

Abstract
Objectives:
We asked whether autoantibodies against neurofascin (NF)186 or NF155, both localized at the nodes of Ranvier, are present in serum of patients with inflammatory neuropathy, and whether NF-specific monoclonal antibodies are pathogenic in vivo.
Methods:
We cloned human NF155 and NF186, and developed an ELISA and cell-based assay to screen for antibodies to human NF in a total of 434 donors including 294 patients with Guillain-Barré syndrome variants acute inflammatory demyelinating polyneuropathy (AIDP), acute motor axonal neuropathy, and chronic inflammatory demyelinating polyneuropathy (CIDP). We characterized reactive samples by isotyping, tissue section staining, and epitope mapping. We also injected NF-specific monoclonal antibodies IV into rats with experimental autoimmune neuritis.
Results:
We detected autoantibodies to NF by ELISA in 4% of patients with AIDP and CIDP, but not in controls. Most positive samples contained immunoglobulin G (IgG)1, IgG3, or IgG4 antibodies directed to only one isoform of NF. Two patients with CIDP showed particularly high (1:10,000 dilution) NF155-specific reactivity in both assays and stained paranodes. Two other patients with CIDP who benefited from plasma exchange exhibited antibodies to NF155 by ELISA, and upon affinity purification, antibodies to both isoforms were observed by both assays. Anti-NF monoclonal antibodies enhanced and prolonged induced neuritis in rats.
Conclusions:
Autoantibodies to NF are detected in a very small proportion of patients with AIDP and patients with CIDP, but may nevertheless be pathogenic in these cases.
An involvement of autoantibodies in Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP) is suggested by the efficacy of treatment with plasma exchange (PE).1,2 While antibodies against gangliosides are found in a proportion of patients with the GBS variant acute motor axonal neuropathy (AMAN),3–6 the antigenic target is unknown for most of the patients with the GBS variant acute inflammatory demyelinating polyneuropathy6 (AIDP) and CIDP. Passive transfer of immunoglobulin (Ig)G from some patients with CIDP induced conduction block and demyelination in rats with experimental autoimmune neuritis (EAN),7 while complement-fixing antibodies against Schwann cell antigens were described in patients with AIDP.8
Antibody binding at the node of Ranvier has been observed in EAN9 and in patients with GBS and CIDP.10 The neuronal isoform of neurofascin (NF)186 exposed at the node is crucial for sodium channel clustering,11 while the glial isoform NF155 at the paranode is necessary for proper paranodal junction formation.11 Human autoantibodies to NF were first detected in multiple sclerosis and anti-NF monoclonal antibodies (mAbs) mediated axonal injury in an experimental autoimmune encephalomyelitis model.12 While subtle differences in nodal structure exist between CNS and peripheral nervous system, both NFs are found at similar sites at the nodes.13
We asked whether anti-NF antibodies are present in patients with inflammatory neuropathies. We cloned human NFs, established ELISA and cell-bound assay, and screened serum from neuropathy patients. Since we found NF reactivity in some patients with AIDP and CIDP, we investigated in vivo effects of anti-NF mAbs in EAN, and report that anti-NF mAbs enhance and prolong disease.
METHODS
Patient material
Our study included 394 serum samples collected from institutes in Japan (n = 200), Sweden (n = 62), and Germany (n = 132). The patient group included CIDP (n = 119), AIDP (n = 65), AMAN (n = 50), and other neuropathies (n = 20). The control group consisted of patients with other neurologic diseases (OND; n = 63) and healthy controls (HC; n = 77). PE material was obtained from patients with CIDP (n = 41) and patients with relapsing-remitting multiple sclerosis (n = 4). Further information can be found in appendix e-1 on the Neurology® Web site at www.neurology.org.
Standard protocol approvals, registrations, and patient consents
These studies were approved by each local ethical committee and the donors gave their informed consent.
Cloning and expression of full-length human NF155 and NF186 and truncated forms
The complete cDNAs of human NF155 and NF186 were generated stepwise. Details of the cloning strategy of all plasmids are found in appendices e-2 and e-3. For cell surface expression in TE671 human rhabdomyosarcoma cell line, we inserted full-length NF cDNAs into plasmid pRSV5neo.14 Further, we made 6 truncated forms of NF155 and NF186 that were fused to super green fluorescent protein15 at the C-terminus. We stably transfected TE671 cells with pRSV5neo constructs containing human NF155, NF186, or NF truncation variants. For production of soluble NF, we extended the constructs by a C-terminal polyhistidine tag, inserted them into plasmid pTT5, and transfected human embryonic kidney 293-EBNA1 (HEK293-EBNA) cells.16 We purified soluble human NF155 and NF186 from the supernatant of transiently transfected HEK293-EBNA cells using Ni-NTA agarose chromatography (Qiagen; Hilden, Germany). Details are provided in appendix e-4.
ELISA
We coated protein antigens on Maxisorp 96-well plates (NUNC; Langenselbold, Germany) or HisSorb plates (Qiagen) at 5 μg/mL at 4°C overnight. We added 100 μL of serum (1:100), PE material (100 μg/mL Ig concentration), or pan-NF mAb (0.2 μg/mL; A4/3.4, mouse hybridoma) to wells coated with bovine serum albumin, human NF155, and NF186. Afterwards, antihuman Ig (1:7,000; JacksonImmuno; Suffolk, UK) or antimouse IgM (1:7,000; JacksonImmuno) conjugated to horseradish peroxidase was used for detection. We also used commercially available NS0-derived antigens rat NF155-NS0, human contactin-2-NS0, and human oligodendrocyte myelin glycoprotein (OMgp-NS0; all from R&D Systems; Wiesbaden-Nordenstadt, Germany). For isotyping, we used biotin-labeled antihuman IgG1, IgG2, IgG3, IgG4, IgM, and IgA secondary antibodies (1:1,000; Southern Biotech; Birmingham, AL; JacksonImmuno) and peroxidase-conjugated streptavidin (1:1,000; Jackson Immuno).
Cell-bound assay by flow cytometry
We used TE671 stable transfectants expressing NF155 and NF186 and mock transfectants to detect antibodies to NFs. For staining, we diluted serum samples 1:50 and PE samples 1:10, and used antihuman IgG or antihuman Ig conjugated to R-Phycoerythrin (1:150; JacksonImmuno) for detection. We resuspended the cells in phosphate-buffered saline with TO-PRO3 (1:4,000; Invitrogen; Darmstadt, Germany) and acquired them on FACS Calibur (BD Biosciences; Heidelberg, Germany). The mean fluorescence intensity ratio between NF-expressing cells and mock transfectants was calculated. We used pan-NF mAb (A12/18.1, mouse hybridoma) to monitor the level of NF expression. For isotyping, we used R-Phycoerythrin-labeled antihuman IgG1, IgG2, IgG3, IgG4, IgM, and IgA secondary antibodies (1:100; Southern Biotech; JacksonImmuno).
Affinity purification of anti-NF antibodies
We covalently conjugated recombinant neurofascin (human NF155, NF186, or ratNF155-NS0) to N-hydroxysuccinimide-activated Sepharose (GE Healthcare; Munich, Germany) at 0.5 mg/mL. After loading with PE material, we eluted the column at pH 5, pH 4, pH 3, and pH 11 (100 mM citrate, acetate, glycine, and ethanolamine, respectively), while bringing the pH of both the eluate and column up to neutral between each pH step. The eluted fractions were dialyzed against phosphate-buffered saline and then concentrated. For NF155-specific rabbit antibodies, we immunized rabbits with NF155-unique peptide17 and purified the antibodies using an NF155-conjugated column.
Immunohistochemistry
We fixed frozen sections of rat spinal cord with cold methanol and blocked with goat IgG (Sigma-Aldrich; Munich, Germany) in antibody diluent (Dako; Eching, Germany) at room temperature. We diluted human serum 1:100 in phosphate-buffered saline with 10% fetal bovine serum and applied it overnight at 4°C. Secondary antibody (antihuman IgG Alexa488, 1:100; Invitrogen) and DAPI (1:1,000; Thermo Scientific; Waltham, MA) were then added. Subsequently, we stained with Caspr-specific rabbit antibody18 (1:1,000; from E. Peles), and used antirabbit IgG Alexa594 (1:1,000; Invitrogen) for detection. We used affinity-purified rabbit anti-NF155 antibodies at 5 μg/mL, and anti-pan-NF mAb (A12/18.1) at 2 μg/mL and antimouse IgG Alexa488 (1:1,000; Invitrogen) for detection. Stained sections were mounted using fluoromount (Invitrogen).
Experimental autoimmune neuritis
We immunized Lewis rats subcutaneously on both hind limbs with 100 μg of P2 peptide19 (AA 53-78) emulsified in complete Freund adjuvant (Difco; Lawrence, KS). Clinical scores were evaluated as 0, normal; 0.5, loss of tail tonus; 1, tail paralysis; 2, gait disturbance; 3, hind-limb paralysis. Upon initial signs of clinical disease, we injected anti-NF antibodies (A12/18.1, A4/3.4) or their respective isotype controls (mouse IgG2a, IgM; eBioscience; Frankfurt, Germany) IV at 500 μg per rat. Animal experiments were approved by the Regierung von Oberbayern.
RESULTS
Development of assays to detect autoantibodies against human neurofascin
We have developed an ELISA with the complete extracellular portion of NF155 and NF186 and a cell-bound assay by flow cytometry using cells stably transfected with NF155 and NF186. The utility of our assays was validated using anti-NF–specific antibodies (figure e-1). We noted that by ELISA both mAbs recognized NF186 strongly, while only A4/3.4 but hardly A12/18.1 recognized NF155 (figure e-1B). NF155-specific rabbit antibodies were reactive by ELISA and stained paranodes on tissue sections where NF155 is localized, but they did not stain the NF transfectants (figure e-1C).
We compared the utility of human NF155 and NF186 expressed in HEK293-EBNA cells with a commercially available ratNF155 derived from NS0 murine myeloma cells (ratNF155-NS0) to detect antibodies against human NF by ELISA. We noted that while only a few samples showed reactivity to NF155 and NF186 (figure 1), virtually all donors showed some response against ratNF155-NS0 (figure 2A). To investigate this discrepancy, we affinity-purified antibodies from 4 donors using a ratNF155-NS0-conjugated column. We took these purified antibodies and probed for reactivity against HEK293-derived human NF155 and NF186, ratNF155-NS0, and unrelated NS0-derived antigens: human contactin-2-NS0 and OMgp-NS0. We found by ELISA and Western blot in all 4 donors that most of the affinity-purified anti-ratNF155-NS0 antibodies did not bind NF155 and NF186; rather, they strongly recognized the unrelated NS0-derived antigens (one example shown in figure 2, B and C). This might be based on features of the NS0 murine myeloma cell line: it is known to produce abnormally glycosylated proteins that are potentially immunogenic depending on culture conditions.20 However, anti-pan-NF mAb A4/3.4 recognized ratNF155-NS0 along with NF155 and NF186, and as expected, not contactin-2-NS0 or OMgp-NS0 (figure 2B). To summarize, although the commercially available ratNF155 produced in NS0 cells is recognized by an anti-NF mAb, it is not a suitable target for detection of human NF-specific reactivity.
Figure 1. Autoantibodies to NF155 and NF186 in a very small proportion of patients with neuropathy.

Serum samples from patients with chronic inflammatory demyelinating polyneuropathy (CIDP) (n = 117), acute inflammatory demyelinating polyneuropathy (AIDP) (n = 65), acute motor axonal neuropathy (AMAN) (n = 50), other neuropathies (ON) (n = 20), and controls with other neurologic diseases (OND) (n = 61) and healthy controls (HC) (n = 77) were tested for autoantibodies to neurofascin (NF)155 and NF186 by ELISA. Plasma exchange samples from a separate cohort of patients with CIDP (CIDP*; n = 40) were tested at 100 μg/mL immunoglobulin concentration. ELISA reactivities to NF155 (A) and NF186 (B) are shown as baseline-subtracted optical density reading at 450 nm (ΔOD450; response to NF minus bovine serum albumin reactivity). The cutoff (dashed line) represents the mean of OND group plus 4 SDs. Two CIDP samples marked with special symbols (▲, ♦) showed reactivity by both ELISA and flow cytometry to NF155 (more details in figures 3 and e-2, C–K). One AIDP sample marked with a special symbol (■) showed reactivity by ELISA to both isoforms of NF. GBS = Guillain-Barré syndrome.
Figure 2. ratNF155-NS0 is not a specific antigen to detect antibodies to human neurofascin (NF).

Serum screening by ELISA using ratNF155-NS0 (A) included samples from patients with chronic inflammatory demyelinating polyneuropathy (CIDP) (n = 82), acute inflammatory demyelinating polyneuropathy (AIDP) (n = 54), acute motor axonal neuropathy (AMAN) (n = 50), and controls with other neurologic diseases (OND) (n = 61) and healthy controls (HC) (n = 77). The response to ratNF155-NS0 minus bovine serum albumin reactivity (delta OD) is shown. Most of the patients and control donors show a response against this antigen. The mean for each group is shown. Closed circles represent positive samples identified by ELISA or flow cytometry using human NF155 and NF186. Antibodies purified over ratNF155-NS0 conjugated column were tested by (B) ELISA and (C) Western blot for reactivity against HEK293–derived NF155 and NF186, ratNF155-NS0, and unrelated NS0-derived antigens: contactin-2-NS0 and oligodendrocyte myelin glycoprotein (OMgp-NS0). The number above each lane in (C) corresponds to the antigen with the same number in (B). Western blot analysis procedure is detailed in appendix e-5. Anti-pan-NF mAb (A4/3.4) was also tested by ELISA as a positive control for reactivity against NF155, NF186, and ratNF155-NS0 (B). GBS = Guillain-Barré syndrome.
Detection of anti-NF reactivity in a very small proportion of patients with inflammatory neuropathies
We tested a total of 394 serum samples by both ELISA and cell-bound assay using flow cytometry with human NF155 and NF186. Using ELISA, we found reactivity in 8/254 patients with inflammatory neuropathies (5 CIDP, 3 AIDP, 0 AMAN), but not in controls (figure 1). Among these 8 anti-NF–positive patients, 7 recognized only one isoform, while one recognized both. Six of these 8 patients showed a clear reactivity while 2 were at the detection limit (figure 1).
Using our cell-based assay, we found very strong reactivity to NF155 in 2 patients with CIDP (figures 3 and e-2). These 2 patients also had reactivity to NF155 by ELISA (figures 1A, 3, and e-2). In addition, 4 controls (1 OND with sensory symptoms, 3 HC) exhibited reactivity to one NF isoform, but did not show reactivity by ELISA (figure e-2, A and B).
Figure 3. Features of antibodies to neurofascin (NF) in a patient with chronic inflammatory demyelinating polyneuropathy (CIDP).

Reactivity to NF155 was seen by flow cytometry (A–C) up to a dilution of 1:10,000 (B). (A) Reactivity to NF155 (blue line) was compared with reactivity to NF186 (orange line) and to control cells (black, filled). (B) The mean fluorescence intensity (MFI) ratio is plotted for level of reactivity above background. (C) The NF155 reactivity was mediated by immunoglobulin (Ig)G4 with minor contribution from IgG1. Reactivity to NF155 was also seen by ELISA (D–F) up to a dilution of 1:10,000 (E) by IgG4 and weakly by IgG1, IgG3, IgM, and IgA (F). Serum staining (G, left) colocalizes with Caspr staining (G, middle) on longitudinal rat spinal cord sections. The scale bar represents 10 μm. (H) NF186 differs from NF155 by substitution of Fn3-Fn4 with Fn4-Mucin-Fn5 (Ig = immunoglobulin-like domain; Fn = fibronectin type III domain). (I) A scheme of super green fluorescent protein (sGFP) fusion truncated NF variants is shown beside the corresponding serum reactivity by flow cytometry. Reactivities to truncated NF variants and to negative control cells are shown as sGFP intensity vs serum reactivity. The fragment recognized by both NF155-reactive serum samples is boxed.
To further investigate the prevalence of human NF reactivity in patients with CIDP, we tested an additional 40 patient samples taken from PE material (figure 1). We found one donor with clear NF155 reactivity by ELISA, but not in the cell-bound assay (figure e-2, A and B).
A previous report found a higher average reactivity to ratNF155-NS0 by ELISA in patients with GBS compared to healthy controls.21 We also used this antigen for serum screening in parallel with the HEK293-derived protein and found broad reactivity to ratNF155-NS0. However, we did not observe a significant difference between the GBS group (AIDP and AMAN) compared to the control group (OND and HC). Furthermore, the patients who showed reactivity to the NF155/NF186 by ELISA were not conspicuous with ratNF155-NS0 (filled circles; figure 2A).
Features of anti-NF antibody reactivity
Two patients with CIDP who showed high IgG reactivities to NF155 by ELISA also showed reactivity in our cell-based assay (figures 3 and e-2, C–K). Both patients had high titers of anti-NF155 antibodies (figures 3, B and E, and e-2, D and G), and in both cases the dominant IgG subclass was IgG4, with minor contribution from IgG1, IgG2, IgG3, IgM, and IgA (figures 3, C and F, and e-2, E and H). We mapped the domains recognized by these autoantibodies using truncation variants of NF155 and NF186, and found that autoantibodies from both patients recognized a fragment unique to NF155 spanning the fibronectin type III (Fn)3-Fn4 domains (figures 3I and e-2K). Furthermore, serum from both patients stained paranodes that were marked using anti-Caspr antibodies on tissue sections (figures 3G and e-2I).
Another patient with CIDP had antibodies to NF155 only by ELISA (figure e-3). Extensive antibody tests for onconeuronal, rheumatologic, autoimmune, neuronal surface antigen, and neuropathy-related antibodies were negative in this patient. He developed progressive relapsing symmetric paralysis that required continuous escalation of routine immunotherapy. In particular, PE was found to be very efficient to rapidly and completely relieve paralysis. Over 4 years the patient eventually remitted completely and was weaned from PE. We monitored the anti-NF155 reactivity throughout the disease course, which persisted throughout this observation period while showing a progressive decline (figure e-3A). The anti-NF antibodies were predominantly IgG3 (data not shown). We affinity-purified anti-NF antibodies against both NF155 and NF186 from PE material, and eluted IgG3, as well as small amounts of IgG1, IgM, and IgA (data not shown). Following purification, we found reactivity to NF155 by ELISA and also by cell-based assay, and additionally to NF186 by both assays (figure e-3, B–D).
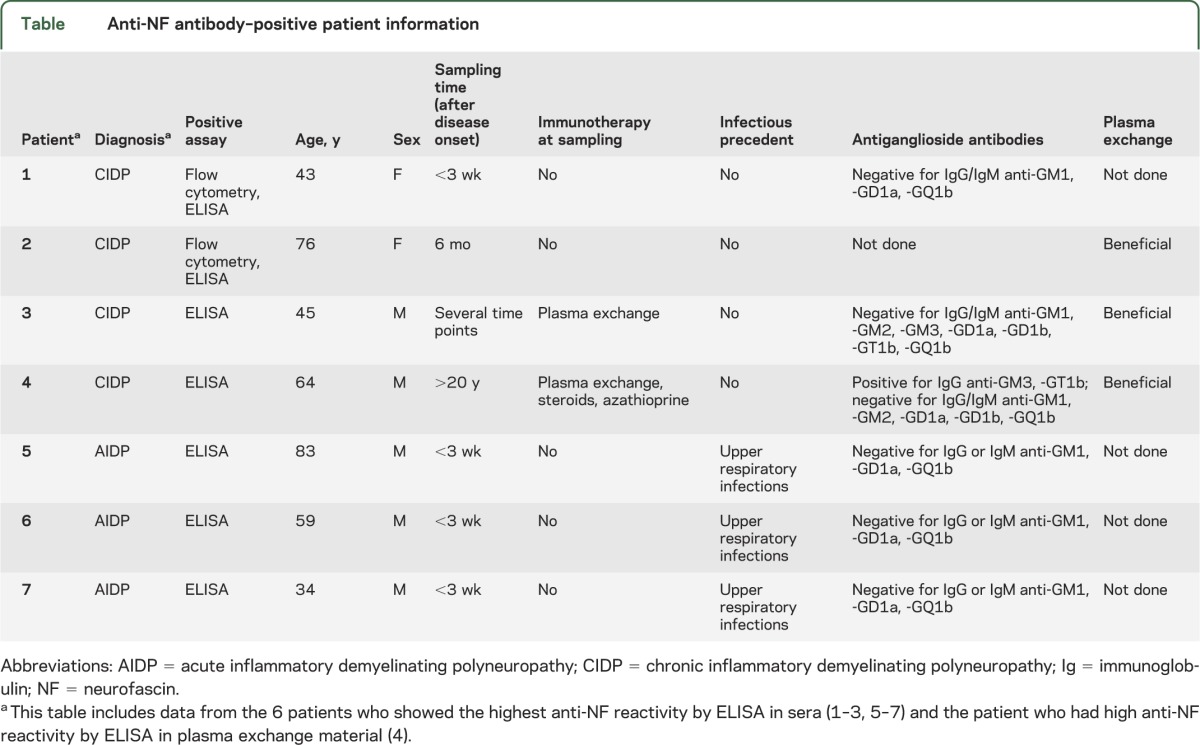
Another patient with CIDP who benefited from PE (from the CIDP* cohort, figure 1) had IgG3, IgM, and IgA antibodies against NF155 by ELISA. Three patients with AIDP showed reactivity to NF at serum dilution of up to 1:1,000 by ELISA. The first exhibited antibodies to only NF155 with IgG1, the second to only NF186 with IgG1, and the third to both NFs with IgG1 and IgG3 (data not shown). These NF-reactive samples were further tested for nodal staining on tissue sections, and only the 2 CIDP samples with the highest reactivities showed clear nodal staining (figures 3G and e-2I). Clinical features of these patients with inflammatory neuropathy with NF reactivity are provided in the table.
Table.
Anti-NF antibody–positive patient information
Anti-NF antibodies enhance and prolong EAN
The 2 pan-NF mAbs (A12/18.1 mouse IgG2a and A4/4.3 mouse IgM) were injected at the beginning of clinical EAN. This enhanced and prolonged the disease (figure 4). Naive rats injected with either anti-NF mAb without prior P2 peptide immunization did not show signs of clinical disease during an observation period of 15 days (data not shown), implying that antibody treatment alone is not enough to cause disease.
Figure 4. Two different monoclonal antibodies to neurofascin enhance and prolong experimental autoimmune neuritis.

Lewis rats immunized with P2 peptide were injected with either anti-pan-neurofascin monoclonal antibodies A12/18.1 mouse immunoglobulin (Ig)G2a (A) or A4/3.4 mouse IgM (B) (closed symbols) or their respective isotype controls (open symbols) at disease onset (day 14 after experimental autoimmune neuritis [EAN] induction, indicated by arrowhead). They were scored blinded daily for EAN disease severity. Values (mean ± SEM) represent EAN clinical scores from 6 rats per group, pooled from 2 independent experiments.
DISCUSSION
We have developed assays to detect antibodies to human NF155 and NF186, and show that such autoantibodies are found in 4% of patients with CIDP and AIDP. Using an EAN model, we have demonstrated that antibody targeting of NF in vivo enhances and prolongs neuritis. Our findings are in harmony with a recent report showing nodal reactivity in a proportion of neuropathy patients, although in that article rodent but not human NF was used as an antigen.10
When comparing our 2 assays, ELISA is most suitable for identifying subjects with anti-NF antibodies among patients with inflammatory neuropathy and distinguishing them from controls. Antibodies that are reactive to NF by ELISA but not by flow cytometry can bind NF in a physiologic setting and may be pathogenic, as exemplified by NF155-specific rabbit antibodies (figure e-1C) that bound to paranodes on tissue sections and caused complement-dependent demyelination in an in vitro myelination culture model (Chris Linington, unpublished data, 2012).
Unexpectedly, almost all the serum reactivities observed were directed against only one isoform. This is surprising because NF155 and NF186 are splice variants such that the 8 extracellular domains from the N-terminus are virtually identical, differing only in the 2 domains closest to the transmembrane region.22 We expressed truncated variants of NF155 and NF186, and identified the Fn3-Fn4 domains as the target for NF155-specific reactivity and Mucin-Fn5 domains for NF186 specificity.
Would the antibodies to NF, which we detect in a minority of patients with CIDP and patients with AIDP, be expected to contribute to the pathology? We investigated the principal pathogenic potential of NF-reactive antibodies with an EAN model, and this yielded 2 main findings. First, antibodies to NF can enhance and prolong an ongoing neuritis. Second, antibodies to NF alone are not pathogenic. Previous studies of this P2 peptide–induced EAN model have demonstrated nerve infiltration by autoreactive T cells and macrophages, yet autoantibodies were not implicated.9 We added anti-pan-NF mAbs into this disease setting and found that NF-targeting exacerbated disease. A similar observation was made in studies using a T-cell transfer EAN model in which addition of antimyelin antibodies greatly exacerbated disease.23 For human antibodies to be pathogenic, the target should be accessible. Inflammatory neuropathies are characterized by pronounced immune cell infiltration24,25 that can open blood–nerve barrier and provide access to autoantibodies.
Would the isoform-specific recognition of NF by human autoantibodies be compatible with pathogenicity? NF186 is displayed on the nodes of Ranvier, and it has been shown that antibody targeting of NF186 in the presence of complement disrupts nerve conduction.12 The anti-NF186 antibodies that we found in 2 patients with AIDP were of complement-activating isotypes IgG1 and IgG3. Furthermore, antibody binding to NF186 might disrupt its ability to perform its normal functions; for example, binding to gliomedin26 or other extracellular ligands.
Antibodies to NF155 found in some patients with AIDP and patients with CIDP could be pathogenic only if their target at the paranodes becomes accessible. Since a disruption of paranodal architecture is a feature of different nerve pathologies such as ischemia and inflammation,27,28 these antibodies could contribute to the pathology. Anti-NF155 antibodies were reported to inhibit myelination by blocking the formation of the Caspr/contactin/NF155 complex, the core structure at paranodal loops for adhesion between axon and glial cell.29 Such a disruption of the paranodal junctions can result in severe reduction of conduction velocity even in the absence of obvious demyelination.30 Thus, it is tempting to speculate that such antibodies interfere with remyelination in patients with AIDP and CIDP.
We made the unexpected observation that in 2 patients with CIDP with remarkably high anti-NF reactivity the anti-NF155 autoantibodies were largely IgG4 with an additional contribution of IgG1, IgG2, IgG3, IgM, and IgA. Anti-NF155 IgG4 in these patients with CIDP may have an antigen-blocking function to impair myelination and thereby nerve conduction, as IgG4 is generally not complement activating nor does it bind Fc receptors on effector cells.31 This blocking effect of IgG4 antibodies may be pathogenic. For example, in endemic pemphigus foliaceus, a blistering skin disease, IgG4 antibodies against desmoglein-1 cause a direct disruption of the epithelial layer, leading to blister formation.32 Apart from IgG4, low levels of complement-activating and immune cell–activating isotypes were also observed in these 2 patients with CIDP. Two other patients with CIDP who benefited from PE had antibodies to NF155. The pathogenic activity of these patient-derived anti-NF antibodies may be shown in further studies.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Ingrid Eiglmeier, Heike Rübsamen, and Martina Sölch (MPI of Neurobiology, Martinsried, Germany) for technical support; Cora Kaiser for support with immunohistochemistry; Drs. Kathrin Doppler and Andreas Weishaupt (Department of Neurology, University Hospital of Würzburg, Germany) for assistance with CIDP PE material; Drs. D. Jenne and G. Krishnamoorthy (MPI of Neurobiology, Martinsried, Germany) for comments on the manuscript; and Prof. H. Wekerle (MPI of Neurobiology, Martinsried, Germany) for support.
Glossary
- AIDP
acute inflammatory demyelinating polyneuropathy
- AMAN
acute motor axonal neuropathy
- CIDP
chronic inflammatory demyelinating polyneuropathy
- EAN
experimental autoimmune neuritis
- GBS
Guillain-Barré syndrome
- HC
healthy control
- HEK
human embryonic kidney
- Ig
immunoglobulin
- mAb
monoclonal antibody
- NF/NF155/NF186
neurofascin (155 kDa/186 kDa isoforms)
- OND
other neurologic diseases
- PE
plasma exchange
Footnotes
Editorial, page 2224
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
J.K.M. Ng: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagent/tools, acquisition of data, statistical analysis. J. Matlotka: analysis or interpretation of data, contribution of vital reagents/tools, acquisition of data. N. Kawakami: study concept or design, analysis or interpretation of data, contribution of vital reagents/tools, acquisition of data, statistical analysis. T. Derfuss: drafting/revising the manuscript, study concept or design, contribution of vital reagent/tools, acquisition of data. M. Khademi: analysis or interpretation of data, contribution of vital reagent/tools. T: Olsson: analysis or interpretation of data, contribution of vital reagent/tools. C. Linington: drafting/revising the manuscript. M. Odaka: analysis or interpretation of data, contribution of vital reagent/tools. B. Tackenberg: analysis or interpretation of data, contribution of vital reagent/tools. H. Prüss: drafting/revising the manuscript, analysis or interpretation of data, contribution of vital reagent/tools. J. Schwab: analysis or interpretation of data, contribution of vital reagent/tools. L. Harms: analysis or interpretation of data, contribution of vital reagent/tools. H. Harms: analysis or interpretation of data, contribution of vital reagent/tools. C. Sommer: analysis or interpretation of data, contribution of vital reagent/tools. M.N. Rasband: drafting/revising the manuscript, contribution of vital reagent/tools. Y. Eshed-Eisenbach: analysis or interpretation of data, acquisition of data. E. Peles: drafting/revising the manuscript, contribution of vital reagent/tools. R. Hohlfeld: drafting/revising the manuscript, study concept or design. N. Yuki: drafting/revising the manuscript, contribution of vital reagent/tools. K. Dornmair: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagent/tools, study supervision or coordination. E. Meinl: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, statistical analysis, study supervision or coordination, obtaining funding.
STUDY FUNDING
Supported by the Deutsche Forschungsgemeinschaft (SFB 571), the Bundesministerium für Bildung und Forschung (“Krankheitsbezogenes Kompetenznetz Multiple Sklerose”), the Excellency Initiative of the Ludwig Maximilians University Munich, and the Gemeinnützige-Hertie Stiftung. C.S. received research support from Genzyme Corp., Bayer, the German Research Foundation (SFB 581), and the Interdisciplinary Center for Clinical Research of the University of Würzburg. E.P. is supported by the National Institutes of Health (NS50220) and the Israel Science Foundation.
DISCLOSURE
J.K.M. Ng, J. Malotka, and N. Kawakami report no disclosures. T. Derfuss serves on scientific advisory boards for Novartis, Merck Serono, Biogen Idec, and Bayer Schering Pharma; has received funding for travel or speaker honoraria from Biogen Idec, Novartis, Merck Serono, and Bayer Schering Pharma; and receives research support from Novartis, Merck Serono, the German Research Foundation, the European Union, and the Swiss MS Society. M. Khademi reports no disclosures. T. Olsson has received unrestricted support for MS research from Biogen Idec, Bayer-Schering Pharma, Sanofi-Aventis, TEVA, Novartis, and Merck Serono. The same companies have given lecture fees or compensations for consultancy. C. Linington, M. Odaka, D.B. Tackenberg, H. Prüss, and J. Schwab report no disclosures. L. Harms received personal compensation from Novartis, Biogen-Idec, Merck-Serono, Bayer, and Biomarin for speaking, and travel support from Bayer, Biomarin, and Biogen-Idec. He received personal compensation from Biomarin for consulting services and from Novartis as a member of the advisory board. H. Harms reports no disclosures. C. Sommer has served on scientific advisory boards for Astellas Pharma Inc., Baxter Inc., Eli Lilly and Company, Grünenthal, Pfizer Inc., and UCB; and has received speaker honoraria from Allergan, Baxter Inc., CSL Behring, Eli Lilly, Genzyme Corp, Grünenthal, GSK, and Pfizer Inc. M.N. Rasband, Y. Eshed-Eisenbach, and E. Peles report no disclosures. R. Hohlfeld received personal compensations and grant support from Teva, Bayer, Merck-Serono, Sanofi-Aventis, Biogen-Idec, and Novartis. N. Yuki is in the Advisory Panel of “Expert Review of Neurotherapeutics” and received research support from the National Medical Research Council, Ministry of Health, Singapore. K. Dornmair reports no disclosures. E. Meinl received honorarium from TEVA and Novartis and grant support from Novartis. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lehmann HC, Meyer zu Horste G, Kieseier BC, Hartung HP. Review: pathogenesis and treatment of immune-mediated neuropathies. Ther Adv Neurol Disord 2009;2:261–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes RAC, Cornblath DR. Guillain-Barré syndrome. Lancet 2005;366:1653–1666 [DOI] [PubMed] [Google Scholar]
- 3.Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain 2002;125:2591–2625 [DOI] [PubMed] [Google Scholar]
- 4.Kaida K, Kusunoki S. Antibodies to gangliosides and ganglioside complexes in Guillain-Barré syndrome and Fisher syndrome: mini-review. J Neuroimmunol 2010;223:5–12 [DOI] [PubMed] [Google Scholar]
- 5.Illa I, Ortiz N, Gallard E, et al. Acute axonal Guillain-Barré syndrome with IgG antibodies against motor axons following parenteral gangliosides. Ann Neurol 1995;38:218–224 [DOI] [PubMed] [Google Scholar]
- 6.Yuki N, Hartung HP. Guillain-Barré syndrome. N Engl J Med 2012;366:2294–2304 [DOI] [PubMed] [Google Scholar]
- 7.Yan WX, Taylor J, Andrias-Kauba S, Pollard JD. Passive transfer of demyelination by serum or IgG from chronic inflammatory demyelinating polyneuropathy patients. Ann Neurol 2000;47:765–775 [PubMed] [Google Scholar]
- 8.Koski CL, Humphrey R, Shin ML. Anti-peripheral myelin antibody in patients with demyelinating neuropathy: quantitative and kinetic determination of serum antibody by complement component 1 fixation. Proc Natl Acad Sci USA 1985;82:905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lonigro A, Devaux JJ. Disruption of neurofascin and gliomedin at nodes of Ranvier precedes demyelination in experimental allergic neuritis. Brain 2009;132:260–273 [DOI] [PubMed] [Google Scholar]
- 10.Devaux JJ, Odaka M, Yuki N. Nodal proteins are target antigens in Guillain-Barré syndrome. J Peripher Nerv Syst 2012;17:62–71 [DOI] [PubMed] [Google Scholar]
- 11.Feinberg K, Eshed-Eisenbach Y, Frechter S, et al. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron 2010;65:490–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathey EK, Derfuss T, Storch MK, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med 2007;204:2363–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Susuki K, Rasband MN. Molecular mechanisms of node of Ranvier formation. Curr Opin Cell Biol 2008;20:616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long EO, Rosen-Bronson S, Karp DR, et al. Efficient cDNA expression vectors for stable and transient expression of HLA-DR in transfected fibroblast and lymphoid cells. Hum Immunol 1991;31:229–235 [DOI] [PubMed] [Google Scholar]
- 15.Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature 1995;373:663–664 [DOI] [PubMed] [Google Scholar]
- 16.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 2002;30:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tait S, Gunn-Moore F, Collinson JM, et al. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol 2000;150:657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peles E, Nativ M, Lustig M, et al. Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J 1997;16:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uyemura K, Suzuki M, Kitamura K, et al. Neuritogenic determinant of bovine P2 protein in peripheral nerve myelin. J Neurochem 1982;39:895–898 [DOI] [PubMed] [Google Scholar]
- 20.Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol 2006;24:1241–1252 [DOI] [PubMed] [Google Scholar]
- 21.Prüss H, Schwab JM, Derst C, Görtzen A, Vermeulen E. Neurofascin as target of autoantibodies in Guillain-Barré syndrome. Brain 2011;134:e173. [DOI] [PubMed] [Google Scholar]
- 22.Basak S, Raju K, Babiarz J, et al. Differential expression and functions of neuronal and glial neurofascin isoforms and splice variants during PNS development. Dev Biol 2007;311:408–422 [DOI] [PubMed] [Google Scholar]
- 23.Spies JM, Pollard JD, Bonner JG, Westland KW, McLeod JG. Synergy between antibody and P2-reactive T cells in experimental allergic neuritis. J Neuroimmunol 1995;57:77–84 [DOI] [PubMed] [Google Scholar]
- 24.Schneider-Hohendorf T, Schwab N, Üçeyler N, et al. CD8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2012;78:402–408 [DOI] [PubMed] [Google Scholar]
- 25.Prineas JW. Pathology of the Guillain-Barré syndrome. Ann Neurol 1981;9:6–19 [DOI] [PubMed] [Google Scholar]
- 26.Eshed Y, Feinberg K, Poliak S, et al. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 2005;47:215–229 [DOI] [PubMed] [Google Scholar]
- 27.Reimer MM, McQueen J, Searcy L, et al. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J Neurosci 2011;31:18185–18194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maier O, Baron W, Hoekstra D. Reduced raft-association of NF155 in active MS-lesions is accompanied by the disruption of the paranodal junction. Glia 2007;55:885–895 [DOI] [PubMed] [Google Scholar]
- 29.Charles P, Tait S, Faivre-Sarrailh C, et al. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol 2002;12:217–220 [DOI] [PubMed] [Google Scholar]
- 30.Sherman DL, Tait S, Melrose S, et al. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 2005;48:737–742 [DOI] [PubMed] [Google Scholar]
- 31.Nirula A, Glaser SM, Kalled SL, Taylora FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 2011;23:119–124 [DOI] [PubMed] [Google Scholar]
- 32.Rock B, Martins CR, Theofilopoulos AN, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (Fogo Selvagem). N Engl J Med 1989;320:1463–1469 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.