

Abstract
The epidermal growth factor receptor (EGFR) signaling pathway is frequently dysregulated in a variety of human malignancies. As a result, agents have been developed to selectively inhibit the tyrosine kinase function of EGFR (EGFR-TKI) for cancer therapy. However, the clinical efficacy of these drugs to date has been limited by both acquired and intrinsic resistance. Macroautophagy, a process of intracellular proteolysis, has been shown to be activated in response to EGFR targeted therapy. However, the specific role of the induction of autophagy remains controversial. Here we show that autophagy is induced in a dose-dependent manner by in vitro treatment of multiple cancer cell lines with EGFR-TKI. Additionally, we find that in cells highly resistant to EGFR-TKI, autophagy is not robustly activated and that co-treatment of these cells with rapamycin, a known inducer of autophagy, can partially restore sensitivity to EGFR-TKI. Finally, we demonstrate that, in resistant cell lines, EGFR-TKI sensitivity can be further inhibited by siRNA-mediated depletion of the critical autophagy protein ATG7. Thus, our data suggests that defective autophagy may be an EGFR-TKI resistance mechanism and that activation of autophagy may be a viable strategy to augment the cytotoxic effect of EGFR-TKIs.
Keywords: EGFR, TKI, autophagy, epidermal, erlotinib, gefitinib, rapamycin
Introduction
Epidermal growth factor receptor (EGFR), a receptor tyrosine kinase of the human epidermal growth factor receptor family, plays a critical role in mediating the relationship between extracellular signals and cellular homeostasis. EGFR signaling has been found to be dysregulated in the majority of epithelial malignancies where increased activation of EGFR signaling results in enhanced cell survival, proliferation and resistance to anti-cancer therapeutics.1 Thus, as a therapeutic target, EGFR has been the focus of extensive research efforts that have culminated in two primary strategies for inhibiting EGFR signaling: inhibition of ligand binding by anti-EGFR antibodies and small molecule inhibition of the tyrosine kinase activity of EGFR. The small molecule EGFR-TKIs, erlotinib and gefitinib, target the intracellular tyrosine kinase activity of EGFR and, while they have been effective in non-small cell lung carcinoma (NSCLC), especially in the tumors with activating EGFR mutations, the TKIs have generally shown limited clinical benefit in the majority of solid tumors.1 Moreover, even in NSCLC, acquired resistance to EGFR-TKI commonly develops via a secondary mutation in EGFR known as T790M or via compensatory MET overactivtity.2 Thus, the limited clinical benefit of EGFR-TKIs is likely due to a confluence of both acquired and intrinsic resistance factors.
Macroautophagy, hereafter known as autophagy, is a catabolic process of intracellular self-digestion that was originally discovered to play a critical protective role in times of metabolic stress while also serving a role in maintaining cellular homeostasis via bulk degradation of proteins and organelles.3 More recently in cancer biology, autophagy has been shown to paradoxically mediate aspects of tumorigenesis, tumor cell survival and tumor cell death.4 Importantly, induction of autophagy has been shown to be cytoprotective in a variety of important processes related to cancer therapy including resistance to chemotherapeutics,5,6 ionizing radiation,7 basement membrane detachment,8 growth factor deprivation9 and hypoxia.10 Conversely, autophagy has been implicated as a causal mechanism of cell death in anoxia10 and has also been shown to contribute to increased radiosensitivity11,12 and chemosensitivity.12 Thus, the specific role of autophagy in cancer has been postulated to be highly contextual and dependent on many factors including tumor origin, treatment type and tumor stage.4 Despite this ambiguity, autophagy has emerged as a focus of intense research efforts, representing a potential therapeutic target to augment existing therapies.
The anti-EGFR antibody cetuximab was shown to induce autophagy in several cancer cell lines via inhibition of EGFR signaling and subsequent downregulation of the mammalian target of rapamycin (mTOR) and hypoxia inducible factor 1-α signaling pathways.13,14 Moreover, Li, et al. further demonstrated that the induction of autophagy served mainly a cytoprotective function in response to cetuximab treatment. Likewise, treatment of lung cancer cells with EGFR TKIs has also been shown to induce autophagy.15 However, it remains unclear whether autophagy is a cytoprotective response in cells resistant to the effects of EGFR tyrosine kinase inhibition. Some authors have suggested that in certain cancer cells, autophagy may delay cell death when EGFR signaling is interrupted and that blocking autophagy may increase the efficacy of EGFR targeted therapies.4
Here we report that, in multiple solid tumor derived cell lines, autophagy is indeed upregulated by treatment with EGFR TKI erlotinib. Surprisingly, we found that in cells most resistant to EGFR TKI induced cell death, autophagy is not upregulated and that the induction of autophagy by mTOR inhibitor rapamycin can increase sensitivity of resistant cancer cells to EGFR tyrosine kinase inhibition. This finding suggests that induction of autophagy may be an effective strategy in restoring sensitivity to EGFR TKI resistant cells.
Results
EGFR tyrosine kinase inhibition induces LC3 lipidation
Erlotinib has been shown to induce apoptosis16 and metabolic oxidative stress.17 These processes are known to be associated with the induction of autophagy4 and thus, we hypothesized that autophagy would be activated in cancer cells that were treated with erlotinib. Furthermore, autophagy has been shown to be activated, to varying degrees, as a response to EGFR targeted therapies.13,15 To investigate the degree of activity in the autophagic pathway in several different cancer cell lines, we first assessed the lipidation of a critical autophagy protein, MAP1LC3A (LC3). LC3 lipidation with a phosphatidylethanolamine residue is a critical step in the autophagic pathway and this conversion from unlipidated (LC3-I) to a faster migrating, lipidated form (LC3-II) is an indicator of increased autophagic activity that can be monitored via immunobloting.18 We assessed LC3 conversion as a marker of increased autophagy in cell lysates of several cancer cell lines that were treated with 10 μM erlotinib for 24 h (Fig. 1A). Erlotinib treatment resulted in the increased detection of LC3-II in 686LN, Hela-R30, T24 and PCI-15B cells. Significant LC3-II conversion was not detected in erlotinib resistant, Hela-R30 cells. To further assess the effect of erlotinib on the conversion of LC3-I to LC3-II we treated SCC-1 and Hela-R29 cells with a range of erlotinib doses from 0.1 μM to 10 μM (Fig. 1B). We observed a dose dependent increase in LC3-II conversion in SCC-1 cells, but not erlotinib resistant Hela-R29 cells. To further determine dose dependence, we treated SCC-1 and T24 cells with erlotinib at doses ranging from 1 μM to 20 μM (Fig. 1C). We observed that at these doses, erlotinib continues to induce a robust, dose-dependent conversion of LC3. In order to confirm that the increased LC3 conversion was not limited to treatment with erlotinib, we next treated T-24 cells with micromolar range doses of gefitinib (2 to 32 μM), a similar EGFR targeted TKI. We found that gefitinib also induced a dose-dependent accumulation of LC3-II in T-24 cells (Fig. 1D).

Figure 1. EGFR tyrosine kinase inhibition induces LC3 lipidation. (A) 686LN, HeLa-R30, T24 and PCI-15B cells were treated with 10 μM erlotinib or carrier control for 24 h. Whole cell lysates were subjected to immunoblotting for LC3 and β-tubulin. Erlotinib treatment induces the accumulation of faster migrating phosphatidylethanolamine conjugated LC3 (LC3-II). (B) SCC-1 and HeLa-R29 were treated with erlotinib doses ranging from 0.1 to 10.0 μM for 24 h. A dose-dependent increase in LC3-II was observed in SCC-1 cells but not HeLa-R29 (or R30) cells. (C) SCC-1 and T-24 cells were treated with erlotinib doses in the micromolar (2.5–20 μM) range for 24 h. Dose-dependent increases in LC3-II were observed in both SCC-1 and T-24 cells. (D) T-24 cells treated with gefitinib in the micromolar (2–32 μM) range for 24 h also induce a dose-dependent increase in LC3-II accumulation. (E) Treatment with 10 μM erlotinib or 10 μM gefitinib inhibit Akt and Erk phosphorylation in 686LN but not HeLa-R30 cells. For all immunoblots, image shown is representative of three experimental replicates.
We observed that in the EGFR TKI resistant Hela-R29 and Hela-R30 cells, erlotinib treatment did not induce significant LC3-II accumulation (Fig. 1A and B). To confirm the resistance of these cells to erlotinib we treated EGFR-TKI sensitive 686LN cells and EGFR-TKI resistant Hela-R30 cells with pharmacologically relevant doses of erlotinib and gefitinib and assayed the levels of downstream components of the EGFR pathway. Both erlotinib and gefitinib significantly inhibited Erk and Akt phosphorylation in 686LN cells while there was little to no effect in the EGFR-TKI resistant Hela-R30 cells (Fig. 1E). This confirms that EGFR-TKI does not inhibit downstream elements of the EGFR pathway in Hela-R30 cells.
Erlotinib induces autophagosome formation and increased autophagic flux
To further assess the effect of erlotinib on autophagy, we assessed the formation of autophagosomes in response to erlotinib treatment. During the formation of these double-membraned organelles, LC3 is relocated from a diffuse distribution in the cytoplasm to a subcellular, punctate distribution corresponding to autophagosomes. This relocation can be visualized in cells expressing a GFP-LC3 construct as a conversion from diffuse cytoplasmic fluorescence to a punctate or “dot-like” pattern.18 Accordingly, we created cell lines stably expressing GFP-LC3 and measured the formation of GFP-LC3 positive puncta in response to erlotinib treatment. In T-24 cells stably expressing GFP-LC3 (T-24 GFP-LC3), erlotinib treated cells displayed more punctate GFP fluorescence when compared with untreated cells (Fig. 2A) suggesting that LC3 sequestration in autophagosomes is actively occurring. Moreover, the number of GFP fluorescent puncta per cell were quantified to estimate the level of autophagosome formation (Fig. 2B). We observed a dose dependent increase in average GFP puncta per cell (P value < 0.0001) when T-24 GFP-LC3 cells were treated with 1–10 μM erlotinib as compared with carrier control. Overall, the induction of LC3 conversion and dose-dependent increase in GFP-LC3 puncta by erlotinib treatment is strongly suggestive of the increased formation of autophagosomes.

Figure 2. Erlotinib induces autophagosome formation and increased autophagic flux (A) T-24 cells stably expressing GFP-LC3 were treated with carrier control or 1, 5 or 10 μM erlotinib for 24 h, fixed and imaged using epifluorescence microscopy. T-24 cells treated with 10 μM erlotinib (right) were observed to have higher punctate or “dot-like” fluorescence when compared with untreated (left) cells. Representative images of two independent experiments are shown. Bar = 50 μm. (B) Puncta per cell were quantified in cells treated with 0, 1, 5 and 10 μM erlotinib and erlotinib was found to induce a dose dependent increase in GFP-LC3 puncta. Statistically significant dose dependence was observed between all erlotinib doses. Error bars shown are standard error of the mean. *One-way ANOVA P value < 0.0001. (B) T-24, SCC-1, 686LN and HeLa-R30 cells were treated with 10 μM erlotinib for 24 h and lysosomal protease inhibitors E64D and pepstatin A (E/P) were added to medium 2 h before collection of whole cell lysates which were immunoblotted for LC3 and β-tubulin. Increased LC3-II was observed in cells treated with E/P and erlotinib suggesting an increased level of autophagic flux in cells treated with erlotinib. Images shown are representative of three experimental replicates.
Proteins and organelles contained within autophagosomes, including LC3, are dynamically degraded in lysosomes. Thus, LC3-II protein levels only reflect a static assessment of the number of autophagosomes and not the level of flux or turnover through the autophagic pathway over time. Since LC3-II can be degraded in the lysosome along with autophagosomal cargo, measuring true autophagic flux is difficult with standard western blot technique. To better assess the dynamic process of autophagic flux induced by erlotinib, we treated T24, SCC-1, 686LN and Hela-R30 cells with lysosomal protease inhibitors E64D and pepstatin A two hours prior to harvesting of lysates, thereby inhibiting lysosomal degradation of LC3-II and causing an accumulation of LC3-II which is proportional to the level of flux. We observed that LC3-II accumulation, in the presence of E64D and pepstatin A, was higher in T24, SCC-1, and 686LN cells treated with erlotinib than in carrier controls (Fig. 2C). We did not observe this increase in LC3-II in Hela-R30 cells. This suggests that cells sensitive to erlotinib undergo increased autophagic flux while cells that are resistant to erlotinib do not exhibit an increase in autophagic flux.
Rapamycin treatment of EGFR-TKI resistant cells induces autophagy and increases cell death when combined with erlotinib treatment
Previous studies have shown that the induction of autophagy serves as a cell survival pathway in cancer cells treated with a variety of drugs, while also serving as a potential alternative cell death pathway.19 We observed that in erlotinib resistant cell lines the induction of autophagy by erlotinib treatment was minimal. This finding suggests that in our model, autophagy may be function as a cell death mechanism that is induced by EGFR blockade. Thus, we hypothesized that inducing autophagy by pharmacologic means in conjunction with erlotinib would sensitize these cells to erlotinib. We cotreated EGFR-TKI resistant Hela-R30 cells with rapamycin, a known inducer of autophagy,4 and erlotinib and assayed the induction of autophagy by LC3 immunoblotting. We observed that an accumulation of LC3-II occurred when erlotinib resistant Hela-R30 cells were treated with both erlotinib and rapamycin but not either agent alone (Fig. 3A). In erlotinib sensitive 686LN cells, a higher level of LC3-II accumulation was observed in cells treated with both agents as opposed to either alone. Additionally, we assayed ATP levels as a proxy for cell viability in 686LN and Hela-R30 cells treated with erlotinib, rapamycin and both agents together (Fig. 3B). We observed that in erlotinib resistant Hela-R30 cells, combination treatment with erlotinib and rapamycin resulted in an additive cell death effect while in erlotinib sensitive 686LN cells this effect was minimal. Moreover, rapamycin alone did not induce significant cell death in 686LN cells. In EGFR-TKI resistant Hela-R30 cells, cotreatment with both rapamycin and erlotinib significantly reduced the phosphorylation of ERK, a downstream activator of the EGFR signaling cascade, more than either agent alone (Fig. S1).
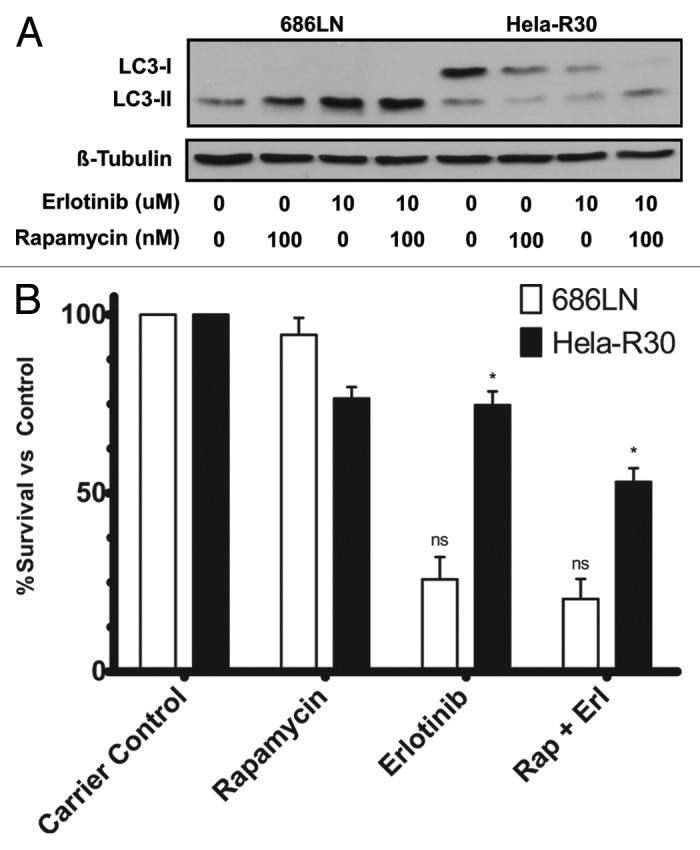
Figure 3. Rapamycin treatment of EGFR-TKI resistant cells induces autophagy and increases cell death when combined with erlotinib treatment. (A) 686LN, a TKI sensitive cell line, and Hela-R30, an in vitro generated TKI resistant cell line, cells were treated with carrier control (1:1000 DMSO), 100 nM rapamycin, 10 μM erlotinib or both 100 nM rapamycin and 10 μM erlotinib for 24 h. Whole cell lysates were subjected to immunoblotting for LC3 and β-tubulin. Both rapamycin and erlotinib alone induce LC3-II accumulation in 686LN cells but neither agent alone induces significant LC3-II accumulation in Hela-R30 cells. Conversely, combination treatment with both rapamycin and erlotinib induces LC3-II accumulation in Hela-R30 cells while causing more significant accumulation in 686LN cells. Images shown are representative of three experimental replicates. (B) 686LN and Hela-R30 cells were treated with with carrier control (1:1000 DMSO), 100 nM rapamycin, 10 μM erlotinib or both 100 nM rapamycin and 10 μM erlotinib for 72 h and assayed for viability using Cell-Titer-Glo luminescence assay. In Hela-R30 cells, rapamycin and erlotinib cotreatment significantly increased cell death relative to either agent alone. Conversely, rapamycin treatment exhibited only minimal effects in 686LN cells either alone or when combined with erlotinib. Graph shown is percent survival normalized to carrier control. nsUnpaired t-test P value = 0.32. *Unpaired t-test P value = 0.0024.
Induction of autophagy in EGFR-TKI resistant cells sensitizes them to erlotinib
In order to further investigate the effect of combination rapamycin and erlotinib treatment on EGFR-TKI resistant cells, we assayed the erlotinib IC50 of Hela-R30 cells when they were cotreated with 100 nM rapamycin (Fig. 4A). We observed that the erlotinib IC50 was significantly decreased with rapamycin cotreatment. Additionally, we treated EGFR-TKI sensitive 686LN cells with rapamycin and erlotinib and did not observe a similar decrease in the erlotinib IC50 (Fig. 4B). As a potent autophagy inducer, we postulated that this sensitization to erlotinib by rapamycin cotreatment was due to an increase in autophagy and conversely, that the resistance to erlotinib observed in Hela-R30 cells was attributable to poor autophagy induction . Thus, we inhibited autophagy using siRNA directed against ATG7, a critical autophagy protein. We observed that inhibition of autophagy by siATG7 in resistant Hela-R30 cells, increased the IC50 of these cells (Fig. 4C and D). This suggests that the induction of autophagy facilitates erlotinib-induced cell death and thus, provides a mechanism whereby cancer cells can gain resistance to EGFR tyrosine kinase inhibition via suppression of autophagy.

Figure 4. Induction of autophagy in EGFR-TKI resistant cells sensitizes cells to erlotinib. (A) Erlotinib resistant Hela-R30 cells or (B) erlotinib sensitive 686LN, were treated with erlotinib at doses ranging from 0.03 to 30 μM and cell viability was measured with MTT reduction assay at 72 h of treatment. Erlotinib IC50 of Hela-R30 cells was reduced by cotreatment with 100 nM rapamycin. Representative nonlinear curve fit IC50 estimation is shown on left. Average IC50 of six experimental replicates is shown on right. Error bars are standard deviation (SD). *Unpaired t-test P value = 0.0029. (B) 686LN cells sensitive to erlotinib were treated with erlotinib as above. Erlotinib IC50 was not significantly altered by rapamycin treatment. Representative nonlinear curve fit IC50 estimation is shown on left. Average IC50 of four experimental replicates is shown on right. Error bars are SD nsUnpaired t-test P value = 0.77. (C) Hela-R30 cells that were transfected with siATG7 or siControl were treated as above and assayed for IC50. Knockdown of ATG7 increased erlotinib IC50 when compared with cells transfected with non-targeting siRNA control. Nonlinear curve fitting is shown on left. Average IC50 of four experimental replicates is shown on right. Error bars are SD *Unpaired t-test P value = 0.0303. (D) Whole cell lysates of Hela-R30 cells were collected 48 h after transfection of siRNA targeted against ATG7 (siATG7) or non-targeted siControl and immunoblotted for ATG7, LC3 or β-tubulin. Greater than 80% knockdown was achieved with siATG7 when compared with siControl. LC3-I accumulation was noted in cells with ATG7 knockdown.
Discussion
Here, we report that EGFR-TKI induces autophagy in multiple EGFR-TKI sensitive cancer cell lines derived from epithelial malignancies of the head and neck, cervix and bladder. We demonstrate that treatment of these cells with micromolar doses of erlotinib and gefitinib causes increased autophagy as measured by the accumulation of lipidated of LC3. We also show that treatment of cancer cells with erlotinib causes a dose-dependent accumulation of GFP-LC3 positive autophagosomes across a wide range of erlotinib doses. Surprisingly, in our initial survey of erlotinib treated cancer cell lines, we found that cells resistant to erlotinib exhibited poor induction of autophagy in response to erlotinib treatment. Thus, we explored the potential role of autophagy in these erlotinib resistant, autophagy deficient cells. Unfortunately, there are currently no squamous cell carcinoma of the head and neck derived cell lines that are resistant to erlotinib. Therefore, we employed a resistant cell line derived from Hela cells, which is an inherent limitation of the current study.
We report that in Hela-R30 cells, a clonal Hela cell line selected for erlotinib resistance, combination treatment with erlotinib and rapamycin can cause a mild increase autophagic flux while also causing an increase in cell death when compared with either agent alone. Moreover, the induction of autophagy by rapamycin sensitizes these cells to erlotinib while the depletion of ATG7, a critical autophagy protein, by siRNA imparts increased erlotinib resistance in these already resistant cells suggesting that autophagy mediates a component of resistance to EGFR tyrosine kinase inhibition. Also, our finding that cotreatment of resistance cells with rapamycin and erlotinib may, at least in part, be due to disruption of downstream EGFR signaling molecules such as ERK. Our data suggests that the loss of autophagy or the inability to increase autophagic flux above basal levels may contribute to the development of acquired erlotinib resistance. This represents a potential new pathway by which cancer cells can acquire resistance to biological therapies. Consequently, the addition of autophagy inducing drugs may be a viable strategy for maximizing the efficacy of EGFR tyrosine kinase inhibition.
Recently, erlotinib and gefitinib, in micromolar doses, were shown to induce autophagy in lung cancer cells.15 These authors also demonstrated that autophagy serves a cytoprotective role in response to TKI treatment and accordingly, that inhibition of autophagy greatly increased the cytotoxic effect of both gefitinib and erlotinib. Other studies have also demonstrated that autophagy can also be induced by EGFR tyrosine kinase inhibition when used in combination with other chemotherapeutics.20,21 In these reports, autophagy was shown to serve a predominantly cytoprotective role.
Conversely, our study seems to suggest that, at least in EGFR-TKI resistant and autophagy deficient cells, restoration of autophagy via rapamycin can partially restore erlotinib sensitivity to these cells and thus contribute to cell death. This finding is supported by recent studies that have shown that rapamycin, in cells that show mild or absent apoptosis in response to cetuixmab, can cause non-apoptotic cell death.14 Similarly, Gorzalczany, et al. reported that the combination of erlotinib and rapamycin increased the cytotoxicity of erlotinib in lung cancer cells that were resistant to erlotinib.22 In this study, the authors demonstrated that the combination of erlotinib and rapamycin could increase autophagy in cells without a significant induction of autophagy in response to erlotinib alone. Indeed, the combination of rapamycin with erlotinib has previously been shown to produce a synergistic cytotoxic effect in a variety of cancer cell lines.23 Our data and the results from other groups suggest that the addition of rapamycin to erlotinib may be an effective strategy to combat EGFR TKI resistance via the induction of autophagy and thus, implies that autophagy is contributing to cell death. The question of whether autophagy contributes to the death or survival of cancer cells remains a controversial topic24 and the likely answer is that the role of autophagy is highly context dependent. We have shown that in at least one highly resistant cancer cell line, in vitro acquired resistance to erlotinib is mediated by a lack of autophagy and that downstream restoration of autophagy via mTOR inhibition can restore sensitivity to erlotinib.
Materials and Methods
Cell lines and culture conditions
SCC-1 (UM-SCC1), 15B (PCI-15B), a pyriform sinus-derived squamous cell carcinoma and 686LN (MDA-686LN) human oral squamous cell carcinoma cell lines and the T-24 transitional cell carcinoma cell line were used as previously described.25-28 Hela-R29 and Hela-R30 cell lines are in vitro-derived isogenic erlotinib resistant Hela cells.29 All cell lines were maintained in DMEM (Invitrogen, #11965) or DMEM:F12 (Invitrogen, #11320) medium supplemented with 10% fetal bovine serum and cultured at 37°C in humidified air and 5% CO2. Hela-R29 and Hela-R30 media was additionally supplemented with 250 nM erlotinib to maintain resistance. All cell lines were routinely tested for mycoplasma and found to be mycoplasma free. Cell line genetic identities were verified by short tandem repeat profiling and matched to ATCC database. GFP-LC3 T-24 stable cell line was created using retroviral transduction of transgenes as previously described.8 The pBabe GFP-LC3 plasmid was a kind gift of Dr. Jayanta Debnath.
Reagents and chemicals
Erlotinib (Cayman Chemical, #10483) was dissolved in DMSO and used at concentrations ranging from 0.03 μM to 30 μM. Gefitinib (Cayman Chemical, #13166) was dissolved in DMSO and used at concentrations ranging from 2 to 32 μM. Rapamycin (Sigma, #R0395) was dissolved in DMSO and used at 100 nM. For siRNA transfection, 100 pmoles of non-targeting and ATG7 targeted siRNA pools (Dharmacon, #L-020112) were transfected using Lipofectamine-2000 (Invitrogen, #11668) per 250,000 cells according to manufacturer’s instructions. E64D (Sigma, #E8640) and pepstatin A (Sigma, #P5318) were both used at 10 ug/ml and dissolved in DMSO.
Cell Viability Assays and Calculation of IC50
ATP level was assayed using Cell-titer-glo luminescent cell viability assay (Promega, #G7572) according to manufacturer’s protocol. Briefly, cells were seeded at 5,000 cells per well in opaque walled 96-well tissue culture plates and after attachment, cells treated with rapamycin and/or erlotinib for 72 h. Cells were lysed in Cell-titer-glo reagent buffer and luminescence was assayed. Five wells per condition were assayed and the high and low values for dropped for subsequent calculations. Percent survival was normalized relative to carrier control (1:1000 DMSO). For IC50 measurements, cells were seeded at 3 x 10^4 per well of 24-well tissue culture plates and allowed to attach to plastic for 24 h. After attachment, cells were treated, in triplicate, with increasing doses of erlotinib (0.03 μM to 30 μM) or vehicle control. Cell viability was assessed using MTT reduction assay (Sigma, #M2128) after 72 h of treatment. Fifty percent inhibition of proliferation (IC50) compared with untreated controls was determined using GraphPad Prism software (GraphPad Software).
Immunoblotting
Whole cell lysates were subjected to SDS-PAGE and transferred to PVDF membranes (Biorad, #162–177) according to manufacturers instructions. Membranes were blocked in 5% non-fat dry milk and probed with primary antibodies for LC3 (Nanotools, #0260), ATG7 (Rockland, #600–401–487), phospho-Erk (Cell Signaling, #4370), Erk (Cell Signaling, #4695), phospho-Akt (Cell Signaling, #4060), Akt (Cell Signaling, #4691) and B-tubulin (Abcam, #ab15568). Membranes were washed in 0.2% Tween-Tris buffered saline and incubated with horseradish-peroxidase conjugated secondary antibody (Biorad, #170–5046 and #170–5047). Visualization of immunoblots was performed using Luminol (Santa Cruz, #sc-2048) and autoradiography film (Bioexpress, #F-9023).
Assessment of autophagy via fluorescence microscopy
T-24 cells stably expressing GFP-LC3 were seeded on poly-l-lysine coated glass coverslips one day prior to the start of assays. Cells were treated with erlotinib at 1 to 10 μM for 24 h and coverslips were fixed in 4% paraformaldehyde and washed several times with phosphate buffered saline. Coverslips were mounted using Vectashield Hardset (Vector Laboratories, H-1400) and images were obtained at room temperature by widefield epifluorescent microscopy. GFP-LC3 puncta were quantified by counting the number of GFP positive puncta per cell in 50–100 cells over multiple microscopic fields.
Supplementary Material
Acknowledgments
These studies were supported by grants 57006715 (C.F.) from the Howard Hughes Medical Institute, 5P50CA097190 and 2R01CA098372 (J.G.) from the National Institutes of Health, CRP-08–229–01 (J.G.) from the American Cancer Society, and CDA-2–057–10S (U.D.) from the United States Department of Veteran’s Affairs. The ideas in this article do not represent the views of the Department of Veteran's Affairs.
Glossary
Abbreviations:
- EGFR
epidermal growth factor receptor
- TKI
tyrosine kinase inhibitor
- NSCLC
non-small cell lung cancer
- mTOR
mammalian target of rapamycin
- LC3
microtubule-associated proteins 1A/1B light chain 3A
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/22002
References
- 1.Fung C, Grandis JR. Emerging drugs to treat squamous cell carcinomas of the head and neck. Expert Opin Emerg Drugs. 2010;15:355–73. doi: 10.1517/14728214.2010.497754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–7. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 4.Mathew R, White E. Role of autophagy in cancer. Autophagy. 2007;7:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren J-H, He W-S, Nong L, Zhu Q-Y, Hu K, Zhang R-G, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm. 2010;25:75–80. doi: 10.1089/cbr.2009.0701. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta. 2010;1806:220–9. doi: 10.1016/j.bbcan.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125:717–22. doi: 10.1002/ijc.24402. [DOI] [PubMed] [Google Scholar]
- 8.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 10.Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22:177–80. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer. 2011;129:2720–31. doi: 10.1002/ijc.25975. [DOI] [PubMed] [Google Scholar]
- 12.Lin C-I, Whang EE, Donner DB, Du J, Lorch J, He F, et al. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through Met inhibition in papillary thyroid cancer. Mol Cancer Res. 2010;8:1217–26. doi: 10.1158/1541-7786.MCR-10-0162. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010;70:5942–52. doi: 10.1158/0008-5472.CAN-10-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Lu Y, Pan T, Fan Z. Roles of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy. 2010;6:1066–77. doi: 10.4161/auto.6.8.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011;6:e18691. doi: 10.1371/journal.pone.0018691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang PA, Tsao M-S, Moore MJ. A review of erlotinib and its clinical use. Expert Opin Pharmacother. 2006;7:177–93. doi: 10.1517/14656566.7.2.177. [DOI] [PubMed] [Google Scholar]
- 17.Orcutt KP, Parsons AD, Sibenaller ZA, Scarbrough PM, Zhu Y, Sobhakumari A, et al. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res. 2011;71:3932–40. doi: 10.1158/0008-5472.CAN-10-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu Q, Amano O, Kanda Y, Kunii S, Wang Q, Sakagami H. Tumor-specific cytotoxicity and type of cell death induced by gefitinib in oral squamous cell carcinoma cell lines. Anticancer Res. 2009;29:5023–31. [PubMed] [Google Scholar]
- 21.Schmid K, Bago-Horvath Z, Berger W, Haitel A, Cejka D, Werzowa J, et al. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br J Cancer. 2010;103:622–8. doi: 10.1038/sj.bjc.6605761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorzalczany Y, Gilad Y, Amihai D, Hammel I, Sagi-Eisenberg R, Merimsky O. Combining an EGFR directed tyrosine kinase inhibitor with autophagy-inducing drugs: a beneficial strategy to combat non-small cell lung cancer. Cancer Lett. 2011;310:207–15. doi: 10.1016/j.canlet.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Buck E, Eyzaguirre A, Brown E, Petti F, McCormack S, Haley JD, et al. Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol Cancer Ther. 2006;5:2676–84. doi: 10.1158/1535-7163.MCT-06-0166. [DOI] [PubMed] [Google Scholar]
- 24.Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ. 2009;16:1–2. doi: 10.1038/cdd.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krause CJ, Carey TE, Ott RW, Hurbis C, McClatchey KD, Regezi JA. Human squamous cell carcinoma. Establishment and characterization of new permanent cell lines. Arch Otolaryngol. 1981;107:703–10. doi: 10.1001/archotol.1981.00790470051012. [DOI] [PubMed] [Google Scholar]
- 26.Heo DS, Snyderman C, Gollin SM, Pan S, Walker E, Deka R, et al. Biology, cytogenetics, and sensitivity to immunological effector cells of new head and neck squamous cell carcinoma lines. Cancer Res. 1989;49:5167–75. [PubMed] [Google Scholar]
- 27.Liu TJ, el-Naggar AK, McDonnell TJ, Steck KD, Wang M, Taylor DL, et al. Apoptosis induction mediated by wild-type p53 adenoviral gene transfer in squamous cell carcinoma of the head and neck. Cancer Res. 1995;55:3117–22. [PubMed] [Google Scholar]
- 28.Bubeník J, Baresová M, Viklický V, Jakoubková J, Sainerová H, Donner J. Established cell line of urinary bladder carcinoma (T24) containing tumour-specific antigen. Int J Cancer. 1973;11:765–73. doi: 10.1002/ijc.2910110327. [DOI] [PubMed] [Google Scholar]
- 29.Muller S, Su L, Tighiouart M, Saba N, Zhang H, Shin DM, et al. Distinctive E-cadherin and epidermal growth factor receptor expression in metastatic and nonmetastatic head and neck squamous cell carcinoma: predictive and prognostic correlation. Cancer. 2008;113:97–107. doi: 10.1002/cncr.23557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.