe DNA Gel Stain (Invitrogen). The oligonucleotides used and the schematics of the model DNA substrates constructed are presented in Tables 1 and 2, respectively.
Table 1. List of oligonucleotides used to construct model DNA substrates.
| Oligonucleotide no. | Oligonucleotide name | Length (nt) | Sequence (5′–3′) | Reference or source |
| 1 | A | 40 | CGTGACATGCCGTGACTAGCTTTTTTTTTTTTTTTTTTTT | Biswas et al. (2009) |
| 2 | B | 20 | GCTAGTCACGGCATGTCACG | Biswas et al. (2009) |
| 3 | C | 40 | TTTTTTTTTTTTTTTTTTTTCGTGACATGCCGTGACTAGC | Biswas et al. (2009) |
| 4 | HJSO1 | 49 | GACGCTGCCGAATTCTGGCTTGCTAGGACATCTTTGCCCACGTTGACCC | Lloyd & Sharples (1993) |
| 5 | HJSO2 | 50 | TGGGTCAACGTGGGCAAAGATGTCCTAGCAATGTAATCGTCTATGACGTT | Lloyd & Sharples (1993) |
| 6 | HJSO3 | 51 | CAACGTCATAGACGATTACATTGCTAGGACATGCTGTCTAGAGACTATCGA | Lloyd & Sharples (1993) |
| 7 | HJSO4 | 50 | ATCGATAGTCTCTAGACAGCATGTCCTAGCAAGCCAGAATTCGGCAGCGT | Lloyd & Sharples (1993) |
| 8 | HJSO5 | 49 | CGGGTCAACGTGGGCAAAGATGTCCTAGCAAGCCAGAATTCGGCAGCGT | This study |
| 9 | RFSO1 | 50 | GTCGGATCCTCTAGACAGCTCCATGATCACTGGCACTGGTAGAATTCGGC | McGlynn & Lloyd (2001) |
| 10 | RFSO2 | 50 | CAACGTCATAGACGATTACATTGCTACATGGAGCTGTCTAGAGGATCCGA | McGlynn & Lloyd (2001) |
| 11 | RFSO3 | 25 | TAGCAATGTAATCGTCTATGACGTT | McGlynn & Lloyd (2001) |
| 12 | RFSO4 | 26 | TGCCGAATTCTACCAGTGCCAGTGAT | McGlynn & Lloyd (2001) |
| 13 | DLO1 | 61 | GGGTGAACCTGCAGGTGGGCGGCTGCTCATCGTAGGTTAGTTGGTAGAATTCGGCAGCGTC | McGlynn et al. (1997) |
| 14 | DLO2 | 61 | GACGCTGCCGAATTCTACCAGTGCCTTGCTAGGACATCTTTGCCCACCTGCAGGTTCACCC | McGlynn et al. (1997) |
| 15 | DLO3 | 41 | TAAGAGCAAGATGTTCTATAAAAGATGTCCTAGCAAGGCAC | McGlynn et al. (1997) |
| 16 | DLO4 | 41 | AAAGATGTCCTAGCAAGGCACGATCGACCGGATATCTATGA | McGlynn et al. (1997) |
| 17 | DLO5 | 61 | TATAGAACATCTTGCTCGTTTTCGAGCAAGATGTTCTATAAAAGATGTCCTAGCAAGGCAC | This study |
| 18 | DLO6 | 61 | AAAGATGTCCTAGCAAGGCACGATCGACCGGATATCTACTTTTGTAGATATCCGGTCGATC | This study |
| 19 | DLO7 | 21 | AAAGATGTCCTAGCAAGGCAC | McGlynn et al. (1997) |
| 20* | 2-Methyl RNA-1 | 41 | UAAGAGCAAGAUGUUCUAUAAAAGAUGUCCUAGCAAGGCAC | This study |
| 21* | 2-Methyl RNA-2 | 41 | AAAGAUGUCCUAGCAAGGCACGAUCGACCGGAUAUCUAUGA | This study |
| 22 | HJSO6 | 66 | TGGGTCAACGTGGGCAAAGATGTCCTAGCAATGTAATCGTCTATGACGTTGTTTTTTTTTTTTTTT | This study |
2′-O-Methyl-RNA.
Table 2. Summary of RecGMtb DNA binding and unwinding activity.
Minus and plus symbols indicate the absence or presence of RecGMtb activity on the indicated substrate, respectively.

γ-32P-Labelled 5′ end.
DNA binding assays.
DNA binding assays were carried out as described by Whitby & Lloyd (1998) with some modifications. Assays were performed in reactions (20 µl) containing binding buffer [50 mM Tris/HCl (pH 8.0), 5 mM EDTA, 100 µg BSA ml−1, 6 % (w/v) glycerol, 2 mM DTT, 50 ng poly(dI-dC) µl−1 (Thermo Scientific)] and 0.1 nM of the indicated DNA substrate. Where indicated, poly(dI-dC) was omitted from the binding buffer. Reactions were initiated by adding indicated concentrations of RecGMtb. After 15 min incubation on ice, 4 µl of 60 % (w/v) glycerol was added and immediately loaded onto a pre-cooled and pre-run (30 min) 4 % non-denaturing polyacrylamide gel (29 : 1). Electrophoresis was performed using low-ionic-strength buffer at 200 V for 5 min and at 160 V for an additional 85 min in an ice-water bath with buffer recirculation. Gels were dried, exposed to a phosphorimaging screen, visualized using a phosphorimager (Typhoon 9410, Amersham Biosciences) and quantified by ImageQuant TLv 2003.02 software (GE Healthcare).
Helicase assays.
Unless otherwise specified, all helicase assays were conducted in a 20 µl reaction containing helicase buffer [20 mM Tris/HCl (pH 7.5), 2 mM MgCl2, 2 mM ATP, 100 µg BSA ml−1, 2 mM DTT], 0.5 nM DNA substrate and the indicated concentrations of RecGMtb. For divalent metal cofactor studies, MgCl2 in the aforementioned buffer was replaced by 2 mM of the indicated metal chloride. Similarly, in fuel preference studies, 2 mM of the tetrasodium salt of the indicated NTP/dNTP was used in place of ATP in the helicase buffer. Helicase reactions were initiated by adding RecGMtb and, after incubating at 37 °C for 30 min, were terminated with 10 µl of 3× helicase stop solution [50 mM EDTA, 40 % (w/v) glycerol, 0.9 % SDS, 0.1 % bromophenol blue, 0.1 % xylene cyanol] containing a 10-fold molar excess of trap oligonucleotide. For helicase time-course assays, the reaction was scaled up to 140 µl and RecGMtb was added into pre-incubated (3 min) reaction mixture at 37 °C. An aliquot (10 µl) of the reaction mixture was withdrawn at the indicated time points and mixed with 5 µl 3× helicase stop solution. All helicase reaction products were resolved by 10 % non-denaturing polyacrylamide (19 : 1) gel electrophoresis at 150 V for 2 h at room temperature using 1× Tris/borate-EDTA buffer. Gels were dried and analysed as described for DNA binding assays. The proportion of helicase substrate unwound (%) was calculated as described by Brosh et al. (2006).
ATPase assays.
RecGMtb ATP hydrolysis activity was monitored by TLC, as described by Kornberg et al. (1978) with some modifications. Briefly, RecGMtb was added to initiate a 10 µl reaction in ATPase buffer [20 mM Tris/HCl (pH 7.5), 2 mM MgCl2, 100 µg BSA ml−1, 25 µM cold ATP, 0.023 nM [γ-32P]ATP, 2 mM DTT] and the indicated DNA cofactor. The reaction was incubated at 37 °C for the indicated times and terminated by adding 5 µl 0.5 M EDTA (pH 8.0). Samples (2 µl) were spotted onto TLC plates (Cellulose PEI F, Merck) at 1.5 cm intervals and resolved using a solution containing 1 M formic acid and 0.5 M LiCl. The TLC plates were air-dried, exposed to a phosphorimaging screen, imaged and quantified as described above for the DNA binding assays. The proportion of hydrolysed ATP (%) was calculated as {counts for γ-32Pi/(counts for γ-32Pi+counts for [γ-32P]ATP)}×100. The values obtained from samples lacking RecGMtb were subtracted from the samples containing RecGMtb to account for background ATP hydrolysis. An unpaired Student’s t test was used to determine statistical significance.
To determine the steady-state kinetic parameters of ATP hydrolysis, a 20 µl reaction was set up with ATPase buffer [20 mM Tris/HCl (pH 7.5), 4 mM MgCl2, 100 µg BSA ml−1, cold ATP, 0.023 nM [γ-32P]ATP, 2 mM DTT], 125 ng plasmid DNA (pET28b) µl−1 and 150 nM RecGMtb. The cold ATP concentration was varied between 100 and 800 µM. The reactions were incubated at 37 °C for 10 min and quenched with 10 µl 0.5 M EDTA (pH 8.0). The concentration of hot ATP was negligible and thus not considered in the calculations. The velocity data points versus cold ATP concentrations were non-linearly fitted to Michaelis–Menten and Hill equations using Prism 5 software (GraphPad).
Results
RecG is conserved in bacteria and is present in vascular plants
The full-length E. coli RecG protein sequence was used as a query to search NCBI protein sequence databases (Sayers et al., 2012) for conserved homologues in bacteria, archaea, and plants and other eukaryotes. Homologues of recG were found to be present in the genomes of most bacteria, except Chlamydiae and Mollicutes, as reported earlier (Rocha et al., 2005; Sharples et al., 1999). However, recG homologues were not found in any of the >90 archaeal genomes in the current version of the database (Sayers et al., 2012). Notably, full-length recG was also present in the genomes of most vascular plants, including Arabidopsis thaliana, the castor oil plant (Ricinus communis), common grape vine (Vitis vinifera), California poplar (Populus trichocarpa) and the lycophyte Selaginella moellendorffii, as well as in green algae such as Ostreococcus tauri and Nannochloris bacillaris. The recG gene was not detected in eukaryote species outside the kingdom Plantae. The plant genes are localized to the nuclear genomes and do not appear to be recently acquired from bacteria. For example, A. thaliana (Arabidopsis Genome Initiative, 2000) and V. vinifera (Jaillon et al., 2007) have intron-rich recG genes encoded on chromosomes 2 and 1, respectively, with all 16 introns shared between the two species. The RecG homologues, including RecGMtb, have two RecA-like helicase domains, an N-terminal wedge-containing domain and a C-terminal TRG (translocation in RecG) motif (Mahdi et al., 2003) (Fig. 1).
Fig. 1.

RecG, with its unique wedge-containing domain, is found in bacteria and plants. Domain organization showing the N-terminal wedge-containing domain that is unique to the RecG helicase family, the N- and C-terminal RecA-like helicase domains, and the C-terminal TRG motif for RecG from the bacteria M. tuberculosis, E. coli and T. maritima, and the vascular plant Arabidopsis thaliana. Numbers indicate approximate domain boundaries.
RecGMtb binds a wide variety of DNA substrates
RecGMtb bound to a wide variety of model DNA substrates, including partial and complete replication forks, HJ, bubble, and D- and R-loop substrates (Fig. 2a, b, Table 2), with the highest affinity for HJs (Fig. 2c). In contrast, the affinity of RecGMtb for a linear DNA duplex (49 bp) was very low, and it did not bind DNA substrates containing 20 nt 5′ or 3′ overhangs, in the presence of poly(dI-dC) competitor (Fig. 2a, c, Table 2). When we analysed the binding affinity of RecGMtb to homopolymeric nucleotides (40 nt) in the absence of poly(dI-dC) competitor, poly(dA) showed very weak binding compared with poly(dC), poly(dG), poly(dT) and random nucleotides (dN) (Fig. 2d). However, the binding affinity of RecGMtb to poly(dA) appeared to increase with increasing length of nucleotides as for poly(dT) and poly(dA : dT), yet with less stable protein–DNA complexes (Fig. 2e; data not shown). The binding activity of RecGMtb was not influenced by the presence or absence of ATP, ADP or ATPγS (see Fig. S1 available with the online version of this paper).
Fig. 2.

RecGMtb DNA binding specificity. (a) Titrations of the DNA binding activity of RecGMtb using 0.25, 0.5, 1.0, 1.5, 2.0 and 2.5 µM RecGMtb (lanes 2–7, respectively) and 0.1 nM linear DNA duplex (substrate C), lagging strand replication fork (substrate E), leading strand replication fork (substrate F) and HJ (substrate H). (b) Gel shift assay on a bubble and a variety of D- and R-loop substrates in the presence of 2 µM RecGMtb. Lanes: (−), absence of RecGMtb; (+), presence of RecGMtb. (c) Quantification of the gel images in (a). (d) Shift assays using 0.1 nM of 40 nt poly(dA), poly(dC), poly(dG), poly(dT) and random nucleotide (dN), and 1 µM RecGMtb in the absence of poly(dI-dC). (e) ssDNA and dsDNA binding activity of RecGMtb using 0.1 nM poly(dA) (A), poly(dT) (T) and poly(dA : dT) (A : T) of increasing length and 1 µM RecGMtb in the absence of poly(dI-dC). Data presented in (c–e) are means±sd from at least three independent experiments.
RecGMtb unwinds DNA replication forks, D-loops, R-loops and HJ substrates
The unwinding activity of RecGMtb was examined using a variety of DNA substrates, including flayed DNA duplex, and lagging, leading and complete replication fork substrates. RecGMtb did not exert any unwinding activity on flayed DNA duplex, but demonstrated weak and strong unwinding activities on leading and lagging strand replication forks, respectively (Fig. 3a–c). Moreover, RecGMtb unwound both strands of a complete replication fork substrate (Fig. 3d). These results suggested that RecGMtb, like E. coli RecG, requires more than one duplex arm to unwind a three-way junction (Whitby & Lloyd, 1998). Interestingly, both RecGMtb and E. coli RecG (McGlynn & Lloyd, 2001) unwound the lagging strand replication fork more efficiently than the leading strand replication fork substrate (Fig. 3b, c; see also Fig. 5c).
Fig. 3.
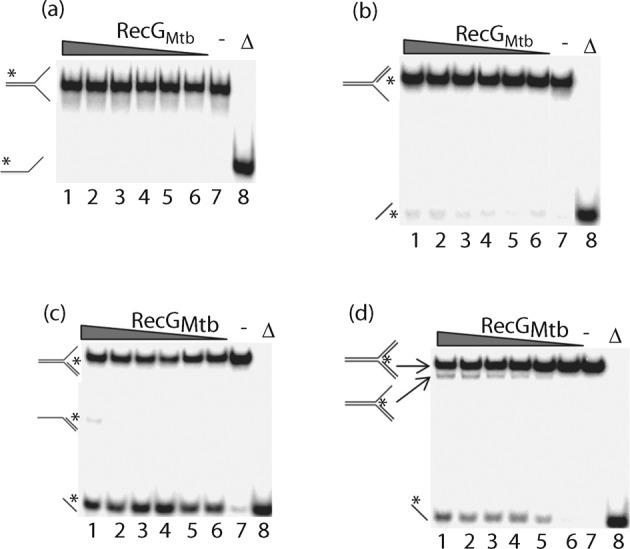
RecGMtb unwinds nascent DNA strands from partial and complete replication fork substrates. Titration of helicase activity using 0.5 nM flayed DNA duplex (a), leading strand replication fork (b), lagging strand replication fork (c) and complete replication fork (d) substrates, and 250, 200, 150, 100, 50 and 25 nM RecGMtb (lanes 1–6, respectively). Lanes: (−), reaction lacking RecGMtb; (Δ), heat-denatured substrate.
RecGMtb also unwound an HJ substrate with a 12 bp central homologous ‘movable core’, producing flayed duplexes (Fig. 4a). To determine the direction of RecGMtb-mediated branch migration of the HJ, RecGMtb was challenged with an HJ substrate containing a 16 nt extension on one of the four duplex arms (Fig. 4b). Interestingly, RecGMtb appeared to drive branch migration bidirectionally. The time course of HJ substrate unwinding indicated that nearly half (47 %) of the substrate was converted to flayed duplex by RecGMtb within 1 min (Fig. 4c).
Fig. 4.
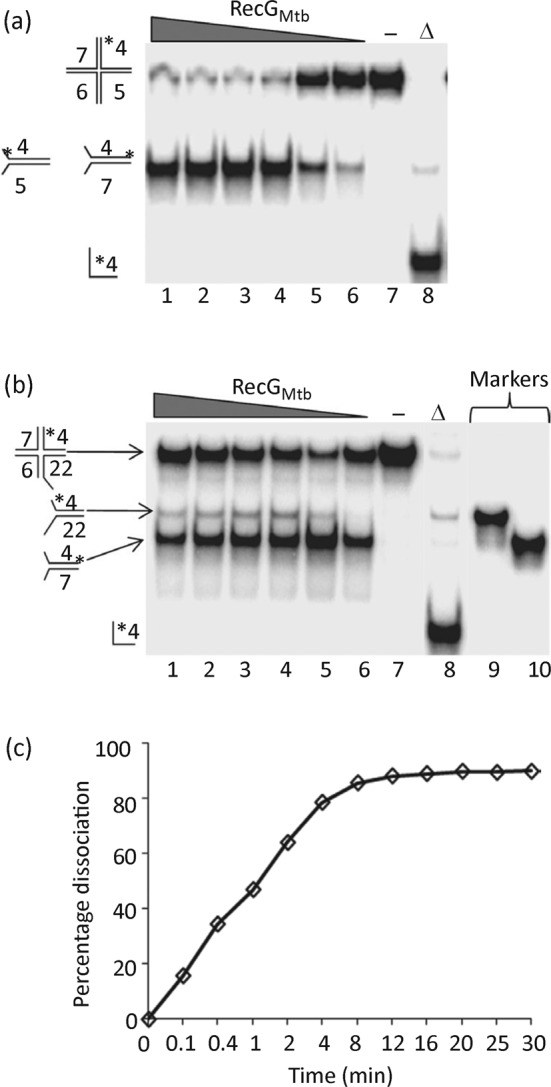
RecGMtb dissociates HJ substrates to flayed duplexes. (a) Titration of branch migration activity using 250, 200, 150, 100, 50 and 25 nM RecGMtb (lanes 1–6, respectively) and 0.5 nM HJ substrate. Lanes: (−), reaction lacking RecGMtb; (Δ), heat-denatured substrate. (b) Direction of branch migration of HJs. Reactions were conducted in the same way as in (a) except that a modified HJ substrate in which oligonucleotide 5 was replaced with oligonucleotide 22 was used (Tables 1 and 2). Lanes 9 and 10 show size markers for the expected branch migration products. (c) Time course of dissociation of HJ substrate by RecGMtb. The helicase reaction (140 µl) was carried out using 150 nM RecGMtb and 0.5 nM HJ substrate. Data in (c) are the average of two independent experiments.
RecGMtb also unwound a variety of synthetic D- and R-loop structures. These included D-loops without tail (substrate J), D-loops with 5′ or 3′ tails (substrates L and K, respectively), D-loops with a hairpin end at the 5′ or 3′ tail (substrates N and M, respectively), and R-loops with 5′ and 3′ (2′-O-methyl-RNA) tails (substrates P and O, respectively) (Fig. 5a, b). The 2′-O-methyl modification was introduced to protect the RNA against nuclease degradation (Inoue et al., 1987). The ability of RecGMtb to unwind D- and R-loops regardless of the absence or presence of a tail suggests that RecGMtb may preferentially interact with such DNA structures at the junction. RecGMtb might also pull one side or a segment of the duplex arm of the D- or R-loop structure through the wedge-containing domain, thereby stripping the invading strand off the loop structure, as previously proposed for unwinding of the lagging strand from a partial replication fork (Rudolph et al., 2010b; Singleton et al., 2001).
Fig. 5.
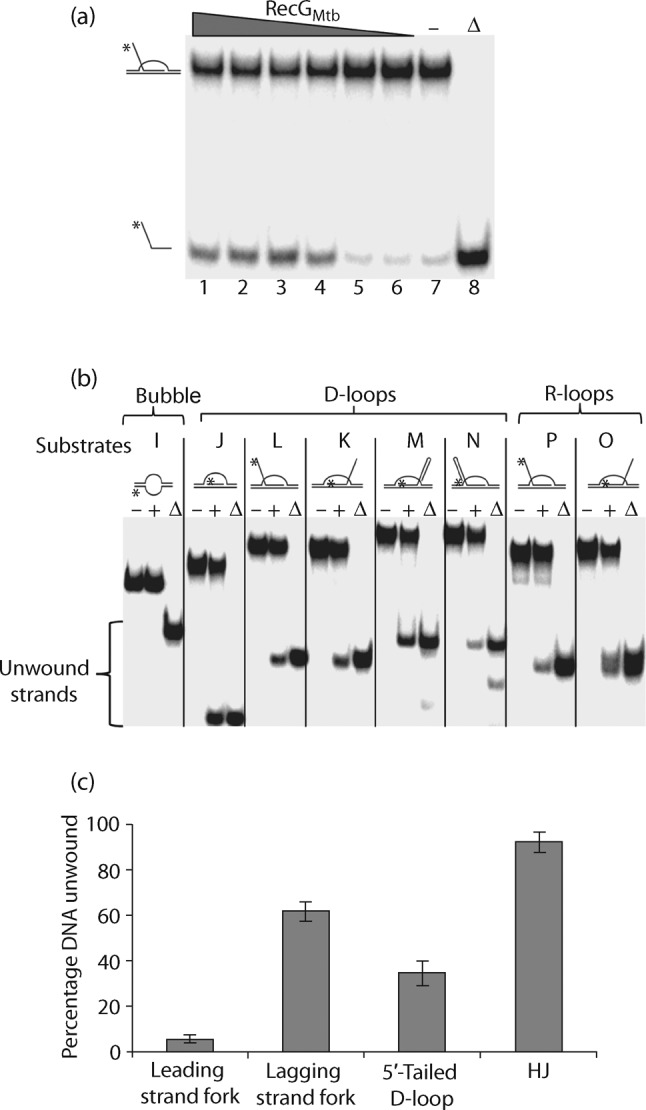
RecGMtb unwinds the invading strands from a variety of synthetic D- and R-loop structures. (a) Titration of helicase activity using 250, 200, 150, 100, 50 and 25 nM RecGMtb (lanes 1–6, respectively) and 0.5 nM 5′-tailed D-loop (substrate L, Table 2). (b) Helicase assay using 150 nM RecGMtb and 0.5 nM of the indicated substrates (substrates I–P, Table 2). Lanes: (−), absence of RecGMtb; (+), presence of RecGMtb; (Δ), heat-denatured samples. (c) RecGMtb has a predilection for dissociating HJ compared with D-loop and replication fork substrates. The helicase assay was conducted using 150 nM RecGMtb and 0.5 nM of the indicated DNA substrates. Data are means±sd from three independent experiments.
We determined the efficiency with which RecGMtb unwound HJ, replication fork and D-loop substrates, and identified HJ as the preferred DNA substrate of RecGMtb helicase, followed by the lagging strand replication fork (Fig. 5c). These two DNA substrates were also preferentially bound by RecGMtb (Fig. 2c). On the other hand, RecGMtb did not unwind blunt-end DNA duplex (Fig. S2a), 3′- or 5′-tailed DNA duplex (Fig. S2b, c), or a bubble substrate (Fig. 5b).
Divalent metal ion and nucleotide requirements for RecGMtb unwinding activity
The RecGMtb helicase was active in the presence of magnesium, manganese, copper, iron or cobalt ions, but completely inactive in the reactions lacking divalent cations (Fig. 6a). No significant difference was observed in RecGMtb unwinding activity in the presence of Mn2+ or Mg2+ (P = 0.125). RecGMtb unwound DNA substrates in the presence of ATP or dATP, but was inactive in the presence of other NTP/dNTPs (Fig. 6b). RecGMtb unwinding activity was significantly higher in the presence of ATP than dATP (P = 0.023). Furthermore, ADP and the slowly hydrolysable ATP analogue ATPγS did not support the unwinding activity of RecGMtb (Fig. S3).
Fig. 6.
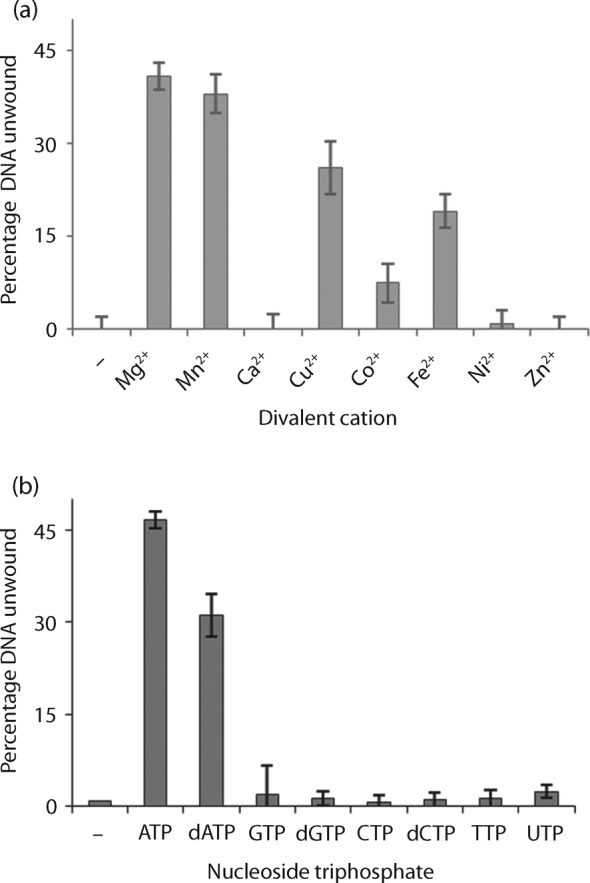
Divalent cation and NTP/dNTP specificity of RecGMtb for its helicase activity. (a) Effects of divalent metal cofactors on RecGMtb helicase activity. The helicase assay was performed in the presence of 2 mM of the indicated divalent metal cofactor, RecGMtb (150 nM) and lagging strand replication fork substrate (0.5 nM). (b) Fuel specificity for RecGMtb helicase activity. The helicase assay was performed in the presence of 2 mM of the indicated NTP/dNTP, RecGMtb (150 nM) and lagging strand replication fork substrate (0.5 nM). Data are means±sd from four independent experiments.
RecGMtb is a DNA-dependent ATPase
The ATPase activity of RecGMtb was measured in the presence of 50 nM ssDNA (49 nt), dsDNA (49 bp) or an HJ substrate (assembled from four ~49 nt oligos). The efficiency of ATP hydrolysis was 68, 32 and 0 % in the presence of HJ, dsDNA and ssDNA, respectively (Fig. 7a). Moreover, no ATP hydrolysis was observed in the reactions containing 20–100 nt poly(dT) (data not shown). We also tested ATP hydrolysis in the presence of 10 nM circular DNA cofactors, M13mp18 (ssDNA) or pET28b (dsDNA); pET28b (51 %) stimulated threefold higher ATP hydrolysis than M13mp18 (16 %) (Fig. 7b). To avoid intramolecular duplex formation, all the ssDNA cofactors tested in this study were heated to 95 °C for 5 min immediately before use.
Fig. 7.

ATP hydrolysis by RecGMtb. (a) Effects of DNA cofactors on RecGMtb ATPase activity. The assays were conducted in the presence of 300 nM RecGMtb and 50 nM of the indicated unlabelled DNA cofactor (a) or 10 nM of the indicated circular plasmid DNA (b). Plasmid pET28b was purified using Qiagen Midi kits, whereas M13mp18 (NEB) was used as obtained. Data are means±sd from at least three independent experiments.
Steady-state kinetic analysis of RecGMtb ATPase activity was performed by titrating ATP in the presence of saturating circular dsDNA (pET28b). Under these conditions, ATP hydrolysis was linear for at least 15 min (data not shown). Michaelis–Menten (hyperbolic) curve-fitting analysis showed that the mean (±se) Vmax, Km and Kcat of RecGMtb were 477 (±27) µM min−1, 202 (±35) µM and 3180 (±59) min−1, respectively. Using the Hill (allosteric sigmoidal) equation to model the data, the mean (±se) Hill coefficient (H) obtained was 1.0±0.3, which suggests a monomeric state during ATP hydrolysis.
Discussion
The M. tuberculosis genome is susceptible to the effects of genotoxic and general cellular stress, including nitrosative and oxidative damage to DNA, RNA and other biomolecules (Stallings & Glickman, 2010; Warner & Mizrahi, 2006), owing to the harsh internal environment, the human macrophage, in which it usually resides. Mechanisms that promote genome maintenance and function are likely to be essential for M. tuberculosis survival and virulence, because persistent unrepaired DNA damage can completely block replication of the genome (Masai et al., 2010; McGlynn & Lloyd, 2001; Mirkin & Mirkin, 2007). RecG is an important enzyme widely thought to play a role in remodelling replication forks stalled at DNA lesions, mediating replication restart via fork regression (McGlynn & Lloyd, 2001, 2002). In the present study, the biochemical activities of RecGMtb are characterized, providing considerable insight into the potential role(s) of RecG in DNA/nucleic acid metabolism in an intracellular pathogen.
This study demonstrated that RecGMtb binds and unwinds a variety of DNA substrates that mimic intermediates in DNA replication, recombination and repair, like its E. coli orthologue. Among the substrates examined here, RecGMtb had the highest affinity for HJ, while 3′- and 5′-overhang DNA substrates and blunt-end duplex DNA substrates were bound very poorly (Table 2). In general, the binding and unwinding activity of RecGMtb required protein concentrations that were considerably higher than those described for E. coli RecG. Whereas E. coli RecG shifted an HJ substrate at protein concentrations of 0.1 nM (Briggs et al., 2005), the same shift was only observed with 0.25 µM RecGMtb. This might be due to suboptimal assay conditions. Generally, M. tuberculosis helicases exerted their activities at concentrations >100 nM (Balasingham et al., 2012; Biswas et al., 2009; Curti et al., 2007).
Unexpectedly, RecGMtb had very low affinity for poly(dA), and much higher affinity for poly(dT), poly(dG) and poly(dC). Generally, RecGMtb and other superfamily 2 helicases make extensive contact with the sugar-phosphate DNA backbone and this interface is the dominant functional mode of interaction (Büttner et al., 2007; Kim et al., 1998; Pyle, 2008; Singleton et al., 2001). DNA helicases also tend to exhibit low DNA sequence specificity, presumably because higher sequence specificity might hinder the translocation and/or processivity of the helicase (Rocak & Linder, 2004; Tanner & Linder, 2001; Tuteja & Tuteja, 2004). However, a recent report has revealed that Vaccinia viral helicase NPH-II, a superfamily 2 helicase, favours purine-rich over pyrimidine-rich dsDNA helicase substrates (Taylor et al., 2010). The sequence bias of RecGMtb reported here might be an intrinsic property of this helicase; however, this conclusion is preliminary and requires additional investigation.
Notably, another interesting observation reported here is that the affinity of RecGMtb for ssDNA and dsDNA is length-dependent. RecGMtb had higher affinity for longer oligomers (≥40 nt) than for shorter oligomers (20 nt). This suggests that 20 nt/bp DNA substrates are not long enough to form a stable complex with RecGMtb. The relatively more stable binding of RecGMtb ≥40 nt poly(dT) is consistent with the site size determined for E. coli RecG for poly(dT), 36 nt (Slocum et al., 2007). RecGMtb may be recruited to stalled replication forks via an interaction with ssDNA binding protein (SSB) (as for E. coli RecG) (Buss et al., 2008; Lecointe et al., 2007; Liu et al., 2011). Nevertheless, the observed interaction between RecGMtb and ssDNA may represent an alternative mechanism for targeting RecG to stalled replication forks or other branched DNA substrates.
Even though RecGMtb binds a variety of DNA substrates, including ssDNA, it only unwinds branched DNA substrates, including HJs, replication forks, and D- and R-loops. This specificity suggested a potential involvement of RecGMtb in multiple DNA metabolic pathways of mycobacteria, including recombination, replication and repair. The relatively high affinity of RecGMtb for the HJ and the fact that HJs maximally stimulate RecGMtb ATPase activity indicate that HJs could be a relevant in vivo substrate for RecGMtb.
RecGMtb required Mg2+ for its optimal activity, although unwinding activity is also supported by Mn2+, Cu2+, Co2+ and Fe2+. A similar observation was also reported for another mycobacterial helicase, UvrD (Curti et al., 2007). This property, shared by these two M. tuberculosis helicases, could contribute to the pathogenicity of M. tuberculosis, because Mg2+ is scarce in the phagosomes of macrophages (Groisman, 1998). The observation that the ATPase activity of RecGMtb was stimulated to a greater extent in the presence of dsDNA than of ssDNA suggests that the enzyme may translocate on dsDNA, as does E. coli RecG (Mahdi et al., 2003).
Evidence shows that expression of recG in M. tuberculosis is upregulated in infected human cells and mouse macrophages, suggesting that RecG may actively promote virulence and/or pathogenicity during infection of mammalian cells (Davis & Forse, 2009; Rachman et al., 2006; Schnappinger et al., 2003). It is also interesting that recG is conserved in the related human pathogen Mycobacterium leprae, in which there is an extreme case of reductive evolution (Vissa & Brennan, 2001). This suggests a potentially important metabolic role for RecG in other mycobacteria also. The genotoxic stress that M. tuberculosis encounters inside the macrophage with reactive nitrogen and oxygen species is very different from that to which E. coli cells are exposed in their various environmental niches. Thus, the metabolic conditions inside an intracellular pathogen such as M. tuberculosis might be considerably different from those of E. coli cells and other model species. This is exemplified by the facts that the genome of M. tuberculosis comprises an unusually high number of genes involved in lipid metabolism (>233), that its genome has a high G+C content (Cole et al., 1998), and that there is a lack of MutS-based mismatch repair in M. tuberculosis (Mizrahi & Andersen, 1998). The existence of a non-homologous end-joining pathway (Della et al., 2004) as well as an alternative regulatory mechanism for DNA damage-inducible genes (Davis et al., 2002) have also been indicated in M. tuberculosis. A recent study further has indicated that RuvAB of M. tuberculosis, unlike E. coli RuvAB, can convert replication forks to HJs (Khanduja & Muniyappa, 2012). Moreover, biochemical characterization of DNA repair components indicates that oxidative DNA glycosylases of M. tuberculosis exhibit substrate preferences different from their E. coli counterparts (Guo et al., 2010). Taken together, these findings suggest that the DNA metabolism of M. tuberculosis might differ considerably from that of E. coli.
Conclusions
The novel findings presented here that RecG exists in vascular plants and algae, in addition to eubacteria, and that RecGMtb preferentially binds relatively long ssDNA, exhibiting a higher affinity for poly(dT), poly(dG) and poly(dC) than for poly(dA), shed new light on the occurrence and role of RecG in nature. Furthermore, the finding that the preferred helicase substrate for RecGMtb is HJ, a key intermediate in DNA repair, recombination and replication fork restart (Kepple et al., 2005; Liu & West, 2004), may suggest that RecGMtb is preferentially involved in such processes in vivo. However, future studies involving M. tuberculosis recG-null mutants are needed to clarify the precise role of RecG in the DNA metabolism, survival, fitness and virulence of M. tuberculosis and possibly of other related mycobacteria.
Acknowledgements
The financial support from the EU 6th frame work project TBadapt, the Norwegian State Educational Loan Fund and the Research Council of Norway (FRIMEDBIO and Centre of Excellence-grant, Centre for Molecular Biology and Neuroscience, CMBN) are gratefully acknowledged.
Abbreviations:
- HJ
Holliday junction
- RecGMtb
M. tuberculosis RecG
Footnotes
Three supplementary figures are available with the online version of this paper.
References
- Ambur O. H., Davidsen T., Frye S. A., Balasingham S. V., Lagesen K., Rognes T., Tønjum T. (2009). Genome dynamics in major bacterial pathogens. FEMS Microbiol Rev 33, 453–470. 10.1111/j.1574-6976.2009.00173.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. 10.1038/35048692 [DOI] [PubMed] [Google Scholar]
- Balasingham S. V., Zegeye E. D., Homberset H., Rossi M. L., Laerdahl J. K., Bohr V. A., Tønjum T. (2012). Enzymatic activities and DNA substrate specificity of Mycobacterium tuberculosis DNA helicase XPB. PLoS ONE 7, e36960. 10.1371/journal.pone.0036960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas T., Pero J. M., Joseph C. G., Tsodikov O. V. (2009). DNA-dependent ATPase activity of bacterial XPB helicases. Biochemistry 48, 2839–2848. 10.1021/bi8022416 [DOI] [PubMed] [Google Scholar]
- Briggs G. S., Mahdi A. A., Wen Q., Lloyd R. G. (2005). DNA binding by the substrate specificity (wedge) domain of RecG helicase suggests a role in processivity. J Biol Chem 280, 13921–13927. 10.1074/jbc.M412054200 [DOI] [PubMed] [Google Scholar]
- Brosh R. M., Jr, Opresko P. L., Bohr V. A. (2006). Enzymatic mechanism of the WRN helicase/nuclease. Methods Enzymol 409, 52–85. 10.1016/S0076-6879(05)09004-X [DOI] [PubMed] [Google Scholar]
- Buss J. A., Kimura Y., Bianco P. R. (2008). RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res 36, 7029–7042. 10.1093/nar/gkn795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner K., Nehring S., Hopfner K. P. (2007). Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol 14, 647–652. 10.1038/nsmb1246 [DOI] [PubMed] [Google Scholar]
- Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S. & other authors (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- Curti E., Smerdon S. J., Davis E. O. (2007). Characterization of the helicase activity and substrate specificity of Mycobacterium tuberculosis UvrD. J Bacteriol 189, 1542–1555. 10.1128/JB.01421-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E. O., Forse L. N. (2009). DNA repair: key to survival? In Mycobacteria: Genomics and Molecular Biology, p. 214 Edited by Paris T., Brown A. Norwich: Caister Academic Press. [Google Scholar]
- Davis E. O., Springer B., Gopaul K. K., Papavinasasundaram K. G., Sander P., Böttger E. C. (2002). DNA damage induction of recA in Mycobacterium tuberculosis independently of RecA and LexA. Mol Microbiol 46, 791–800. 10.1046/j.1365-2958.2002.03199.x [DOI] [PubMed] [Google Scholar]
- Della M., Palmbos P. L., Tseng H. M., Tonkin L. M., Daley J. M., Topper L. M., Pitcher R. S., Tomkinson A. E., Wilson T. E., Doherty A. J. (2004). Mycobacterial Ku and ligase proteins constitute a two-component NHEJ repair machine. Science 306, 683–685. 10.1126/science.1099824 [DOI] [PubMed] [Google Scholar]
- Dos Vultos T., Mestre O., Tønjum T., Gicquel B. (2009). DNA repair in Mycobacterium tuberculosis revisited. FEMS Microbiol Rev 33, 471–487. 10.1111/j.1574-6976.2009.00170.x [DOI] [PubMed] [Google Scholar]
- Fonville N. C., Blankschien M. D., Magner D. B., Rosenberg S. M. (2010). RecQ-dependent death-by-recombination in cells lacking RecG and UvrD. DNA Repair (Amst) 9, 403–413. 10.1016/j.dnarep.2009.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorna A. E., Bowater R. P., Dziadek J. (2010). DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: varying activities at different stages of infection. Clin Sci (Lond) 119, 187–202. 10.1042/CS20100041 [DOI] [PubMed] [Google Scholar]
- Groisman E. A. (1998). The ins and outs of virulence gene expression: Mg2+ as a regulatory signal. Bioessays 20, 96–101. [DOI] [PubMed] [Google Scholar]
- Guo Y., Bandaru V., Jaruga P., Zhao X., Burrows C. J., Iwai S., Dizdaroglu M., Bond J. P., Wallace S. S. (2010). The oxidative DNA glycosylases of Mycobacterium tuberculosis exhibit different substrate preferences from their Escherichia coli counterparts. DNA Repair (Amst) 9, 177–190. 10.1016/j.dnarep.2009.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H., Hayase Y., Imura A., Iwai S., Miura K., Ohtsuka E. (1987). Synthesis and hybridization studies on two complementary nona(2′-O-methyl)ribonucleotides. Nucleic Acids Res 15, 6131–6148. 10.1093/nar/15.15.6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O., Aury J. M., Noel B., Policriti A., Clepet C., Casagrande A., Choisne N., Aubourg S., Vitulo N. & other authors (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. 10.1038/nature06148 [DOI] [PubMed] [Google Scholar]
- Kepple K. V., Boldt J. L., Segall A. M. (2005). Holliday junction-binding peptides inhibit distinct junction-processing enzymes. Proc Natl Acad Sci U S A 102, 6867–6872. 10.1073/pnas.0409496102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanduja J. S., Muniyappa K. (2012). Functional analysis of DNA replication fork reversal catalyzed by Mycobacterium tuberculosis RuvAB proteins. J Biol Chem 287, 1345–1360. 10.1074/jbc.M111.304741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. L., Morgenstern K. A., Griffith J. P., Dwyer M. D., Thomson J. A., Murcko M. A., Lin C., Caron P. R. (1998). Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 6, 89–100. 10.1016/S0969-2126(98)00010-0 [DOI] [PubMed] [Google Scholar]
- Kornberg A., Scott J. F., Bertsch L. L. (1978). ATP utilization by rep protein in the catalytic separation of DNA strands at a replicating fork. J Biol Chem 253, 3298–3304. [PubMed] [Google Scholar]
- Kosa J. L., Zdraveski Z. Z., Currier S., Marinus M. G., Essigmann J. M. (2004). RecN and RecG are required for Escherichia coli survival of bleomycin-induced damage. Mutat Res 554, 149–157. 10.1016/j.mrfmmm.2004.04.011 [DOI] [PubMed] [Google Scholar]
- Lecointe F., Sérèna C., Velten M., Costes A., McGovern S., Meile J. C., Errington J., Ehrlich S. D., Noirot P., Polard P. (2007). Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J 26, 4239–4251. 10.1038/sj.emboj.7601848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., West S. C. (2004). Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol 5, 937–944. 10.1038/nrm1502 [DOI] [PubMed] [Google Scholar]
- Liu J., Choi M., Stanenas A. G., Byrd A. K., Raney K. D., Cohan C., Bianco P. R. (2011). Novel, fluorescent, SSB protein chimeras with broad utility. Protein Sci 20, 1005–1020. 10.1002/pro.633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd R. G., Sharples G. J. (1993). Dissociation of synthetic Holliday junctions by E. coli RecG protein. EMBO J 12, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi A. A., Briggs G. S., Sharples G. J., Wen Q., Lloyd R. G. (2003). A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J 22, 724–734. 10.1093/emboj/cdg043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H., Tanaka T., Kohda D. (2010). Stalled replication forks: making ends meet for recognition and stabilization. Bioessays 32, 687–697. 10.1002/bies.200900196 [DOI] [PubMed] [Google Scholar]
- McGlynn P., Lloyd R. G. (2000). Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101, 35–45. 10.1016/S0092-8674(00)80621-2 [DOI] [PubMed] [Google Scholar]
- McGlynn P., Lloyd R. G. (2001). Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci U S A 98, 8227–8234. 10.1073/pnas.111008698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P., Lloyd R. G. (2002). Genome stability and the processing of damaged replication forks by RecG. Trends Genet 18, 413–419. 10.1016/S0168-9525(02)02720-8 [DOI] [PubMed] [Google Scholar]
- McGlynn P., Al-Deib A. A., Liu J., Marians K. J., Lloyd R. G. (1997). The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol 270, 212–221. 10.1006/jmbi.1997.1120 [DOI] [PubMed] [Google Scholar]
- McGlynn P., Lloyd R. G., Marians K. J. (2001). Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc Natl Acad Sci U S A 98, 8235–8240. 10.1073/pnas.121007798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin E. V., Mirkin S. M. (2007). Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71, 13–35. 10.1128/MMBR.00030-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi V., Andersen S. J. (1998). DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol Microbiol 29, 1331–1339. 10.1046/j.1365-2958.1998.01038.x [DOI] [PubMed] [Google Scholar]
- Müller B., West S. C. (1994). Processing of Holliday junctions by the Escherichia coli RuvA, RuvB, RuvC and RecG proteins. Experientia 50, 216–222. 10.1007/BF01924004 [DOI] [PubMed] [Google Scholar]
- Niga T., Yoshida H., Hattori H., Nakamura S., Ito H. (1997). Cloning and sequencing of a novel gene (recG) that affects the quinolone susceptibility of Staphylococcus aureus. Antimicrob Agents Chemother 41, 1770–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner U. A., Vasil M. L., Alsabbagh E., Parvatiyar K., Hassett D. J. (2000). Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol 182, 4533–4544. 10.1128/JB.182.16.4533-4544.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle A. M. (2008). Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37, 317–336. 10.1146/annurev.biophys.37.032807.125908 [DOI] [PubMed] [Google Scholar]
- Qiagen (2003). The QIA Expressionist: a Handbook for High-Level Expression and Purification of 6× His-Tagged Proteins. Valencia, CA: Qiagen. [Google Scholar]
- Rachman H., Strong M., Ulrichs T., Grode L., Schuchhardt J., Mollenkopf H., Kosmiadi G. A., Eisenberg D., Kaufmann S. H. (2006). Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect Immun 74, 1233–1242. 10.1128/IAI.74.2.1233-1242.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocak S., Linder P. (2004). DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5, 232–241. 10.1038/nrm1335 [DOI] [PubMed] [Google Scholar]
- Rocha E. P., Cornet E., Michel B. (2005). Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet 1, e15. 10.1371/journal.pgen.0010015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph C. J., Upton A. L., Harris L., Lloyd R. G. (2009a). Pathological replication in cells lacking RecG DNA translocase. Mol Microbiol 73, 352–366. 10.1111/j.1365-2958.2009.06773.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph C. J., Upton A. L., Lloyd R. G. (2009b). Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol 74, 940–955. 10.1111/j.1365-2958.2009.06909.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph C. J., Mahdi A. A., Upton A. L., Lloyd R. G. (2010a). RecG protein and single-strand DNA exonucleases avoid cell lethality associated with PriA helicase activity in Escherichia coli. Genetics 186, 473–492. 10.1534/genetics.110.120691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph C. J., Upton A. L., Briggs G. S., Lloyd R. G. (2010b). Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 9, 210–223. 10.1016/j.dnarep.2009.12.014 [DOI] [PubMed] [Google Scholar]
- Sayers E. W., Barrett T., Benson D. A., Bolton E., Bryant S. H., Canese K., Chetvernin V., Church D. M., Dicuccio M. & other authors (2012). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 40 (Database issue), D13–D25. 10.1093/nar/gkr1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D., Ehrt S., Voskuil M. I., Liu Y., Mangan J. A., Monahan I. M., Dolganov G., Efron B., Butcher P. D. & other authors (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198, 693–704. 10.1084/jem.20030846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples G. J., Ingleston S. M., Lloyd R. G. (1999). Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG, and RusA. J Bacteriol 181, 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton M. R., Scaife S., Wigley D. B. (2001). Structural analysis of DNA replication fork reversal by RecG. Cell 107, 79–89. 10.1016/S0092-8674(01)00501-3 [DOI] [PubMed] [Google Scholar]
- Slocum S. L., Buss J. A., Kimura Y., Bianco P. R. (2007). Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J Mol Biol 367, 647–664. 10.1016/j.jmb.2007.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings C. L., Glickman M. S. (2010). Is Mycobacterium tuberculosis stressed out? A critical assessment of the genetic evidence. Microbes Infect 12, 1091–1101. 10.1016/j.micinf.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. H., Exley R., Li Y., Goulding D., Tang C. (2005). Identification and characterization of genes required for competence in Neisseria meningitidis. J Bacteriol 187, 3273–3276. 10.1128/JB.187.9.3273-3276.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamae C., Liu A., Kim K., Sitz D., Hong J., Becket E., Bui A., Solaimani P., Tran K. P. & other authors (2008). Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J Bacteriol 190, 5981–5988. 10.1128/JB.01982-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner N. K., Linder P. (2001). DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell 8, 251–262. 10.1016/S1097-2765(01)00329-X [DOI] [PubMed] [Google Scholar]
- Taylor S. D., Solem A., Kawaoka J., Pyle A. M. (2010). The NPH-II helicase displays efficient DNA RNA helicase activity and a pronounced purine sequence bias. J Biol Chem 285, 11692–11703. 10.1074/jbc.M109.088559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja N., Tuteja R. (2004). Unraveling DNA helicases. Motif, structure, mechanism and function. Eur J Biochem 271, 1849–1863. 10.1111/j.1432-1033.2004.04094.x [DOI] [PubMed] [Google Scholar]
- Vincent S. D., Mahdi A. A., Lloyd R. G. (1996). The RecG branch migration protein of Escherichia coli dissociates R-loops. J Mol Biol 264, 713–721. 10.1006/jmbi.1996.0671 [DOI] [PubMed] [Google Scholar]
- Vissa V. D., Brennan P. J. (2001). The genome of Mycobacterium leprae: a minimal mycobacterial gene set. Genome Biol 2, REVIEWS1023. 10.1186/gb-2001-2-8-reviews1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner D. F. (2010). The role of DNA repair in M. tuberculosis pathogenesis. Drug Discov Today Dis Mech 7, e5–e7. 10.1016/j.ddmec.2010.08.002 [DOI] [Google Scholar]
- Warner D. F., Mizrahi V. (2006). Tuberculosis chemotherapy: the influence of bacillary stress and damage response pathways on drug efficacy. Clin Microbiol Rev 19, 558–570. 10.1128/CMR.00060-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby M. C., Lloyd R. G. (1998). Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J Biol Chem 273, 19729–19739. 10.1074/jbc.273.31.19729 [DOI] [PubMed] [Google Scholar]
- Whitby M. C., Vincent S. D., Lloyd R. G. (1994). Branch migration of Holliday junctions: identification of RecG protein as a junction specific DNA helicase. EMBO J 13, 5220–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2011). Global tuberculosis control report. Geneva: WHO.
- Wu Y., Chen W., Zhao Y., Xu H., Hua Y. (2009). Involvement of RecG in H2O2-induced damage repair in Deinococcus radiodurans. Can J Microbiol 55, 841–848. 10.1139/W09-028 [DOI] [PubMed] [Google Scholar]