

Abstract
The incidence of pancreatic adenocarcinoma is increasing with more than 43,000 predicted new cases in the US and 65,000 in Europe this year. Pancreatic cancer patients have a short life expectancy with less than 3–4% 5-y survival, which results in an equivalent incidence and mortality rate. One of the major challenges in pancreatic cancer is the identification of pharmacological approaches that overcome the resistance of this cancer to therapy. Intensive research in the past decades has led to the classification of pancreatic cancers and the identification of the driver key genetic events. Despite the advances in understanding the molecular mechanisms responsible for pancreatic cancer pathogenesis, this knowledge had little impact on significantly improving the treatment for this dismal disease. In particular, we know today that the lack of therapeutic response in pancreatic cancer is due to the intrinsic high resistance of these tumors to chemotherapy and radiation, rather than to the inappropriate design of these therapeutic approaches. Thus, in order to ensure a better outcome for pancreatic cancer patients, there is a strong need for research focused on the mechanism that determines this resistant phenotype and the means that might drive enhanced response to therapy.
Keywords: NUPR1, pancreas cancer, hypoxia, glucose starvation, cannabinoids, AURKA
Pancreatic tumors exhibit a paradoxical composition, with few epithelial tumor cells embedded in a fibrotic stromal microenvironment with poor vascularization. While the hypoxigenic stroma within the tumor might be perceived as a lethal challenge for the pancreatic cancer cells, the chronic deprivation of nutrients and oxygen supply in the tumor results in the establishment of a metabolic selective force that modifies the genetic and epigenetic landscape of cancer cells and promotes the survival and propagation of cells with unique metabolic properties. The metabolic reprogramming in cancer is a concept that has emerged in recent years after a century of lethargy from the initial observations by Otto Warburg. The joined effort of classical biochemists working on metabolism, and cancer researchers, provided a renewed view of the mechanisms and consequences of the “metabolic switch.” Cancer cells adapt their metabolism to fulfill different objectives throughout the process of tumor formation and cancer progression. First, cancer cells increase the uptake of nutrients (particularly glucose and glutamine) and modify their catabolic route, redirecting the ATP-generating pathways into less energy-efficient reaction chains that allow the generation of intermediary metabolites for anabolism, in order to obtain building blocks for new cells. On the other hand, cancer cells in adverse environments, such as the loss of contact with the extracellular matrix or the colonization of foreign organs, undergo the inhibition of nutrient uptake and the loss of ATP and redox potential, an event that can be compensated by the enforcement of fatty acid oxidation. Since the ability of pancreatic cancer cells to adapt to this hostile metabolic environment is crucial for the selection of the most resistant and aggressive clones, it is tempting to speculate that the identification of a factor(s) modifying the response to metabolic stress will aid the design of better anticancer approaches. This idea has fueled a large number of ongoing studies, which altogether have established the fertile field of pancreatic cancer metabolism.
NUPR1, also known as p8 and COM1, is a stress-induced chromatin-associated factor, which is systematically overexpressed in cancer tissues, particularly in tumors with bad prognosis and remarkable resistance to antitumoral treatments. NUPR1 is required for pancreatic cancer progression through several mechanisms, and recent data from our laboratories demonstrates that the function of this protein is critical to modulate the metabolic stress response to glucose and oxygen deprivation. NUPR1 is also required for the optimal expression of metabolic stress-response genes, particularly those involved in DNA repair, cell cycle regulation, apoptosis, entosis and autophagy. Notably, Aurora Kinase A (AURKA) rises as an important component in NURP1-mediated resistance to metabolic stress. This novel NUPR1-AURKA pathway counteracts the autophagy-dependent, CASP3-CASP7-independent cell death that is induced by glucose starvation and hypoxia. Thus, until small molecules against NUPR1 are developed, our data provide a strong rationale for the use of AURKA inhibitors in cancers with high NUPR1 expression. AURKA is overexpressed in several cancers and inhibitors against this kinase have been developed and are undergoing clinical evaluation. In the context of pancreatic cancer, our data suggest that AURKA inhibitors will target primarily intrapancreatic tumor cells located near hypovascularized regions which house the resistant cells selected by the adverse microenvironment. Combined, all the available data lead us to propose a model in which NUPR1 participates in a positive-feedback mechanism that contributes to the pathophysiology of pancreatic cancer. According to this model, NUPR1 is activated by glucose and oxygen deprivation in order to initiate a transcriptional program, which ultimately aids cells to adapt to the metabolic stress through the inhibition of autophagy-mediated cell death.
To note, the role of autophagy in cancer is extremely complex. On the one hand, different studies have shown that several autophagy-related genes play a tumor suppressor role, which suggests that inhibition of this cellular process is beneficial for cancer. On the other hand, recent work by different laboratories supports the idea that autophagy has a protumor growth effect in some cancers, including pancreatic adenocarcinoma. Moreover, additional studies support the idea that autophagy plays opposite roles at early and late stages of tumor generation/progression. Furthermore, although autophagy is primarily considered a cell survival mechanism, activation of this cellular process can also lead to cancer cell death. In our work we found that NUPR1 negatively regulates autophagy-mediated cell death via AURKA, which is in line with the idea of a tumor suppressor role of autophagy in cancer. Thus, the oncogenic function of NUPR1 under a situation of metabolic stress is based, at least in part, on its ability to inhibit the antineoplasic functions of autophagy.
Intriguingly, previous work by our laboratories showed that NUPR1 can also lead to a positive stimulation of autophagy in a different cellular context. Thus, in response to the treatment with cannabinoids, a novel family of potential anticancer agents, NUPR1 becomes acutely upregulated, which leads in turn to stimulation, rather than inhibition, of autophagy-mediated cell death. However, to produce this effect in response to cannabinoids, NUPR1 activates a completely different set of genes (the transcription factors ATF4 and DDIT3/CHOP in combination with the pseudokinase TRIB3, which stimulates autophagy via inhibition of the AKT1-MTORC1 axis). These observations indicate that, depending on the cellular situation, NUPR1 can regulate autophagy via two separate signaling pathways (Fig. 1).
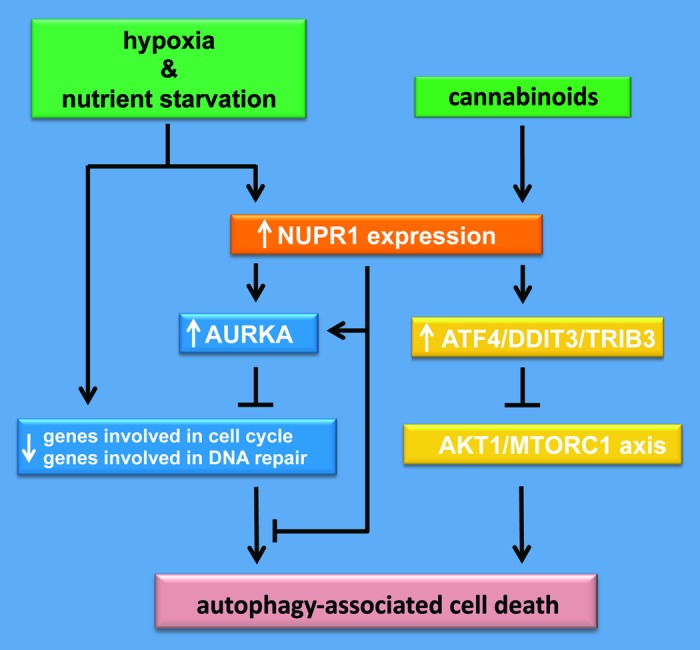
Figure 1. NUPR1 is involved against metabolic stress-associated cell death. Pancreatic ductal adenocarcinoma is hypovascularized as a consequence of its high stroma content. Thus, pancreatic cancer cells become nutrient starved and subjected to long periods of hypoxia. Glucose starvation and hypoxia mediate, on the one hand, inhibition of DNA repair- and cell cycle-associated gene expression, resulting in an autophagy-associated cell death, and, on the other hand, the induction of NUPR1 expression to maintain DNA repair- and cell cycle-associated gene expression and therefore promote cell survival by inhibiting autophagy-associated cell death. However, cannabinoids induce NUPR1-mediated autophagy-dependent cell death by controlling a different set of genes.
In summary, on the basis of our data, further research is warranted for the design and evaluation of Nupr1-AURKA pathway directed therapies in order to tackle the pancreatic cancer cells hosted in the metabolically adverse microenvironment.
Acknowledgments
This work was supported by grants from INSERM, La Ligue Contre le Cancer, EGIDE, Canceropole PACA and INCA to J.I. The work of AC is supported by the Ramón y Cajal award (Spanish Ministry of Education), the ISCIII (PI10/01484), the Marie Curie IRG grant (277043) and the Basque Government of education (PI2012–03). The work of G.V. is supported by the ISCIII (PS09/01401) and MICINN (FR2009–0052). The work of R.U. is supported by the NIH (DK52913).
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/22258