

Abstract
Mutations in the LMNA gene, which encodes lamin A and C (lamin A/C), cause a diverse spectrum of tissue-selective diseases termed laminopathies. The most prevalent form affects striated muscles as dilated cardiomyopathy with variable skeletal muscle involvement, which includes autosomal Emery-Dreifuss muscular dystrophy. Mechanisms underlying the disease pathogenesis are beginning to be understood and they point toward defects in cell signaling. We therefore assessed putative signaling defects in a mouse model carrying a point mutation in Lmna (LmnaH222P/H222P) that faithfully recapitulates human Emery-Dreifuss muscular dystrophy. We found that AKT-mechanistic target of rapamycin (MTOR) signaling was hyperactivated in hearts of LmnaH222P/H222P mice and that reducing MTOR activity by pharmacological intervention ameliorated cardiomyopathy. Given the central role of MTOR in regulating autophagy, we assessed fasting-induced autophagic responses and found that they were impaired in hearts of these mice. Moreover, the improved heart function associated with pharmacological blockade of MTOR was correlated with enhanced autophagy. These findings demonstrated that signaling defects that impair autophagy underlie pathogenesis of dilated cardiomyopathy arising from LMNA mutation.
Keywords: LMNA, dilated cardiomyopathy, laminopathy, autophagy, nuclear lamina, cell signaling
A-type lamins (lamin A/C) are type V intermediate filament proteins that form the nuclear lamina, a proteinaceous meshwork lining the inner surface of the nucleus. Previously relegated to a role as relatively inert structural proteins providing mechanical support to the nucleus, interest in them intensified when several human diseases, collectively termed laminopathies, were linked to mutations in LMNA. Although ubiquitously expressed in most differentiated mammalian somatic cells, site and sequence-specific mutations in LMNA lead to tissue-selective diseases, the most common of which affects striated muscle in the form of dilated cardiomyopathy (hereinafter referred to as LMNA cardiomyopathy). While native functions of lamin A/C have been studied for decades, how disease-causing point mutant lamin A/C variants alter basic cellular processes to cause disease is just beginning to be elucidated. Emerging evidence suggests that lamin A/C plays a direct and dynamic role in regulating signal transduction, and that the expression of variants alters cell signaling pathways that underlie disease pathogenesis.
A recent study from our group lends further credence to this hypothesis. To elucidate the consequence of lamin A/C mutation on cell signaling, we examined various signaling pathways in ventricular tissue from LmnaH222P/H222P mice and found that AKT-MTOR signaling was hyperactivated. This enhanced signaling occurs at an early age and is sustained; phosphorylated AKT and MTOR levels are already elevated by 4 weeks of age, which is prior to detectable signs of cardiomyopathy, and remain elevated up to 16 weeks. Hyperactivation of AKT is also observed in hearts from human subjects with LMNA cardiomopathy, demonstrating that similar signaling defects occur in the human disease. To ascertain whether enhanced AKT-MTOR signaling contributes to the pathogenesis of LMNA cardiomyopathy, we reduced MTOR signaling in vivo by systemic administration of a rapamycin analog, temsirolimus. LmnaH222P/H222P mice treated with temsirolimus exhibit improved cardiac function with reduced expression of genes associated with ventricular dilatation relative to those treated with placebo. This suggested that enhanced MTOR signaling is a pathogenic trigger and not elicited as a protective response.
In light of the protective effect of MTOR inhibition in LMNA cardiomyopathy, we sought to identify the putative pathogenic mechanism(s) triggered by hyperactivated MTOR. We focused on autophagy, an evolutionarily conserved catabolic process that maintains cellular homeostasis by clearing damaged or toxic proteins/organelles and promotes cell survival under periods of starvation or increased energy demand by recycling its own cellular components. We showed that the expression of lipidated LC3B, which we used as an indirect measure of autophagosomes, is progressively reduced relative to controls as the mice aged. This reduction is coincident with increased expression of SQSTM1/p62 and BECN1, suggesting that despite the impetus for activation, autophagosome formation under basal conditions is impaired. Even under experimentally-induced energy deficit, hearts of LmnaH222P/H222P mice and cardiomyocytes isolated from them are refractory to autophagy induction. Notably, a similar pattern of expression indicative of defective autophagy is observed in hearts from human subjects with LMNA cardiomyopathy, suggesting that concordant pathogenic mechanisms underlie the disease in humans.
Given the defective autophagy in LMNA cardiomyopathy, we reasoned that the beneficial effect of temsirolimus is mediated by its ability to stimulate autophagy. Indeed, systemic administration of temsirolimus to LmnaH222P/H222P mice increases cardiac expression of lipidated LC3B with a reciprocal reduction in SQSTM1 expression, indicative of enhanced autophagic activity. Furthermore, increased autophagic responses are also associated with selumetinib, a MAP2K1/MEK1-MAP2K2/MEK2 inhibitor that had previously been shown to be effective in treating LMNA cardiomyopathy. Temsirolimus also reduces expression of ubiquitinated protein in hearts of LmnaH222P/H222P mice but does not reduce total levels of lamin A/C expression, suggesting that the pathogenesis and amelioration of LMNA cardiomyopathy is not mediated by accumulation and autophagic clearance of mutant lamin A/C variants.
Our study revealed that mutations in LMNA establish a cell signaling environment that antagonizes autophagy and that activation of autophagy through pharmacological means can be used as a therapeutic strategy to treat LMNA cardiomyopathy (Fig. 1). Although these results implicate defective autophagy in the progression of LMNA cardiomyopathy, additional studies are required to address several unanswered questions. For example, are both basal and starvation-induced autophagy affected? Although we demonstrated that starvation-induced autophagy is impaired (MTOR hyperactivation and impaired autophagy under energy deficit), accumulation of ubiquitinated proteins suggests that basal housekeeping autophagy is also affected. Lamin A/C are physically connected to the actin cytoskeleton through the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex, a series of connected inner and outer nuclear membrane proteins that traverse the nuclear envelope. Given the essential role of actin cytoskeleton in establishing the infrastructure of autophagosome trafficking machinery, it is plausible that both types of autophagy are impaired in LMNA cardiomyopathy. Answering this question will help address an analogous question: is the pathogenic effect emanating from impaired autophagy due to cell damage from energy deficit or from accumulation of toxic proteins and/or organelles? The answers to these questions will ultimately help determine whether LMNA cardiomyopathy is a metabolic disease or a cardiac proteinopathy and assist in tailoring mechanism-based therapeutic strategies.
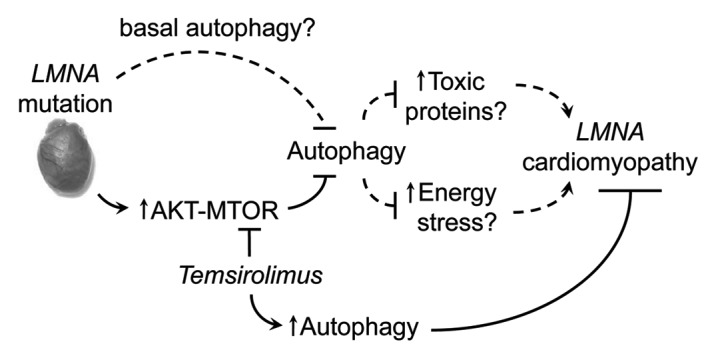
Figure 1. Proposed pathogenesis model of LMNA cardiomyopathy. LMNA mutation activates AKT-MTOR and impairs autophagy. This impairment results in energy stress and/or accumulation of toxic proteins that ultimately leads to cell damage and cardiomyopathy. Temsirolimus inhibits MTOR and mitigates pathogenic effects of impaired autophagy. Dashed lines indicate hypothetical connections that require further studies to validate.
Acknowledgments
This work was supported by NIH Ruth L. Kirschstein National Research Service Award (F32-HL094037), NIH/NIAMS (R01AR048997) and Muscular Dystrophy Association (MDA172222).
Glossary
Abbreviations:
- MTOR
mechanistic target of rapamycin
- A-type lamins
lamin A/C
- LINC
Linker of Nucleoskeleton and Cytoskeleton
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/22403