

Abstract
Defects in motile cilia and sperm flagella cause primary ciliary dyskinesia (PCD), characterized by chronic airway disease, infertility, and left-right laterality disturbances, usually as a result of loss of the outer dynein arms (ODAs) that power cilia/flagella beating. Here, we identify loss-of-function mutations in CCDC114 causing PCD with laterality malformations involving complex heart defects. CCDC114 is homologous to DCC2, an ODA microtubule-docking complex component of the biflagellate alga Chlamydomonas. We show that CCDC114 localizes along the entire length of human cilia and that its deficiency causes a complete absence of ciliary ODAs, resulting in immotile cilia. Thus, CCDC114 is an essential ciliary protein required for microtubular attachment of ODAs in the axoneme. Fertility is apparently not greatly affected by CCDC114 deficiency, and qPCR shows that this may explained by low transcript expression in testis compared to ciliated respiratory epithelium. One CCDC114 mutation, c.742G>A, dating back to at least the 1400s, presents an important diagnostic and therapeutic target in the isolated Dutch Volendam population.
Main Text
Motile cilia are found on the epithelial surface of the upper and lower respiratory airway systems, the brain ependyma, and fallopian tubes. Their core structure (axoneme), shared with sperm flagella, comprises nine peripheral outer doublet microtubules surrounding a central microtubular pair (“9+2” arrangement), except in the case of motile embryonic node cilia that lack the central pair (“9+0”). Microtubule-associated protein complexes are attached along its length at regularly repeating intervals, which contribute to axonemal stability and the coordinated beating movement of cilia/flagella. These include paired inner and outer dynein arms (IDA and ODA), dynein motor protein complexes that provide the ATP-driven force for self-propagating axonemal beating,1 in addition to radial spoke complexes and nexin-dynein regulatory complexes. In the biflagellate alga Chlamydomonas, a well-established model organism for human ciliary motility research because of its highly similar axonemal structure, the outer dynein arms are preassembled in the cytoplasm, transported to the axoneme, and then attached to the axonemal microtubules via outer dynein arm docking complexes.2,3
Primary ciliary dyskinesia (PCD [MIM 244400]) is a recessively inherited ciliary disorder affecting an estimated 1 per 15,000–30,000 live births,4–6 with an increased disease frequency in some isolated and inbred populations.7,8 In PCD, abnormal cilia/flagella motility leads to a number of symptoms. Ineffective mucociliary clearance caused by respiratory epithelial cilia dysmotility gives rise to chronic, destructive upper and lower airway disease manifesting with recurrent respiratory infections, chronic sinusitis, and otitis media, usually evident from the first year of life and progressing to permanent lung damage (bronchiectasis).4,9 Individuals affected by PCD are often subfertile and occasionally manifest hydrocephalus, and their left-right axis determination is randomized with about half having situs abnormalities (Kartagener syndrome [combined PCD and situs inversus] [MIM 270100]) resulting from embryonic node cilia dysfunction during development.10,11 This causes complex malformations in ∼6% of cases, often associated with congenital heart disease.12–14
PCD is genetically heterogeneous and associated with a variety of axonemal ultrastructural defects. Mutations causing PCD have been defined in 17 genes, in addition to RPGR (MIM 312610), which causes syndromic disease.15 Loss of the outer dynein arms is the most common ciliary defect observed in PCD (>65% of cases), caused by mutations in ODA components (DNAH5 [MIM 603335], DNAI1 [MIM 6043661], DNAI2 [MIM 605483], DNAL1 [MIM 602135], TXNDC3 [MIM 607421])16–20 or in genes encoding proteins involved in ODA assembly and stability causing accompanying inner dynein arm defects (LRRC50/DNAAF1 [MIM 613190], KTU/DNAAF2 [MIM 612517], DNAAF3 [MIM 614566], CCDC103 [MIM 614677], HEATR2 [MIM 614864], LRRC6 [MIM 614930]).3,21–26 An exception is DNAH11 (MIM 603339), which encodes an ODA protein but is associated with a normal ultrastructure.27,28 Mutations have also been reported in radial spoke genes (RSPH4A [MIM 612647] and RSPH9 [MIM 612648]),29 nexin-dynein regulatory complex genes (CCDC39 [MIM 613798] and CCDC40 [MIM 613799]),30,31 and central pair apparatus genes (HYDIN [MIM 610812]).32
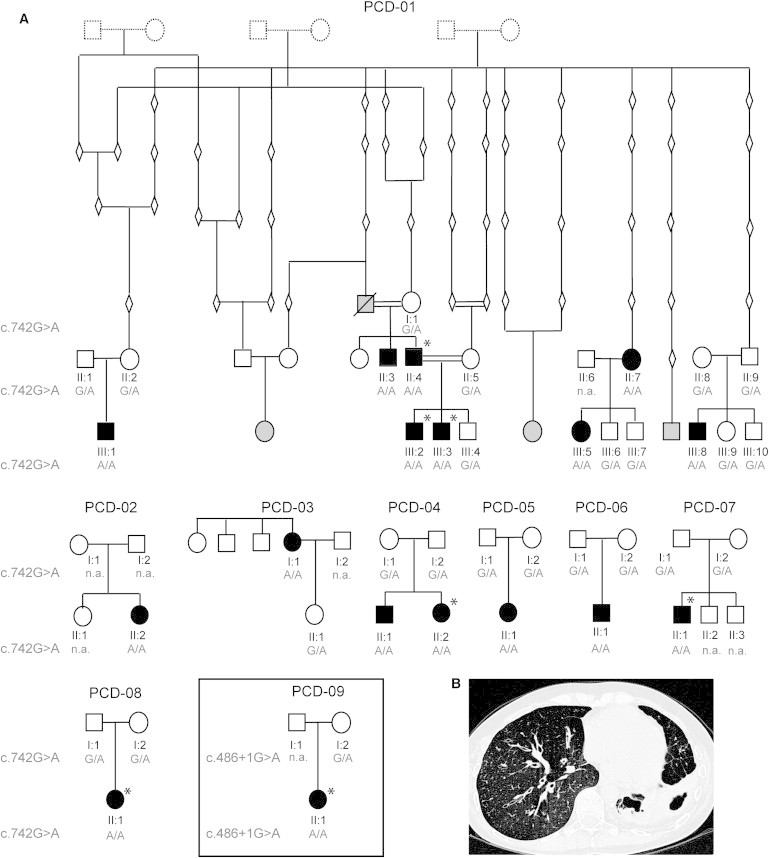
Here, we first sought to identify the genetic defect in PCD-affected families from Volendam, a fishing village in North Holland that has been genetically isolated for geographic and religious reasons since the 15th century.33 This genetic bottleneck effect has increased by 50- or 100-fold the risk of PCD to at least 1 per 400, as shown by the fact that we have recorded >56 individuals (among the current population of approximately 22,000) affected by PCD in Volendam who are registered at family physicians. The carrier frequency of the mutation in this population can thus be estimated at 1 in 10. For genetic studies, signed and informed consent was obtained from all participants according to protocols approved by the institutional ethics review boards. We used genomic DNA isolated from peripheral blood samples from a total of eight Volendam families. PCD-01 is a large multigeneration family with eight affected individuals that was shown from genealogical studies via available church records to originate from three ancestral marriages, with extensive inbreeding throughout the subsequent generations (Figure 1A). The seven other families included eight affected individuals (PCD-02 to PCD-08, Figure 1A). These families were not aware of immediate blood connections to each other, but surnames were shared among the family of PCD-01 III:8 and three of the smaller families, suggesting that historical relationships do exist.
Figure 1.
Segregation Analysis of CCDC114 Mutations
(A) Pedigree structure of Volendam families PCD-01–PCD-08 showing the segregation of the c.742G>A mutation and of UK family PCD-09 (boxed) showing segregation of the c.486+1G>A mutation. The genealogy of PCD-01 is derived from available church records. Not all ascertained individuals have been shown in the pedigrees, for reasons of space. Filled symbols indicate affected individuals, clear symbols indicate unaffected individuals, gray indicates affected individuals for whom samples could not be obtained, diamonds and dashed symbols indicate confirmed older individuals where samples are unavailable. Asterisks indicate situs abnormalities were reported.
(B) High-resolution computed tomography (HRCT) chest scan of an affected Volendam individual showing bronchiectasis of the right and left lower lobes of the lung.
All 16 individuals affected with PCD from Volendam share a similar disease course including typical PCD symptoms of early neonatal respiratory symptoms (cough, increased mucus production, and shortness of breath), pneumonia, and/or atelectasis (partial lung collapse) (Table 1). During the course of disease, these individuals variously manifested with otitis media, chronic respiratory infections, chronic cough, and pneumonia. This induces haemoptysis and requires hospital visits because of infections with a variety of pathogens, including Pseudomonas aeruginosa. Six affected individuals from Volendam (38%) have situs-related abnormalities, either complete left-right organ reversal or isolated thoracic/abdominal complications, with complex heart malformations in two cases (Table 1). Where information is available, all affected individuals had documented bronchiectasis or the early signs of it (Table 1, Figure 1B). The high disease incidence in Volendam is intriguing because infertility is often associated with PCD. It is therefore notable that five affected individuals from Volendam had children, with offspring that included affected individuals in two cases (PCD-01 II:4 and II:7) (Table 1). Fertility problems were not reported by any Volendam families. One male affected individual homozygous for the mutation with children underwent fertility testing in the past, but it showed a normal sperm count and motility; paternity was confirmed by marker analysis (Powerplex system, Promega).
Table 1.
Clinical Phenotype of the PCD Cases from the Volendam and UK Population
| ID | Cilia Dysmotile | Neonatal Symptoms | Situs | CHD | Chronic Wet Cough | Serous Otitis Media | Sinusitis | BX on CT | Chronic Abnormalities on CXR | Lobectomy | Hemoptysis | Recurrent Bacterial Presence | Recurrent Pneumonia | P. aeruginosa | Fertility Defect |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PCD-01 II:3 | n.a. | dyspnoea, feeding problems | situs solitus | N | Y | Y | Y | Y | Y | N | n.a. | Y | N | n.a. | n.a. |
| PCD-01 II:4 | n.a. | n.a. | isolated dextrocardia | N | Y | N | Y | Y | Y | N | N | Y | Y | N | N; 3 children |
| PCD-01 II:7 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | N; 3 children |
| PCD-01 III:1 | n.a. | RDS, atelectasis | situs solitus | Ya | Y | Y | Y | n.a. | n.a. | N | Y | no cultures obtained | N | no cultures obtained | n.a. |
| PCD-01 III:2 | n.a. | dyspnoea | situs inversus totalis | N | N | Y | Y | n.a. | n.a. | N | Y | no cultures obtained | N | no cultures obtained | untested, too young |
| PCD-01 III:3 | n.a. | N | situs inversus totalis | N | Y | Y | N | n.a. | n.a. | N | N | no cultures obtained | N | no cultures obtained | untested, too young |
| PCD-01 III:5 | n.a. | wet cough | situs solitus | N | Y | Y | N | Y | Y | LLL | Y | Y | Y | Y | N; 2 children |
| PCD-01 III:8 | n.a. | pneumonia | situs solitus | N | Y | Y | N | n.a. | n.a. | N | N | N | N | N | N |
| PCD-02 II:2 | Y | pneumonia | situs solitus | N | Y | Y | Y | Y | Y, signs of BX | MRL, LLL | Y | Y | Y | N | N; 2 children |
| PCD-03 I:1 | n.a. | n.a. | situs solitus | N | Y | Y | Y | Y | Y | N | N | Y | N | yes, recurrent | N; 2 children |
| PCD-04 II:1 | Y | pneumonia | situs solitus | N | Y | Y | N | n.a. | Y, signs of BX | N | N | Y | N | Y, 1× 2007, cleared | untested, too young |
| PCD-04 II:2 | Y | pneumonia | abdominal situs inversus | N | Y | Y | N | n.a. | Y, signs of BX | N | N | Y | N | N | untested, too young |
| PCD-05 II:1 | Y | wet cough | situs solitus | N | Y | Y | n.a. | n.a. | Y, signs of BX | N | N | Y | Y | Y, 1× 2010, cleared | untested, too young |
| PCD-06 II:1 | Y | rhinorrhoea | situs solitus | N | Y | Y | Y | n.a. | Y, recurrent atelectasis, wall thickening | N | Y | Y | N | Y, 1× 2006, cleared | untested, too young |
| PCD-07 II:1 | Y | sputum during feedings | situs inversus totalis with medial heart position | Yb | Y | Y | N | n.a. | Y, moderate wall thickening | N | N | Y | Y | N | untested, too young |
| PCD-08 II:1 | n.a. | RDS, atelectasis | situs inversus totalis | N | Y | N | N | n.a. | n.a. | N | N | N | N | N | untested, too young |
| PCD-09 II:1 | Y | dyspnoea | situs inversus totalis | Y | n.a. | Y | Y | Y | Y | Y | n.a. | Y | n.a. | Y | N; has children |
Abbreviations: RDS, respiratory distress syndrome; CHD, congenital heart disease; BX, bronchiectasis; CT, computed tomography; CXR, chest X-ray; LLL, left lower lobe; MRL, middle right lobe; P. aeruginosa, Pseudomonas aeruginosa; fertility defect: i.e., ectopic pregnancy, unable to conceive, ever received in vitro fertilization or intracytoplasmic sperm injection.
double outlet right ventricle, ventricular septal defect, aortic stenosis, persistent left vena cava superior, tricuspid valve insufficiency, pulmonary arterial hypertension.
atrial situs inversus with double discordance, pulmonary artery stenosis, ventricular septal defect.
We performed exome sequencing at the Wellcome Trust Sanger Institute (Cambridge, UK) as part of the UK10K project in two distantly related affected individuals from the extended Volendam pedigree: PCD-01 III:3 and PCD-01 III:8 (Figure 1). Approximately 3 μg of genomic DNA was sheared to 100–400 bp by sonication (Covaris). Fragments were subjected to Illumina paired-end DNA library preparation and enriched for target sequences (Agilent SureSelect All Exon 50 Mb kit), which were sequenced with 75 bp paired-end reads on the HiSeq platform (Illumina). Sequencing reads that failed QC were removed with the Illumina GA Pipeline, and the rest were aligned to the reference human genome (GRCh37) by BWA (v0.5.9-r16). GATK (v1.1.5) was used to realign around known indels from the 1000 Genomes project34 and recalibrate base quality scores. Alignments for a single sample were merged and duplicates marked. Variants were called per-sample by both SAMtools (v0.1.17) and GATK UnifiedGenotyper (v1.1.5), filtered on variant quality metrics separately, and the resulting data sets were merged. More than 6.60 Gb of sequence was generated per sample, such that >77% of the target exome in both cases was present at greater than 20-fold coverage (Table S1 available online).
Analysis of the exome variant profiles was performed with EVAR software tool v.0.2.2 beta. We filtered variants for novelty by comparing them to 181 UK10K non-PCD exomes and by excluding those that were present in the 1000 Genomes Project polymorphism database with a minor allele frequency >0.005.34 Because the Volendam population is isolated and the PCD-01 III:3 individual is the offspring of a consanguineous marriage, we followed a model of rare autosomal-recessive inheritance. Therefore we focused on homozygous nonsynonymous and splice-site substitutions and indels that were shared by both members of the extended pedigree. This revealed CCDC114 (RefSeq accession number NM_144577.3) as the only gene harboring low-frequency variants meeting this criteria that were compatible with recessive inheritance (Table S2).
CCDC114, located on chromosome 19q13.3, represented an excellent functional candidate, being the human gene orthologous to Chlamydomonas DCC2, which encodes an axonemal outer dynein arm microtubule-docking complex subunit.35 Furthermore, in situ hybridization images of mouse embryos generated as part of the Eurexpress project and available within the Mouse Genome Informatics pages showed a strong pattern of gene expression in motile ciliated tissues, including the nasal cavity epithelium and brain ventricles.36 Both affected Volendam individuals were homozygous for a c.742G>A substitution affecting the final G nucleotide of CCDC114 exon 7, one of the consensus splice donor bases essential to the mRNA splicing machinery. This base change is therefore predicted to cause a frameshift in the CCDC114 protein resulting from loss of the conserved donor splice site. The c.742G>A substitution is also predicted to create a missense change p.Ala248Thr, but the p.Ala248 amino acid is not well conserved across species, and the missense change was predicted to be nondeleterious to protein structure according to programs that assess nonsynonymous SNPs (Polyphen-2, SIFT). We therefore concluded that the putative splicing defect predicted by this substitution was the more likely mutation mechanism.
The c.742G>A variant was confirmed by capillary sequencing (Figure S1). Segregation analysis of the c.742G>A substitution in all available members of the PCD-01 pedigree confirmed the recessive inheritance of the variant (Figure 1A). The same variant was then confirmed to segregate with disease in all available Volendam pedigrees, PCD-02 to PCD-08 (Figure 1A). In total, all 16 affected Volendam individuals carry the c.742G>A variant as a homozygous change. This variant is reported in dbSNP v135 to be present at a very low frequency in heterozygous state in European descent controls from the NHLBI Exome Variant Server (rs147718607). The A allele is present at a frequency of less than 1 in 3,200 alleles, well below DNA polymorphism levels because the homozygous genotype would be extremely rare; this variant frequency would give a prevalence of homozygous cases of less than 1 in 40,000,000 (107).
We proceeded to sequence the CCDC114 exons and flanking intronic regions in a larger cohort of 44 individuals affected with PCD resulting from ODA and combined ODA/IDA defects (primers listed in Table S3). Signed and informed consent was obtained from all participants according to protocols approved by the institutional ethics review boards. Mutational analysis resulted in the identification of an additional homozygous splice-site variant, c.486+1G>A, in one UK family, PCD-09 (Figure S1). This substitution affects the CCDC114 exon 5 consensus splice donor site, predicted to cause a frameshift in the CCDC114 protein product. This individual’s disease was consistent with the Volendam cases, with typical features of PCD including bronchiectasis requiring lobectomy (Table 1). The affected individual PCD-09 II:1 has situs inversus with congenital heart disease and had children with no reported fertility problems. Segregation analysis in PCD-09 family members confirmed a consistent recessive pattern of inheritance (Figure 1A). The c.486+1G>A variant was absent from all the control sequence databases (dbSNP v135, 1000 Genomes Project, NHLBI Exome Variant Server).
We next used RNA isolated from ciliated cells of affected individuals and controls to assess the functional impact of the two CCDC114 splice donor site mutations. Nasal brushings or curette biopsies were obtained from PCD-02 II:2 (c.742G>A homozygote) and PCD-09 II:1 (c.486+1G>A homozygote) and healthy volunteers. For PCD-02 II:2, the RNA was isolated from the cells after culture in standard conditions37 and for PCD-09 II:1 the RNA was isolated from noncultured cells. RNA was extracted from the cells via TRIzol (Invitrogen) or the Quick RNA Miniprep Kit (Zymogen), and first-strand complementary DNA was synthesized with random nonamers (Sigma-Aldrich) or oligo-d(T)20 primer (Invitrogen) and Omniscript transcriptase (QIAGEN) or Superscript II reverse transcriptase (Invitrogen). PCR amplification was carried out with primers in exons 6 and 8 of CCDC114 in PCD-02 II:2 and in exons 2 and 5 of CCDC114 in PCD-09 II:1, in parallel to amplification of the same samples with the control housekeeping genes GAPDH (MIM 138400) and ACTB (MIM 102630), respectively. The RT-PCR primers are listed in Table S4.
A larger RT-PCR product size was amplified from the PCD-02 II:2 c.742G>A Volendam individual, compared to controls (Figure S2). Sequence analysis revealed an intronic insertion of 79 basepairs after the end of exon 7, shifting the protein reading frame and introducing a novel nonsense codon 52 residues downstream (p.Ala248Thrfs52∗). Mutation prediction software (Alamut) suggests that this aberrant message is generated through utilization of a cryptic splice donor site downstream of the mutated site, within intron 7 (Figure 2A). Furthermore, incubation with 0.1 ml of a 50 mg/ml ethanol solution of cycloheximide for 4.5 hr according to standard protocols to block protein translation indicated only a limited effect of nonsense-mediated decay on this mutation (Figure S2). RT-PCR of the PCD-09 II:1 exon 5 c.486+1G>A UK individual’s sample yielded no product despite repeated attempts to amplify, although the control had the expected RT-PCR product size; ACTB RT-PCR confirmed amplification of an equivalent product in both, suggesting that CCDC114 gene expression is specifically disrupted (not shown). The aberrant mRNA in PCD-09 II:1 could be difficult to amplify if it contains a large intronic insertion comprising all or part of the large (5,229 bp) intron 5.
Figure 2.

CCDC114 Splice-Site Mutations Causing Primary Ciliary Dyskinesia
(A) Effect of the c.742G>A Volendam mutation on splicing. The upper panels show the location of the mutation in genomic DNA sequence chromatograms and the splice-site prediction effect according to Alamut. Alamut uses the four different splice prediction software programs listed on the left. In comparison of the reference sequence from a control individual (top) against the mutant genomic DNA (bottom), the software predicts loss of the splice donor site and presence of a cryptic splice site 79 bp into the intron. The bottom panel shows the sequence of cDNA from a person who is homozygous for the mutation, isolated from ciliary cells and amplified via primers in exons 6 and 8. An intronic insertion of 79 basepairs is present in the c.742G>A individual’s cDNA, located between the mutation substitution site (green arrow) and the presumed intronic cryptic splice site (pink arrow). The sequence shows no indication of use of the regular splice donor site. The inclusion of 79 bases leads to a frameshift and a premature stop codon in exon 8 after addition of 52 novel amino acids, at the in-frame TAA codon indicated by the red box with arrow.
(B) Relative expression levels (normalized to ACTB) of CCDC114, CCDC63, and DNAH5 in mRNA from testis and cultured nasal epithelial cells from controls or from Volendam PCD-02 II:2, assessed by qPCR with a Roche Lightcycler as described in Table S5. CCDC114 is expressed at higher levels in cilia-producing cells compared to testis whereas CCDC63 is expressed highly in testis with no detectable expression in cilia-producing cells. In addition, CCDC114 and DNAH5 levels are both reduced in cilia from the Volendam affected individual compared to control. The means ± SEM from triplicate repeat experiments are shown.
(C) Location of the Volendam and UK splice-site mutations in the intron-exon structure shown above, and in a model of the CCDC114 protein shown below. Black boxes indicate coding exons, white boxes noncoding exons. The green boxes indicate coiled-coil domains as detected by Paircoil2 run with a minimum window size of 28. Homology was also detected identifying an SMC (structural maintenance of chromosomes protein) domain in CCDC114 indicated by the blue box (SMC_prok_B TIGR02168) and a putative prokaryotic phosphodiesterase domain indicated by the orange box (PRK12704).
By qPCR we were able to investigate the effect of the c.742G>A mutation on CCDC114 expression levels in the Volendam individual, PCD-02 II:2, by using a Roche Lightcycler to quantify mRNA in nasal epithelial cells cultured from PCD-02 II:2 compared to controls. The primers, probe design, and method are described in Table S5, with all gene expression normalized to the housekeeping gene ACTB. CCDC114 ciliary transcript levels were significantly reduced in these actively ciliated cells compared to that of controls, but a low transcript level was retained in the affected individual (Figure 2B). Thus, although the splicing defect severely abrogates gene expression, these data do not exclude the possibility of some remnant functional expression of CCDC114 transcripts.
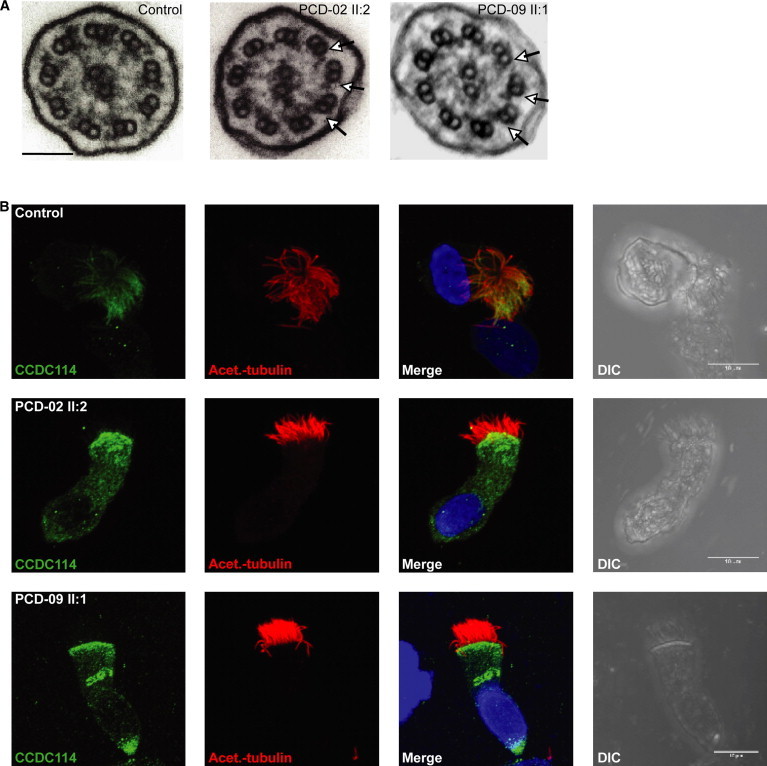
High-speed video imaging of respiratory epithelial cells demonstrated the effect of CCDC114 deficiency on ciliary beat frequency, as well as amplitude and coordination of movement (Bassler A602F-2 camera and Image Pro software, and as previously described38). Both c.742G>A (Movie S1) and c.486+1G>A (Movie S3) epithelia compared to normal controls filmed in identical conditions (Movies S2 and S4, respectively) showed abnormal ciliary motility comprising large areas of static cilia, with occasionally 1–2 cilia having a twitching or flickering movement that was stiff, slow, and ineffective for mucus transport across the epithelial surface. This is consistent with findings in other PCD individuals lacking the ODAs (with or without accompanying IDA loss).39,40 Transmission electron microscopy of respiratory cilia cross-sections showed that all CCDC114 mutant samples shared a common ciliary ultrastructural defect, a loss of the outer dynein arms (ODA) (Figure 3A). This is consistent with EM findings in the Chlamydomonas strain oda1 carrying null mutations in the CCDC114 ortholog DCC2.41 Interestingly, despite this lack of ODAs, the flagella of the Chlamydomonas oda1 strain retain some ability to beat; however, they beat slowly and without the correct effective waveform.42 This species difference when ODA components are deficient has been reported before.2
Figure 3.
CCDC114 Splice-Site Mutations Are Associated with Ciliary Axoneme Defects
(A) Transmission electron micrographs of cross-sections of respiratory epithelial cell cilia demonstrates loss of outer dynein arms in both the PCD-02 II:2 and PCD-09 II:1 individuals carrying the c.742G>A and c.486+1G>A splice donor mutations, respectively. All nine peripheral doublets showed loss and reduction of the outer dynein arms (arrows) compared to controls. Scale bar represents 100 nm.
(B) Subcellular localization of CCDC114 protein (green) in respiratory epithelial cells via a rabbit polyclonal antibody (Sigma HPA042524). In healthy individuals (top), CCDC114 is localized along the length of the axoneme of the ciliated cells, whereas in both PCD-02 II:2 and PCD-09 II:1, CCDC114 is markedly reduced (middle and bottom). Axoneme-specific anti-acetylated-α-tubulin antibody (Sigma) was used as a control to stain the entire axoneme (red). DNA (blue) was stained with DAPI (Invitrogen). Scale bars represent 10 μm.
The Chlamydomonas ortholog of CCDC114 (DCC2/ODA1) is a component of the ODA docking complex (ODA-DC) required for the assembly of ODAs onto the flagella peripheral doublet microtubules.35 In Chlamydomonas, ODA-DCs are transported and assembled onto the peripheral microtubules independently from the ODAs that attach to them, and the ODAs cannot attach in their absence.43 In oda1 DCC2-null mutant strains, ODA-DCs are not assembled onto the axoneme’s microtubules, and consequently neither are the ODAs.43 We modeled the comparative protein structure of CCDC114 to investigate the potential functional impact of the identified splice-site mutations. CCDC114, like Chlamydomonas DCC2, has three coiled-coil domains (Figure 2C). Mutations in coiled-coil domain proteins are already associated with PCD, playing an important role in axonemal organization and cilia ultrastructure.24,30,31 Coiled-coils were proposed as likely to be important for interactions between DCC2 and other docking complex subunits, and the domain between the second and third DCC2 coiled-coil domain was also proposed to participate in protein-protein interactions.35 A conserved structural maintenance of chromosomes (SMC) domain was also detected in CCDC114, similar to those identified to play a role in microtubule-based ciliary transport processes in the PCD-associated proteins CCDC39 and CCDC40.30 The c.742G>A and c.486+1G>A mutations would lead to the lack of either one or two critical coiled-coil domains, with apparently similarly deleterious consequences (Figure 2C).
To further investigate CCDC114 function, we analyzed protein localization by high-resolution immunofluorescence microscopy in ciliated epithelial cells. In controls, CCDC114 antisera decorates the full length of the cilia (Figure 3B), suggesting that its putative role in tethering of outer dynein arms is required along the entire axoneme. Two different classes of ODAs have been defined by Fliegauf et al. at the distal (DNAH5-positive, DNAH9-positive) and proximal (DNAH5-positive, DNAH9-negative) ends of cilia.40 CCDC114 appears to be a component of ODA docking complexes capable of interacting with both ODA types. In contrast, the individuals carrying c.742G>A and c.486+1G>A mutations had severely reduced levels of CCDC114 along their entire cilia (Figure 3B). By using well-established diagnostic markers of axoneme integrity developed by the Omran lab,22 we confirmed by staining with the ODA component DNAH5 that the ODAs along the cilia length are absent in c.742G>A and c.486+1G>A cells, whereas DNALI1 staining confirmed that the IDAs are present and undisturbed (Figures S3 and S4). It is not known whether human ODA-DCs and ODAs are transported by the same or different mechanisms to the axonemes, but in Chlamydomonas they can be assembled separately in the cytoplasm, and ODAs are assembled in the cytoplasm even without the ODA-DC being present.43 However, the cells deficient for CCDC114 arising from either mutation did not show any noticeable cytoplasmic accumulation of ODAs by DNAH5 staining, indicating a possible species difference in these pathways (Figures S3 and S4). We investigated the expression levels of DNAH5 mRNA by qPCR in cultured ciliated epithelial cells from the Volendam individual PCD-02 II:2 (Roche Lightcycler, Table S5), normalizing to the housekeeping gene ACTB. There was a reduction in DNAH5 levels in PCD-02 II:2 compared to control, although this was less marked than the reduced CCDC114 ciliary expression (Figure 2B). These results are in agreement with the lowered CCDC114 protein expression seen via immunofluorescence; however, the lack of DNAH5 immunofluorescence may reflect enhanced degradation of the DNAH5 protein rather than a lack of its accumulation (Figures 3 and S3).
Our data suggest that a single ancestral CCDC114 mutation, c.742G>A, underlies all Volendam PCD cases, most probably spread by genetic bottleneck founder effect. This village was founded in 1462 by 20 families who established a settlement after the nearby town of Edam dug a new exit to its sea harbor and dammed up the old exit. These families settled on the “filling dam” land or “Vollendam” and because of their isolated site, the church reformation in the late 16th century passed them without effect. The major religion remains Roman Catholic and even after their geographic isolation has lessened, their religious and social distinctions have kept the Volendam population very isolated into the modern era. A review of the unfiltered UK10K whole-exome sequence data, to derive the available SNPs across the CCDC114 locus, shows that the two distantly related individuals (PCD-01 III:3 and III:8) carrying the c.742G>A mutation share a 2 megabase haplotype (not shown). This supports the idea that the Volendam mutation was spread within this inbred population from one original founding ancestor, and its small corresponding haplotype size explains why a single locus was missed in past linkage mapping. In the large PCD-01 pedigree (Figure 1), common ancestors are found six to seven generations back, dating to the early 1800s. However, because not all the Volendam families could be connected and this small common haplotype was found to carry the mutation, presumably reduced by ancient meiotic recombination events, this suggests a more advanced age for the shared mutation than the founding of the Volendam village. According to the genetic maps of Genethon, Marshfield, and DeCode, this 2 Mb region on chromosome 19q13.33 spanning CCDC114 corresponds to a genetic distance of 3.4–4.6 centiMorgans. Te Meerman et al.44 have shown that around a new mutation, a mean haplotype sharing length of 5 cM is reached after ∼70 generations. This preceeds the founding of the village of Volendam, which occurred an estimated 22 generations ago. Consequently, most likely, two or more carriers with the CCDC114 mutation were present among the original founding families of Volendam. The finding of four heterozygous European carriers in the NHLBI Exome Variant Server supports the hypothesis that this variant arose prior to the founding of Volendam and was brought in by original settlers.
We found that fertility was not greatly affected among individuals carrying CCDC114 mutations. The reasons for this are not clear, but there may be some functional redundancy of CCDC114 in sperm. In Chlamydomonas the ODA-DC consists of the CCDC114 ortholog DCC2/ODA1 and two other proteins, DCC1/ODA3 and DLE3/ODA14.35 However, the human ODA-DC seems to be differently structured, because no definitive human homolog can be found for DCC1/ODA3 or DLE3/ODA14. Furthermore, there is a second human protein apart from CCDC114 with significant homology to DCC2: CCDC63, which is 26% identical to CCDC114. CCDC63 is also 21% identical to the Chlamydomonas ODA5 protein that is associated with the axoneme and is required for outer dynein arm assembly but independent from the ODAs and ODA-DCs.45 Whether CCDC63 plays an orthologous role to DCC2/CCDC114 or to ODA5 is not yet clear,2,46 but CCDC63 represents an excellent candidate gene for an overlapping phenotype to that associated with CCDC114 mutations. The relative levels of CCDC114 and CCDC63 proteins in the axoneme of sperm is not well understood, but available evidence from public expression databases such as Unigene suggests that the CCDC63 transcript is more highly sperm specific in its expression than CCDC114, and thus it is not impossible that CCDC114 function could be partially replaced by CCDC63 in sperm.
To test this hypothesis, we used qPCR (Table S5) on mRNA from testis (Life Technologies), the source of sperm cells (used because in sperm there is no active transcription), and from cultured nasal epithelial cells that were actively producing cilia. The nasal cells were derived both from controls and from the Volendam individual PCD-02 II:2. A high expression of CCDC63 was detected in control testis, with no detectable expression in control cilia-producing cells, even after adding 10 cycles to the qPCR, whereas CCDC114 is expressed at >100 times higher levels in cilia-producing cells compared to testis (Figure 2B). Without a testis biopsy from an affected person, we cannot exclude the possibility that affected individuals could retain some testis expression of CCDC114; however, we can conclude that the level of CCDC63 expression in control testis is comparable to CCDC114 expression in control ciliary cells and >100 times higher than CCDC114 in testis.
In summary, we report mutations within conserved CCDC114 splice donor sites affecting a total of 17 individuals with PCD, all homozygous for either c.742G>A or c.486+1G>A substitutions, conferring PCD with outer dynein arm loss, cilia immotility, and laterality defects including complex cardiac malformations. Recent large-scale studies show the importance of this phenotype, estimating that 65%–67% of PCD cases have outer dynein arm deficiencies,47,48 either alone (33%–43%) or with other structures involved. We reveal that CCDC114 has a highly conserved role in ODA microtubular attachment, with a likely role as an integral protein of the ODA-DC, the loss of which prevents ODAs from binding onto axonemal microtubules. We identified a difference in relative expression levels of CCDC114 that might suggest it has a more prominent role in cilia compared to testis. Identification of the Volendam founder mutation c.742G>A highlights CCDC114 as an important target for future therapeutic intervention, particularly in this at-risk population that has a high prevalence of PCD.
Acknowledgments
We would like to thank all the PCD families for their participation in the study, Fiona Copeland, and the U.K. PCD Family Support Group. We thank Maggie Meeks and R. Mark Gardiner for patient recruitment and their past involvement in the project. We thank Paul Griffin for electron microscopy processing. We thank all the participants of the UK10K RARE group, as listed in the Supplemental Data file, which is part of the UK10K Consortium, in particular Matthew Hurles, Saeed Al Turki, and Philip Beales. The UK10K project is funded by the Wellcome Trust (award WT091310). T.P., D.M., B.K., and G.P. were supported by the Dutch patient organization PCD Belangengroep, funded by “It Krystteam” (Friesland). P.J.S. is supported by the Wellcome Trust and British Heart Foundation. M.S. is supported by an Action Medical Research UK Clinical Training Fellowship. A.O., D.A., M.S., S.P., E.M.K.C., and H.M.M. are supported by the Milena Carvajal Pro-Kartagener Foundation, Action Medical Research UK, the Henry Smith Charity, and Newlife Foundation for Disabled Children UK.
Contributor Information
Gerard Pals, Email: g.pals@vumc.nl.
Hannah M. Mitchison, Email: h.mitchison@ucl.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
Mouse Genome Informatics, http://www.informatics.jax.org/
NHLBI Exome Variant Server/Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
STRING 9.0, http://www.string-db.org/
UK10K Consortium, http://www.uk10k.org/
References
- 1.Mitchison T.J., Mitchison H.M. Cell biology: how cilia beat. Nature. 2010;463:308–309. doi: 10.1038/463308a. [DOI] [PubMed] [Google Scholar]
- 2.Pazour G.J., Agrin N., Walker B.L., Witman G.B. Identification of predicted human outer dynein arm genes: candidates for primary ciliary dyskinesia genes. J. Med. Genet. 2006;43:62–73. doi: 10.1136/jmg.2005.033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbato A., Frischer T., Kuehni C.E., Snijders D., Azevedo I., Baktai G., Bartoloni L., Eber E., Escribano A., Haarman E. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 2009;34:1264–1276. doi: 10.1183/09031936.00176608. [DOI] [PubMed] [Google Scholar]
- 5.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M. Primary ciliary dyskinesia: current state of the art. Arch. Dis. Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Afzelius B.A. Genetics and pulmonary medicine. 6. Immotile cilia syndrome: past, present, and prospects for the future. Thorax. 1998;53:894–897. doi: 10.1136/thx.53.10.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeganathan D., Chodhari R., Meeks M., Faeroe O., Smyth D., Nielsen K., Amirav I., Luder A.S., Bisgaard H., Gardiner R.M. Loci for primary ciliary dyskinesia map to chromosome 16p12.1-12.2 and 15q13.1-15.1 in Faroe Islands and Israeli Druze genetic isolates. J. Med. Genet. 2004;41:233–240. doi: 10.1136/jmg.2003.014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Callaghan C., Chetcuti P., Moya E. High prevalence of primary ciliary dyskinesia in a British Asian population. Arch. Dis. Child. 2010;95:51–52. doi: 10.1136/adc.2009.158493. [DOI] [PubMed] [Google Scholar]
- 9.Coren M.E., Meeks M., Morrison I., Buchdahl R.M., Bush A. Primary ciliary dyskinesia: age at diagnosis and symptom history. Acta Paediatr. 2002;91:667–669. doi: 10.1080/080352502760069089. [DOI] [PubMed] [Google Scholar]
- 10.Kosaki K., Ikeda K., Miyakoshi K., Ueno M., Kosaki R., Takahashi D., Tanaka M., Torikata C., Yoshimura Y., Takahashi T. Absent inner dynein arms in a fetus with familial hydrocephalus-situs abnormality. Am. J. Med. Genet. A. 2004;129A:308–311. doi: 10.1002/ajmg.a.30177. [DOI] [PubMed] [Google Scholar]
- 11.Ibañez-Tallon I., Heintz N., Omran H. To beat or not to beat: roles of cilia in development and disease. Hum. Mol. Genet. 2003;12(Spec No 1):R27–R35. doi: 10.1093/hmg/ddg061. [DOI] [PubMed] [Google Scholar]
- 12.Bush A., Cole P., Hariri M., Mackay I., Phillips G., O’Callaghan C., Wilson R., Warner J.O. Primary ciliary dyskinesia: diagnosis and standards of care. Eur. Respir. J. 1998;12:982–988. doi: 10.1183/09031936.98.12040982. [DOI] [PubMed] [Google Scholar]
- 13.Nakhleh N., Francis R., Giese R.A., Tian X., Li Y., Zariwala M.A., Yagi H., Khalifa O., Kureshi S., Chatterjee B. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012;125:2232–2242. doi: 10.1161/CIRCULATIONAHA.111.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy M.P., Omran H., Leigh M.W., Dell S., Morgan L., Molina P.L., Robinson B.V., Minnix S.L., Olbrich H., Severin T. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 15.Moore A., Escudier E., Roger G., Tamalet A., Pelosse B., Marlin S., Clément A., Geremek M., Delaisi B., Bridoux A.M. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 2006;43:326–333. doi: 10.1136/jmg.2005.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 17.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pennarun G., Escudier E., Chapelin C., Bridoux A.M., Cacheux V., Roger G., Clément A., Goossens M., Amselem S., Duriez B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999;65:1508–1519. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V.C., Aviram M., Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duriez B., Duquesnoy P., Escudier E., Bridoux A.M., Escalier D., Rayet I., Marcos E., Vojtek A.M., Bercher J.F., Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panizzi J.R., Becker-Heck A., Castleman V.H., Al-Mutairi D.A., Liu Y., Loges N.T., Pathak N., Austin-Tse C., Sheridan E., Schmidts M. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horani A., Druley T.E., Zariwala M.A., Patel A.C., Levinson B.T., Van Arendonk L.G., Thornton K.C., Giacalone J.C., Albee A.J., Wilson K.S. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:685–693. doi: 10.1016/j.ajhg.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kott E., Duquesnoy P., Copin B., Legendre M., Dastot-Le Moal F., Montantin G., Jeanson L., Tamalet A., Papon J.-F., Siffroi J.-P. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartoloni L., Blouin J.L., Pan Y., Gehrig C., Maiti A.K., Scamuffa N., Rossier C., Jorissen M., Armengot M., Meeks M. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2002;99:10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knowles M.R., Leigh M.W., Carson J.L., Davis S.D., Dell S.D., Ferkol T.W., Olivier K.N., Sagel S.D., Rosenfeld M., Burns K.A., Genetic Disorders of Mucociliary Clearance Consortium Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax. 2012;67:433–441. doi: 10.1136/thoraxjnl-2011-200301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olbrich H., Schmidts M., Werner C., Onoufriadis A., Loges N.T., Raidt J., Banki N.F., Shoemark A., Burgoyne T., Al Turki S. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012;91:672–684. doi: 10.1016/j.ajhg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madan K., Pieters M.H., Kuyt L.P., van Asperen C.J., de Pater J.M., Hamers A.J., Gerssen-Schoorl K.B., Hustinx T.W., Breed A.S., Van Hemel J.O. Paracentric inversion inv(11)(q21q23) in The Netherlands. Hum. Genet. 1990;85:15–20. doi: 10.1007/BF00276319. [DOI] [PubMed] [Google Scholar]
- 34.The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takada S., Wilkerson C.G., Wakabayashi K., Kamiya R., Witman G.B. The outer dynein arm-docking complex: composition and characterization of a subunit (oda1) necessary for outer arm assembly. Mol. Biol. Cell. 2002;13:1015–1029. doi: 10.1091/mbc.01-04-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diez-Roux G., Banfi S., Sultan M., Geffers L., Anand S., Rozado D., Magen A., Canidio E., Pagani M., Peluso I. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011;9:e1000582. doi: 10.1371/journal.pbio.1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorissen M., Willems T., Van der Schueren B. Ciliary function analysis for the diagnosis of primary ciliary dyskinesia: advantages of ciliogenesis in culture. Acta Otolaryngol. 2000;120:291–295. doi: 10.1080/000164800750001116. [DOI] [PubMed] [Google Scholar]
- 38.Shoemark A., Ozerovitch L., Wilson R. Aetiology in adult patients with bronchiectasis. Respir. Med. 2007;101:1163–1170. doi: 10.1016/j.rmed.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Chilvers M.A., Rutman A., O’Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J. Allergy Clin. Immunol. 2003;112:518–524. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fliegauf M., Olbrich H., Horvath J., Wildhaber J.H., Zariwala M.A., Kennedy M., Knowles M.R., Omran H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005;171:1343–1349. doi: 10.1164/rccm.200411-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamiya R. Mutations at twelve independent loci result in absence of outer dynein arms in Chylamydomonas reinhardtii. J. Cell Biol. 1988;107:2253–2258. doi: 10.1083/jcb.107.6.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamiya R., Okamoto M. A mutant of Chlamydomonas reinhardtii that lacks the flagellar outer dynein arm but can swim. J. Cell Sci. 1985;74:181–191. doi: 10.1242/jcs.74.1.181. [DOI] [PubMed] [Google Scholar]
- 43.Wakabayashi K., Takada S., Witman G.B., Kamiya R. Transport and arrangement of the outer-dynein-arm docking complex in the flagella of Chlamydomonas mutants that lack outer dynein arms. Cell Motil. Cytoskeleton. 2001;48:277–286. doi: 10.1002/cm.1015. [DOI] [PubMed] [Google Scholar]
- 44.te Meerman G.J., Van der Meulen M.A. Genomic sharing surrounding alleles identical by descent: effects of genetic drift and population growth. Genet. Epidemiol. 1997;14:1125–1130. doi: 10.1002/(SICI)1098-2272(1997)14:6<1125::AID-GEPI94>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 45.Wirschell M., Pazour G., Yoda A., Hirono M., Kamiya R., Witman G.B. Oda5p, a novel axonemal protein required for assembly of the outer dynein arm and an associated adenylate kinase. Mol. Biol. Cell. 2004;15:2729–2741. doi: 10.1091/mbc.E03-11-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hom E.F., Witman G.B., Harris E.H., Dutcher S.K., Kamiya R., Mitchell D.R., Pazour G.J., Porter M.E., Sale W.S., Wirschell M. A unified taxonomy for ciliary dyneins. Cytoskeleton (Hoboken) 2011;68:555–565. doi: 10.1002/cm.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papon J.F., Coste A., Roudot-Thoraval F., Boucherat M., Roger G., Tamalet A., Vojtek A.M., Amselem S., Escudier E. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2010;35:1057–1063. doi: 10.1183/09031936.00046209. [DOI] [PubMed] [Google Scholar]
- 48.Shoemark A., Dixon M., Corrin B., Dewar A. Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J. Clin. Pathol. 2012;65:267–271. doi: 10.1136/jclinpath-2011-200415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.